

Significance
With the recent discoveries of large reserves of natural gas, the efficient utilization of one-carbon compounds for chemical synthesis would reduce the raw material cost for the petroleum-based chemical industry. Methanol is produced industrially from methane and is a feedstock chemical for the synthesis of higher carbon compounds. However, current chemical synthesis of higher carbon compounds from methanol requires high temperature and pressure. Natural biological pathways for methanol utilization are carbon and ATP inefficient. Here we constructed a synthetic biocatalytic pathway that allows the efficient conversion of methanol to higher-chain alcohols or other higher carbon compounds without carbon loss or ATP expenditure. The high carbon efficiency and favorable operating conditions are attractive for industrial applications.
Keywords: methanol metabolism, metabolic engineering, cell-free synthesis, bio-ethanol, bio-butanol
Abstract
Methanol is an important intermediate in the utilization of natural gas for synthesizing other feedstock chemicals. Typically, chemical approaches for building C–C bonds from methanol require high temperature and pressure. Biological conversion of methanol to longer carbon chain compounds is feasible; however, the natural biological pathways for methanol utilization involve carbon dioxide loss or ATP expenditure. Here we demonstrated a biocatalytic pathway, termed the methanol condensation cycle (MCC), by combining the nonoxidative glycolysis with the ribulose monophosphate pathway to convert methanol to higher-chain alcohols or other acetyl-CoA derivatives using enzymatic reactions in a carbon-conserved and ATP-independent system. We investigated the robustness of MCC and identified operational regions. We confirmed that the pathway forms a catalytic cycle through 13C-carbon labeling. With a cell-free system, we demonstrated the conversion of methanol to ethanol or n-butanol. The high carbon efficiency and low operating temperature are attractive for transforming natural gas-derived methanol to longer-chain liquid fuels and other chemical derivatives.
Methanol is industrially produced from synthetic gas-derived olefins and alkanes (1–7). These reactions typically involve high temperatures and pressures that require large capital investment (8, 9). The condensation of methanol to higher-chain alcohols such as ethanol or n-butanol is thermodynamically favorable (∆G°′ = −68 and −182 kJ/mol, respectively), but the direct condensation of methanol to higher-chain alcohols has been quite challenging. Using the Guerbet reaction, methanol can upgrade short alcohols (such as n-propanol) to longer alcohols; however, methanol cannot self-couple (10). Metal acetylides can convert methanol to isobutanol, although this process was demonstrated to be noncatalytic (11).
Nature has evolved several distinct ways to assimilate methanol to form metabolites necessary for growth. In principle, metabolites resulting from these methylotrophic pathways can be used to form higher-chain alcohols, although inherent pathway limitations prevent complete carbon conservation (Fig. S1). In the ribulose monophosphate pathway (RuMP), three formaldehydes condense to pyruvate, which is decarboxylated to form acetyl-CoA and CO2, reducing the carbon efficiency to 67%. The serine pathway requires an external supply of ATP to drive otherwise unfavorable reactions. Similarly, oxidation of methanol to CO2 followed by CO2 fixation using the Calvin–Benson–Bassham (CBB) cycle also requires additional ATP. To generate the required ATP input, extra carbon must be spent to drive oxidative phosphorylation. To our knowledge, natural methylotrophs are not capable of using the reductive acetyl-CoA pathway, which can produce acetyl-CoA without carbon loss or ATP requirement through carbon reassimilation after complete oxidation of methanol. This route is extremely oxygen sensitive and difficult to engineer due to the complex cofactors involved, and achieving carbon conservation would require that CO2 produced in methanol oxidation is completely reassimilated. Thus, in all native pathways some carbon must be lost during the production of acetyl-CoA, the precursor for n-alcohols.
Here we constructed an enzymatic cycle to achieve the catalytic condensation of methanol to higher alcohols with complete carbon conservation and ATP independence. This pathway is modified from the combination of RuMP coupled with a nonnative pathway, nonoxidative glycolysis (NOG) (12) shown in Fig. 1A. However, the combined pathway, which we call the methanol condensation cycle (MCC), can be simplified to become completely ATP independent. A thorough list of reactions is given in Table S1. The first step in MCC is the oxidation of methanol to formaldehyde. This reaction can be catalyzed by three classes of enzymes: alcohol oxidase (13), quinone-dependent methanol dehydrogenase (14), and NAD-dependent methanol dehydrogenase (15). Only the last class of enzymes provides the correct reducing equivalents, which can be used to drive the reductive portion of MCC for ethanol or n-butanol formation (Fig. S2).
Fig. 1.
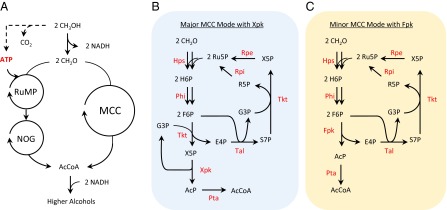
Conversion of methanol to higher n-alcohols. (A) The MCC is the combination of RuMP with NOG that bypasses ATP dependency. See Table S1 for details. (B) The major MCC mode uses the more active X5P-phosphoketolase (Xpk). (C) The minor MCC mode can achieve the same result with the less active F6P-phosphoketolase (Fpk).
The core portion of MCC is the biochemical condensation of two formaldehydes with a CoA to form acetyl-CoA and water (Fig. 1 B and C). Similar to the initial steps in the RuMP pathway, formaldehyde combines with ribulose-5 phosphate (Ru5P) to produce hexulose-6-phosphate, which is isomerized to fructose-6-phosphate (F6P). The formaldehyde assimilation is catalyzed by hexulose-6-phosphate synthase (Hps) and phosphohexulose isomerase (Phi), respectively. A similar conversion could be achieved using dihydroxyacetone synthase (12) and fructose-6-phosphate aldolase (16) as shown in Fig. S2. Once fructose-6-phosphate is formed, half of the molecules are saved for carbon rearrangement to regenerate Ru5P, whereas the rest are cleaved irreversibly by phosphoketolase (F/Xpk). This promiscuous enzyme can cleave either F6P (Fpk) or X5P (Xpk) to acetyl-phosphate and its corresponding sugar phosphate (17). Although Xpk activity is higher, both Xpk and Fpk can be used to achieve the same net conversion. F/Xpk is able to conserve ATP by phosphorylating the two carbon keto group cleaved from F6P or X5P using inorganic phosphate. The produced acetyl-phosphate can be readily converted to acetyl-CoA by the phosphate acetyltransferase (Pta). By avoiding pyruvate decarboxylation to form acetyl-CoA, no carbon is lost. The E4P produced then reacts with F6P through a series of reactions involving transaldolase (Tal), transketolase (Tkt), ribose-5 phosphate isomerase (Rpi), and ribulose 5-phosphate epimerase (Rpe), to regenerate two molecules of Ru5P to complete the cycle. MCC does not involve an essential RuMP enzyme (phosphofructokinase) and avoids three NOG enzymes: triose phosphate isomerase (Tpi), fructose-1,6-bisphosphate aldolase (Fba), and fructose-1,6-bisphosphatase (Fbp).
A unique feature of MCC is the conservation of phosphates in the cycle. The sum of all sugar phosphates remains constant throughout the catalytic cycle as long as no nonenzymatic degradation reactions occur to deplete the sum. No new sugar phosphate intermediates are generated or degraded by pathway reactions. Other pathways like CBB and RuMP do not conserve phosphate groups and instead lose these high energy bonds by the action of phosphatases. The final phase in MCC involves the reduction of acetyl-CoA to alcohols (Fig. S3). Ethanol can be directly produced from acetyl-CoA by an acylating aldehyde dehydrogenase and an alcohol dehydrogenase. The biosynthesis of n-butanol can be accomplished by a pathway previously established (18) that involves reactions similar to the one use by Clostridia.
Results
To demonstrate the feasibility of MCC, we first focused on the core portion from formaldehyde to acetate using purified enzymes (primers listed in Table S2). Each enzyme was demonstrated to have activity in individual assays. Similar to other nonlinear metabolic cycles (like TCA or CBB), an initial pool of intermediates was needed to prime the pathway. 13C-labeled formaldehyde was used to detect the carbon flow. According to MCC, double-labeled acetic acid (molecular weight = 62) was expected if 13C-formaldehyde was catalytically converted and the ribulose-5-phosphate was regenerated. Unfortunately, after buffer optimization, even this core pathway could not be demonstrated. No difference in double labeled acetate was observed with or without carbon rearrangement enzymes.
Robustness of MCC to Enzyme Variation.
The failure of these initial tests prompted us to examine the robustness of the pathway. Because the cycle enzymes involve bifurcating branches, unbalanced enzyme activities may have led to failure of the cycle. We used ensemble modeling for robustness analysis (EMRA) to determine if the cycle is robust against loss of steady state due to nonlinear effects (19). The analysis showed that the MCC cycle is most robust using intermediate levels of phosphoketolase (Fig. 2A) and will produce less acetyl-phosphate as the enzyme amount increases or decreases. At excessive levels of phosphoketolase, a kinetic trap occurs that significantly diminishes the total acetyl-phosphate produced because an accumulation of G3P or E4P occurs. This phenomenon was only predicted for phosphoketolase, whereas other enzymes were immune to this trap at high amounts (Fig. S4). To further investigate the kinetic trap, we simulated the effect of increasing phosphoketolase on conversion of 13C-formaldehyde and unlabeled R5P to acetic acid (Fig. 2B). All enzymes were modeled using Michaelis-Menten kinetics and a batch simulation was solved using a set of ordinary differential equations in Matlab. Enzyme parameters were chosen at random except for Vmax of phosphoketolase, which was varied systematically. The average of 10 parameter sets was calculated, and a maximum amount of acetic acid was predicted at intermediate levels F/Xpk.
Fig. 2.
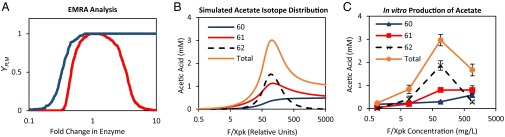
Simulation and in vitro demonstration of a kinetic trap. (A) Ensemble model robustness analysis (EMRA) of the core MCC pathway (19). YR,M is the fraction of robust models constrained to a steady state. The variation of phosphoketolase is shown in red and variation of transaldolase in blue. (B) Simulated distribution of acetic acid isotopes using 13C-formaldehyde as the substrate (C) In vitro production of acetic acid and GC-MS analysis using 13C-formaldehyde as substrate. Seven enzymes (Hps, Phi, Tkt, Tal, Rpe, Rpi, and F/Xpk) were used to produce acetyl-phosphate from 13C-formaldehyde. For GC analysis, the AcP was then converted to acetate by acetate kinase using ATP recycling by glucokinase. An acetate standard curve was established with R2 = 0.998 up to 5 mM to ensure reliable quantitation. Assays were independently run in triplicate (n = 3) with error bars representing SD.
Cell-Free Verification of Kinetic Trap.
To experimentally verify the kinetic trap, we varied the amount of phosphoketolase using 13C-formaldehyde and R5P as substrates to produce acetyl-phosphate. By using glucose phosphorylation to recycle the ADP, acetyl-phosphate was converted to acetate to enable GC-MS analysis (20). GC-MS allowed us to see the distribution of 60, 61, and 62 acetate isotopes. The maximum amount of acetate was observed when F/Xpk was around 90 mg/L (Fig. 2C). Consistent with our previous simulations, increasing the amount of phosphoketolase above this observed maximum caused a twofold decrease in total acetic acid. Single and unlabeled acetic acid both increased at higher F/Xpk values because the initial R5P could isomerize to X5P. The total acetic acid and isotope distribution from the cell-free experiment matched the trend shown in the simulation as the amount of phosphoketolase is varied.
Demonstration of the Catalytic Cycle Using 13C Tracing.
Because sugar phosphates must be added to prime the cycle, it is important to establish whether the carbon in the final product comes from formaldehyde or the initial sugar phosphate pool. Having optimized the amount of F/Xpk in the core portion of MCC, we then extended the pathway further to ethanol using an external source of reducing equivalents. Phosphate acetyltransferase (Pta from Bacillus subtilis) was used to convert acetyl-phosphate to acetyl-coA, which can be reduced by a bifunctional alcohol dehydrogenase (AdhE). However, this enzyme is known to be oxygen labile and forms long rod-like structures (21, 22), making in vitro purification difficult. Instead, the oxygen-tolerant acylating acetaldehyde dehydrogenase (PduP) from Salmonella enterica (23) can be used in a two-step reduction process via an aldehyde intermediate. We used a homolog of PduP from Bacillus methanolicus for converting acetyl-CoA to acetaldehyde and a commercial Adh (Saccharomyces cerevisiae) for producing ethanol. Instead of starting from methanol, here we used formic acid and formate dehydrogenase from Candida boidinii (24) to provide the NADH needed reduce acetyl-CoA to ethanol. This strategy allowed us to optimize the pathway from formaldehyde to ethanol independently without the complication of Mdh. To verify that the carbon rearrangement is essential, we compared the full pathway to a control without Tal. 13Carbon labeled formaldehyde was used to track the carbon flow (Fig. S5). Because there is an initial pool of pentose phosphates, some unlabeled ethanol can form by cleavage of X5P. Additionally, a single pass of formaldehyde assimilation would produce a single labeled [2-13C]-ethanol. However, if the MCC is functional, fully labeled [1,2-13C]-ethanol can be made. Using the complete cycle, the pentose phosphates can be regenerated and fully labeled ethanol can be produced. The fragmentation pattern of ethanol leads to a [M-1]+ ion that is roughly three times more abundant compared with the molecular ion [M]+. This ratio of [M-1]+ to [M]+ ions was consistently observed for all four ethanol isotopes (Fig. 3A). The full pathway produced mostly double-labeled ethanol as determined by the ratio of 48–47 ions (Fig. 3B). In this cell-free system, no 48 ion could be detected in the “No Tal” control (Fig. 3C). Some unlabeled carbon was still present because the 46 ion, which is absent in the double-labeled ethanol standard, could be detected. The presence of the 48 ion demonstrates a catalytic MCC cycle.
Fig. 3.

13C Tracing from 13C-formaldehyde and formate to ethanol. (A) The mass spectra of all four ethanol isotope standards including unlabeled ethanol, [1-13C]-ethanol, [2-13C]-ethanol, and double-labeled [1,2-13C]-ethanol. All spectra were normalized to the most abundant internal peak. Only double-labeled ethanol has a significant 48 ion (red asterisks). (B) Mass spectrum of ethanol experimentally produced from 13C-formaldehyde, unlabeled formate, and unlabeled R5P using the full MCC pathway with formate dehydrogenase. The assays were analyzed after 2 h at room temperature. Formate was oxidized to CO2 by formate dehydrogenase to provide the necessary NADH to reduce acetyl-CoA to ethanol. (C) The “No Tal” control contained the same conditions as the full pathway except for omitting transaldolase. No 48 ion was detected for the control. m/z, mass to charge ratio; R.A., relative abundance.
Continuous Production of Ethanol.
Next, we attempted to show that the production of ethanol can be continuous if there is constant supply of formaldehyde. The optimal productivity was achieved when formaldehyde was added at a rate of 6 mM CH2O/h (Fig. 4A). Although feeding R5P should not be necessary because it can theoretically be replenished, improved production was achieved when also fed at low levels (0.5 mM/h). Because MCC should have conserved metabolites, this suggested that the pool of intermediates was degraded during the course of the reaction. To identify the distribution of metabolites and possible bottlenecks, we used high-performance ion chromatography (HPIC) with pulsed amperometric detection (PAD) (25, 26) to quantify the sugar phosphates (Fig. 4B). Within the first minute, the R5P quickly rearranges to other intermediates. Between the 1st and 20th minutes, the overall pool decreases to a third of the initial point (although relative quantities remain about the same) indicating substrate degradation. G3P is known to be fairly unstable and was not detected in this system (27, 28). The decrease in pool of intermediates explains why feeding low levels of R5P was required to maintain continuous ethanol production from formaldehyde.
Fig. 4.
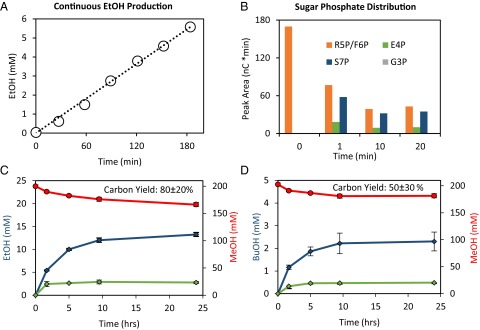
Ethanol and n-butanol production from formaldehyde or methanol using MCC. (A) Steady-state production of ethanol from formaldehyde with MCC using formic acid as electron source; 6 mM formaldehyde, 10 mM formate, and 0.5 mM R5P were added at the 0-, 1-, and 2-h points. (B) Sugar phosphate measurement over time using HPIC-PAD for the batch conversion of formaldehyde to acetyl-phosphate. Most of the carbon rearrangement occurred within the first minute. F6P and R5P standards overlap so the combine area is provided in the full assay. (C) Conversion of methanol to ethanol and (D) to n-butanol over 24 h. The productivity drops after five hours, likely due to instability of intermediates. The alcohol production assays were independently run in triplicate. Methanol consumption (for full assay only) is shown in red circles, whereas ethanol and n-butanol production is shown with diamonds. Blue colors indicates the full MCC pathway, whereas green illustrates the “No Tal” control. Assays were independently run in triplicate (n = 3), with error bars representing SD.
Production of Ethanol and n-Butanol from Methanol.
Finally, we aimed to demonstrate the conversion of methanol to ethanol and n-butanol. Because a NAD-dependent methanol dehydrogenase (EC 1.1.1.244) is only found from B. methanolicus (15, 29), our initial target was to use this unique enzyme. However, its low specific activity (<1 U/mg) and high Km (>100 mM) in the optimized buffer conditions led to trace amounts of alcohols. We purified six methanol dehydrogenases from B. methanolicus and constructed activator-insensitive mutants for each homolog. A single point mutation has been shown to make Mdh 40 times more active in the absence of the Nudix activator (30). Additionally, we bioprospected a wide variety of predicted NAD-dependent alcohol dehydrogenases from other organisms. Unfortunately, all purified enzymes demonstrated relatively poor activity toward methanol with higher specificity toward longer-chain alcohols, consistent with previous results (31). We chose the commercial alcohol dehydrogenase from S. cerevisiae, which is the same enzyme used for ethanol production. Although it does not have the highest activity toward methanol, its availability made it a more reasonable option than purifying large amounts other enzymes. The optimal production was identified by mapping a 2D parameter space (Fig. S5), varying F/Xpk and Adh (S. cerevisiae). This condition was used for a 24-h time course (Fig. 4C). We tested several methanol concentrations, and 200 mM methanol was chosen because it produced the highest carbon yield. After 5 h, the productivity decreases, and this led to a final titer of 610 mg/L (13.3 mM) ethanol from 6,200 mg/L (200 mM) methanol. The carbon yield was 80% (33.5 mM methanol consumed), exceeding the theoretical yield (66%) from the native pathway RuMP followed by EMP.
We then extended the pathway to n-butanol by including the enzymes from acetyl-CoA to n-butanol. These enzymes include thiolase (Escherichia coli), 3-hydroxybutyryl-CoA dehydrogenase (Clostridia acetobutylicium), crotonase (C. acetobutylicum), transenoyl-CoA reductase (Treponema denticola), acylating aldehyde dehydrogenase (PduP from Salmonella enterica), and alcohol dehydrogenase (S. cerevisiae). Starting with 6,200 mg/L (200 mM) of methanol, the final titer for n-butanol was 170 mg/L (2.3 mM). Because only 21.1 mM of methanol was consumed, this represents a 50% carbon yield (Fig. 4D). Remaining carbons were in ethanol (about 15%), acetate and possibly some degradation products of sugar phosphates. Here we used the same alcohol dehydrogenase for methanol oxidation, as well as ethanol and n-butanol production. Interestingly, even though this enzyme is reversible and has higher activity toward n-butanol and ethanol oxidation than methanol oxidation, the thermodynamic driving force (Fig. S6) effectively drives the reaction toward the longer chain alcohol.
Discussion
The above results demonstrate that MCC is indeed functional, although kinetics of the cycle needs to be tuned to avoid the kinetic trap. We expect that with some moderate protein engineering, the activities of Mdh, Fpk, and PduP could be improved to enable substantially higher fluxes (Table S3). Because MCC is completely redox balanced and independent of ATP, a cell-free system could be a viable application for larger-scale production after optimizing the conditions for enzyme and intermediates stability. Unlike microbial systems, cell-free conversion can achieve high theoretical yields, achieve high productivity, and are easier to control (32–34). Alternatively, MCC could be engineered into a variety of hosts because all of the enzymes are oxygen tolerant. Because of the abundance of natural gas, methanol is expected to become an abundant feedstock (35). The building of specific C–C bonds with high carbon and energy efficiency from methanol is of high interest.
Materials and Methods
For details and full list of abbreviations, see SI Text.
Chemicals and Reagents.
All reagents were purchased from Sigma-Aldrich unless otherwise stated. The following enzymes were also purchased from Sigma-Aldrich: hexokinase (S. cerevisiae), phosphoglucose isomerase (S. cerevisiae), glucose-6-phosphate dehydrogenase (S. cerevisiae), and alcohol oxidase (Pichia pastoris). Alcohol dehydrogenase (S. cerevisiae) and formate dehydrogenase (C. boindii) were purchased from Worthington Biochemical Corporation.
Simulation of MCC Robustness and 13C-Tracing Prediction.
For details, see SI Text.
Cloning and Purification MCC Enzymes.
All enzymes were cloned onto the pQE9 (Qiagen) backbone and purified on a Ni-NTA column. Large-scale purification (500 mL) typically produced about 10–50 mg of for each enzyme. For primers and details, see Table S2 and SI Text.
Individual Enzyme Assays.
For details, see SI Text.
Formaldehyde to Acetate Assay with F/Xpk Variation.
A 200-µL reaction contained 50 mM Tris⋅HCl buffer, pH = 7.5, 25 mM potassium phosphate buffer, 10 mM MgCl2, 48 µg Tkt, 76 µg Tal, 24 µg Rpe, 70 µg Hps, 15 µg Phi, 20 µg Rpi, 1 mM R5P, 0.5 mM thiamine pyrophosphate, 5 mM 13C-formaldehyde, 0.1 mM ATP, 5.4 µg Ack, 2.5 U hexokinase, and 5 mM glucose. Various amounts of F/Xpk were used. The reaction time for formaldehyde to acetate assays was 3 h, and the reactions were conducted at room temperature.
Continuous Ethanol Production.
A 200-μL reaction contained 50 mM potassium phosphate, pH 7.5, 0.2 mM NAD+, 0.2 mM CoA, 10 mM MgCl2, and 1 mM TPP. The enzyme amounts were as follows: 30 μg Hps, 10 μg Phi, 100 μg Tkt, 60 μg Tal, 10 μg Rpi, 10 μg Rpe, 15 μg F/Xpk, 10 μg Pta, 50 μg PduP (Bm), 0.01 U Adh, and 0.1 U Fdh. The initial substrates were 6 mM 13C-formaldehyde, 0.5 mM R5P, and 10 mM sodium formate. The same amount of substrates was fed at 1 and 2 h. Tal was excluded for the control. Samples were analyzed by GC-MS every 30 min for 3 h.
For Methanol to Ethanol Assays.
The buffer conditions were changed because methanol oxidation by alcohol dehydrogenase is extremely slow at pH 7.5. Diglycine buffer (pH 8.5) was chosen as a compromise between optimal activity for Mdh and F/Xpk. Because F/Xpk is slower at pH 8.5, more enzyme was added to compensate. A 550-μL reaction contained 100 mM Diglycine buffer, pH = 8.5, 1 mM dipotassium phosphate, 10 mM MgCl2, 1 mM NAD+, 1 mM thiamine pyrophosphate, 0.2 mM CoA, 200 mM 13C-methanol, and 4 mM F6P. The enzyme amounts were as follows: 55 µg Tkt, 431 µg Tal, 53 µg Rpe, 79 µg Rpi, 393 µg Hps, 431 µg Phi, 344 µg Fpk, 55 µg Pta, 297 µg PduP (Bm), and 2.75 mg Adh. A reaction was carried out at 37 °C. At each time point, 120 µL of sample was taken out and mixed with 12 µL of 8 M urea to quench the reaction. Samples were subject to filtration (Costar Centrifuge Devices, cellulose acetate, 0.22 µm; Corning) if precipitation occurs. After 2 min, 120 µL of 1 g/L 1-pentanol was added as an internal standard for GC analysis. The samples were kept at −20 °C before further analysis.
For Methanol to n-Butanol Assays.
The buffer and components of the reaction mixture were identical to the methanol to ethanol assay except for the enzymes. In a 550-µL reaction, the following enzymes were added: 55 µg Tkt, 431 µg Tal, 53 µg Rpe, 79 µg Rpi, 393 µg Hps, 431 µg Phi, 344 µg Fpk, 55 µg Pta, 743 µg AtoB, 88 µg Hbd, 38 µg Crt, 30 µg Ter, 59 µg PduP (Se), and 2.75 mg Adh (Sc). The procedure of sample preparation for GC is identical to the methanol to ethanol assay.
Analytical Methods.
Individual assays were followed spectrophotometrically using a Beckman Coulter DU 800 (Beckman Coulter) or Agilent 8453 UV-Vis spectrophotometer (Agilent Technologies). Acetic acid, ethanol, and n-butanol were analyzed by GC-flame ionization detector (FID) or GC-MS (Agilent Technologies). Details are provided in SI Text.
Sugar Phosphate Analysis.
Sugar phosphates were analyzed using a modified method from Groussac et al. (25). Details are provided in SI Text.
Supplementary Material
Acknowledgments
This work is supported by the Reducing Emissions using Methanotrophic Organisms for Transportation Energy (REMOTE) program of the Advanced Research Projects Agency-Energy (Award DE-AR0000430). This material is based on research performed in a renovated collaboratory by National Science Foundation Grant 0963183, which is an award funded under the American Recovery and Reinvestment Act of 2009.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413470111/-/DCSupplemental.
References
- 1.Fiedler E, Grossman G, Kersebohm D, Weiss G, Witte C. 2012. Methanol. Ullmann's Encyclopedia of Industrial Chemistry, eds Ott J, et al. (John Wiley & Sons, Hoboken, NJ) pp 1–27.
- 2.Summers DP, Leach S, Frese KWJ. The electrochemical reduction of aqueous carbon dioxide to methanol at molybdenym electrodes with low overpotentials. J Electroanal Chem Interfacial Electrochem. 1986;205(1-2):219–232. [Google Scholar]
- 3.Jones JH. The Cativa(TM) process for the manufacture of acetic acid. Platin Met Rev. 2000;44(3):94–105. [Google Scholar]
- 4.Chang CD. Hydrocarbons from methanol. Catal Rev Sci Eng. 1983;25(1):1–118. [Google Scholar]
- 5.Stöcker M. Methanol-to-hydrocarbons: Catalytic materials and their behavior. Microporous Mesoporous Mater. 1999;29(1-2):3–48. [Google Scholar]
- 6.Bjørgen M, et al. Methanol to gasoline over zeolite H-ZSM-5: Improved catalyst performance by treatment with NaOH. Appl Catal A Gen. 2008;345(1):43–50. [Google Scholar]
- 7.McCann DM, et al. A complete catalytic cycle for supramolecular methanol-to-olefins conversion by linking theory with experiment. Angew Chem Int Ed Engl. 2008;47(28):5179–5182. doi: 10.1002/anie.200705453. [DOI] [PubMed] [Google Scholar]
- 8.Haynes CA, Gonzalez R. Rethinking biological activation of methane and conversion to liquid fuels. Nat Chem Biol. 2014;10(5):331–339. doi: 10.1038/nchembio.1509. [DOI] [PubMed] [Google Scholar]
- 9.Conrado RJ, Gonzalez R. Chemistry. Envisioning the bioconversion of methane to liquid fuels. Science. 2014;343(6171):621–623. doi: 10.1126/science.1246929. [DOI] [PubMed] [Google Scholar]
- 10.Kozlowski JT, Davis RJ. Heterogeneous catalysts for the Guerbet coupling of alcohols. ACS Catal. 2013;3(7):1588–1600. [Google Scholar]
- 11.Fox JR, Pesa FA, Curatolo BS. Formation of higher alcohols from methanol in the presence of metal acetylides. J Catal. 1984;90(1-2):127–138. [Google Scholar]
- 12.Bogorad IW, Lin T-S, Liao JC. Synthetic non-oxidative glycolysis enables complete carbon conservation. Nature. 2013;502(7473):693–697. doi: 10.1038/nature12575. [DOI] [PubMed] [Google Scholar]
- 13.Roggenkamp R, Sahm H, Hinkelmann W, Wagner F. Alcohol oxidase and catalase in peroxisomes of methanol-grown Candida boidinii. Eur J Biochem. 1975;59(1):231–236. doi: 10.1111/j.1432-1033.1975.tb02446.x. [DOI] [PubMed] [Google Scholar]
- 14.Anthony C, Zatman LJ. The microbial oxidation of methanol. The prosthetic group of the alcohol dehydrogenase of Pseudomonas sp. M27: A new oxidoreductase prosthetic group. Biochem J. 1967;104(3):960–969. doi: 10.1042/bj1040960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arfman N, et al. Methanol metabolism in thermotolerant methylotrophic Bacillus strains involving a novel catabolic NAD-dependent methanol dehydrogenase as a key enzyme. Arch Microbiol. 1989;152(3):280–288. doi: 10.1007/BF00409664. [DOI] [PubMed] [Google Scholar]
- 16.Schurmann M, Sprenger GA. Fructose-6-phosphate aldolase is a novel class I aldolase from Escherichia coli and is related to a novel group of bacterial transaldolases. J Biol Chem. 2001;276(14):11055–11061. doi: 10.1074/jbc.M008061200. [DOI] [PubMed] [Google Scholar]
- 17.Schramm M, Klybas V, Racker E. Phosphorolytic cleavage of fructose-6-phosphate by fructose-6-phosphate phosphoketolase from Acetobacter xylinum. J Biol Chem. 1958;233(6):1283–1288. [PubMed] [Google Scholar]
- 18.Shen CR, et al. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microbiol. 2011;77(9):2905–2915. doi: 10.1128/AEM.03034-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y, Lafontaine Rivera JG, Liao JC. Ensemble Modeling for Robustness Analysis in engineering non-native metabolic pathways. Metab Eng. 2014;25:63–71. doi: 10.1016/j.ymben.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Kien CL, Chang DH, Murray RD, Ailabouni A, Kepner J. Measurement of stable isotopic enrichment of underivatized acetate by gas chromatography/mass spectrometry: application to in vivo estimation of acetate production. Biomed Environ Mass Spectrom. 1990;19(9):554–558. doi: 10.1002/bms.1200190906. [DOI] [PubMed] [Google Scholar]
- 21.Kessler D, Herth W, Knappe J. Ultrastructure and pyruvate formate-lyase radical quenching property of the multienzymic AdhE protein of Escherichia coli. J Biol Chem. 1992;267(25):18073–18079. [PubMed] [Google Scholar]
- 22.Nnyepi MR, Peng Y, Broderick JB. Inactivation of E. coli pyruvate formate-lyase: Role of AdhE and small molecules. Arch Biochem Biophys. 2007;459(1):1–9. doi: 10.1016/j.abb.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lan EI, Ro SY, Liao JC. Oxygen-tolerant coenzyme A-acylating aldehyde dehydrogenase facilitates efficient photosynthetic n-butanol biosynthesis in cyanobacteria. Energy Environ Sci. 2013;6(9):2672. [Google Scholar]
- 24.Shaked Z, Whitesides G. Enzyme-catalyzed organic synthesis: NADH regeneration by using formate dehydrogenase. J Am Chem Soc. 1980;102(23):7104–7105. [Google Scholar]
- 25.Groussac E, Ortiz M, François J. Improved protocols for quantitative determination of metabolites from biological samples using high performance ionic-exchange chromatography with conductimetric and pulsed amperometric detection. Enzyme Microb Technol. 2000;26(9-10):715–723. doi: 10.1016/s0141-0229(00)00163-0. [DOI] [PubMed] [Google Scholar]
- 26.Park C-S, Yeom S-J, Lim Y-R, Kim Y-S, Oh D-K. Substrate specificity of a recombinant ribose-5-phosphate isomerase from Streptococcus pneumoniae and its application in the production of l-lyxose and l-tagatose. World J Microbiol Biotechnol. 2010;27(4):743–750. [Google Scholar]
- 27.Richard JP. Mechanism for the formation of methylglyoxal from triosephosphates. Biochem Soc Trans. 1993;21(2):549–553. doi: 10.1042/bst0210549. [DOI] [PubMed] [Google Scholar]
- 28.Ye X, et al. Synthetic metabolic engineering-a novel, simple technology for designing a chimeric metabolic pathway. Microb Cell Fact. 2012;11:120. doi: 10.1186/1475-2859-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Vries GE, Arfman N, Terpstra P, Dijkhuizen L. Cloning, expression, and sequence analysis of the Bacillus methanolicus C1 methanol dehydrogenase gene. J Bacteriol. 1992;174(16):5346–5353. doi: 10.1128/jb.174.16.5346-5353.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hektor HJ, Kloosterman H, Dijkhuizen L. Identification of a magnesium-dependent NAD(P)(H)-binding domain in the nicotinoprotein methanol dehydrogenase from Bacillus methanolicus. J Biol Chem. 2002;277(49):46966–46973. doi: 10.1074/jbc.M207547200. [DOI] [PubMed] [Google Scholar]
- 31.Krog A, et al. Methylotrophic Bacillus methanolicus encodes two chromosomal and one plasmid born NAD+ dependent methanol dehydrogenase paralogs with different catalytic and biochemical properties. PLoS ONE. 2013;8(3):e59188. doi: 10.1371/journal.pone.0059188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y-HP, Sun J, Zhong J-J. Biofuel production by in vitro synthetic enzymatic pathway biotransformation. Curr Opin Biotechnol. 2010;21(5):663–669. doi: 10.1016/j.copbio.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y-HP, Evans BR, Mielenz JR, Hopkins RC, Adams MWW. High-yield hydrogen production from starch and water by a synthetic enzymatic pathway. PLoS ONE. 2007;2(5):e456. doi: 10.1371/journal.pone.0000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welch P, Scopes RK. Studies on cell-free metabolism: Ethanol production by a yeast glycolytic system reconstituted from purified enzymes. J Biotechnol. 1985;2(5):257–273. [Google Scholar]
- 35.Caballero A, Pérez PJ. Methane as raw material in synthetic chemistry: The final frontier. Chem Soc Rev. 2013;42(23):8809–8820. doi: 10.1039/c3cs60120j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.