

Significance
Defects in biosynthesis of the signaling lipid phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] are associated with profound neurodegeneration and early mortality in both humans and mice. However, surprisingly little is known about the functions of this lipid in cells, including neurons, where its loss has the most dramatic impact. Prompted by the striking localization of mammalian homolog of yeast vacuole segregation mutant (Vac14), part of the PI(3,5)P2 synthesis complex, to excitatory synapses, we developed new tools to measure and manipulate PI(3,5)P2 synthesis in hippocampal neurons. We find that dynamic changes in PI(3,5)P2 synthesis impose bidirectional changes on synaptic strength by regulating AMPA-type glutamate receptor trafficking and that activity-dependent regulation of this lipid is crucial for enduring forms of synaptic depression, findings that implicate PI(3,5)P2-dependent signaling as a critical synaptic regulatory pathway.
Keywords: PIKfyve, Fab1, phosphatidylinositol lipids, synaptic plasticity, Vac14
Abstract
Dynamic regulation of phosphoinositide lipids (PIPs) is crucial for diverse cellular functions, and, in neurons, PIPs regulate membrane trafficking events that control synapse function. Neurons are particularly sensitive to the levels of the low abundant PIP, phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2], because mutations in PI(3,5)P2-related genes are implicated in multiple neurological disorders, including epilepsy, severe neuropathy, and neurodegeneration. Despite the importance of PI(3,5)P2 for neural function, surprisingly little is known about this signaling lipid in neurons, or any cell type. Notably, the mammalian homolog of yeast vacuole segregation mutant (Vac14), a scaffold for the PI(3,5)P2 synthesis complex, is concentrated at excitatory synapses, suggesting a potential role for PI(3,5)P2 in controlling synapse function and/or plasticity. PI(3,5)P2 is generated from phosphatidylinositol 3-phosphate (PI3P) by the lipid kinase PI3P 5-kinase (PIKfyve). Here, we present methods to measure and control PI(3,5)P2 synthesis in hippocampal neurons and show that changes in neural activity dynamically regulate the levels of multiple PIPs, with PI(3,5)P2 being among the most dynamic. The levels of PI(3,5)P2 in neurons increased during two distinct forms of synaptic depression, and inhibition of PIKfyve activity prevented or reversed induction of synaptic weakening. Moreover, altering neuronal PI(3,5)P2 levels was sufficient to regulate synaptic strength bidirectionally, with enhanced synaptic function accompanying loss of PI(3,5)P2 and reduced synaptic strength following increased PI(3,5)P2 levels. Finally, inhibiting PI(3,5)P2 synthesis alters endocytosis and recycling of AMPA-type glutamate receptors (AMPARs), implicating PI(3,5)P2 dynamics in AMPAR trafficking. Together, these data identify PI(3,5)P2-dependent signaling as a regulatory pathway that is critical for activity-dependent changes in synapse strength.
Phosphorylated phosphoinositide lipids (PIPs) regulate diverse cellular processes (reviewed in refs. 1, 2). These seven interconvertible PIP species are synthesized and turned over by highly regulated lipid kinases and phosphatases. PIPs likely assemble complex protein machines on membrane subdomains through binding of specific downstream protein effectors, which provides tight spatial and temporal control of cellular processes. Such precision is likely critical for complex cellular functions, including regulation of synaptic strength in the CNS.
Pleiotropic defects are associated with impairments in phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] synthesis (reviewed in ref. 3). Mutations in FIG4, the gene that encodes a positive regulator of PI(3,5)P2 (4–10), are linked to several neurological disorders, including Charcot–Marie–Tooth type 4J (CMT4J) (4, 11), ALS, and primary lateral sclerosis (12), familial epilepsy with polymicrogyria (13) and Yunis–Varón syndrome (14). Little is known about how perturbations in PI(3,5)P2 synthesis cause disease.
Fig4 is a member of a protein complex that includes the phosphatidylinositol 3-phosphate (PI3P) 5-kinase (PIKfyve; Fab1 in yeast) (10, 15–18) and the scaffolding protein Vac14 (8, 9, 19–22) (Fig. S1). PIKfyve provides the sole source of PI(3,5)P2 (10, 15, 17, 23–28). The pools of PI3P that are converted to PI(3,5)P2 may derive from the class III PI 3-kinase VPS34 (29) and/or the class II PI 3-kinase C2α (30). In vivo, depletion of PIKfyve affects both PI(3,5)P2 and PI5P pools (10, 21, 24, 28). Identification of PI(3,5)P2 and PI5P protein effectors will likely reveal specific roles for each lipid.
The ability to control PI(3,5)P2 levels dynamically in mammalian cells is likely crucial for cellular function. In yeast, hyperosmotic stress transiently increases and decreases PI(3,5)P2 levels (6, 31). Similarly, in multicellular organisms, diverse external cues, such as hormones, growth factors, or neurotransmitters, may lead to dynamic regulation of PI(3,5)P2 levels. Indeed, analysis of the CMT4J disease mutation Fig4-I>T in yeast showed an impairment in stimulus-induced PI(3,5)P2 synthesis without an effect on basal PI(3,5)P2 levels (4). In cultured cortical neurons, knockdown of PIKfvye impairs the internalization of an AMPA-type glutamate receptor (AMPAR) subunit, HA-tagged GluA2 (32), and loss of Vac14 and/or Fig4 is associated with strengthened synapses (33). Together, these findings suggest that Vac14 and Fig4 regulate synapse strength via positive regulation of PIKfyve.
Here, using multiple approaches, we show that PIKfyve kinase activity negatively regulates postsynaptic strength and plays specialized roles during two distinct forms of synaptic weakening. Chronic down-regulation of PIKfyve activity using shRNA increases postsynaptic strength, whereas brief chemical inhibition of PIKfyve blocks NMDA receptor (NMDAR)-dependent long-term depression (LTD) and reverses homeostatic synaptic weakening (downscaling). Notably, we developed methods to measure the activity-dependent changes in each PIP species in cultured hippocampal neurons and identified that two low abundant PIPs, PI(3,4,5)P3 and PI(3,5)P2, are highly dynamic during LTD. Moreover, PI(3,5)P2 levels increase during homeostatic downscaling, and increasing PI(3,5)P2 via a dominant-active PIKfyve mutant (PIKfyveKYA) is sufficient to weaken postsynaptic strength. We further show that these effects on synapses derive, in part, from PI(3,5)P2-dependent trafficking of AMPARs. Together, these findings demonstrate that PIKfyve lipid kinase activity plays a critical role in regulation of synapse strength.
Results
PIKfyve Is a Regulator of Synapse Strength.
Genetic deletion of Vac14 and/or Fig4, positive regulators of PI(3,5)P2 synthesis, leads to increased basal strength of excitatory synapses (33). These results raise the possibility that Vac14 and/or Fig4 has an impact on synapse function through its known role as a regulator of PIKfyve activity. To determine whether PIKfyve is a regulator of synapse strength, we knocked-down PIKfyve in mouse hippocampal cultured neurons using lentiviral expression of PIKfyve-targeting shRNA and compared miniature excitatory postsynaptic currents (mEPSCs) of shRNA-expressing neurons with sham or nontargeting controls. One week postinfection, PIKfyve shRNA causes small vacuoles to appear, a phenotypic hallmark of reduced PI(3,5)P2 levels (16, 21, 28, 34, 35). We found that the amplitude of mEPSCs in PIKfyve shRNA-transduced neurons is significantly higher than sham or nontargeting control neurons (Fig. 1 A and B). Thus, loss of PIKfyve recapitulates the enhancement of postsynaptic strength observed following genetic deletion of Vac14 or Fig4.
Fig. 1.
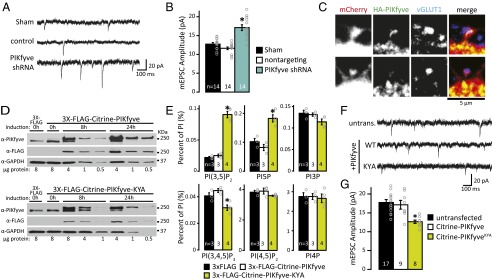
PIKfyve activity regulates synaptic responses. (A) Representative mEPSC recordings from WT mouse neurons 1 wk after lentiviral transduction with vehicle control (sham), nontargeting control shRNA, or PIKfyve shRNA. (B) Mean (±SEM) mEPSC amplitude. Knocking down PIKfyve increased mEPSC amplitude [sham: 12.73 ± 0.40 pA, control shRNA: 11.63 ± 0.35 pA, PIKfyve shRNA: 17.11 ± 0.77 pA; one-way ANOVA: F(2,39) = 29.00, P = 1.9 × 10−8]. (C) Representative confocal image showing HA-PIKfyve in proximity to excitatory synapses. Neurons were transfected with mCherry and HA-PIKfyve before fixing and staining for HA-PIKfyve and presynaptic glutamatergic terminals (vGLUT1). HA-PIKfyve is found inside (Top) and at the base (Bottom) of spines with an opposed presynaptic terminal. (D) Representative Western blots depicting doxycycline-dependent induction of 3× FLAG control, 3× FLAG-Citrine-PIKfyve (Top) or 3× FLAG-Citrine-PIKfyveKYA (Bottom) in stable cell lines (HEK 293). Cells were induced for 0, 8, or 24 h before lysis and analyzed by Western blot. Immunoblotting for PIKfyve shows two bands, consistent with detection of endogenous PIKfyve and 3× FLAG-Citrine-PIKfyve or 3× FLAG-Citrine-PIKfyveKYA. (E) Mean (±SEM) PIP levels relative to total PI. Induction of 3× FLAG-Citrine-PIKfyveKYA for 24 h increases PI(3,5)P2 and PI5P levels [PI(3,5)P2: 3× FLAG: 0.022 ± 0.001%, 3× FLAG-Citrine-PIKfyve: 0.025 ± 0.002%, 3× FLAG-Citrine-PIKfyveKYA: 0.093 ± 0.005%; one-way ANOVA: F(2,7) = 105.44, P = 5.9 × 10−6]. [PI5P: 3× FLAG: 0.102 ± 0.011%, 3× FLAG-Citrine-PIKfyve: 0.082 ± 0.011%, 3× FLAG-Citrine-PIKfyveKYA: 0.185 ± 0.012%; one-way ANOVA: F(2,7) = 21.63, P = 0.001]. (F) Representative recordings from cultured rat hippocampal neurons [21 days in vitro (DIV)] transfected at DIV14 with Citrine-PIKfyve or Citrine-PIKfyveKYA. untrans., untransfected. (G) Mean (±SEM) mEPSC amplitude of untransfected and transfected neurons expressing Citrine-PIKfyve or Citrine-PIKfyveKYA. PIKfyveKYA expression decreased mEPSC amplitude [untransfected: 17.75 ± 0.78 pA, Citrine-PIKfyve: 17.14 ± 1.16 pA, Citrine-PIKfyveKYA: 12.65 ± 0.43 pA; one-way ANOVA: F(2,31) = 17.32, P = 0.0005]. *P < 0.05.
The finding that Vac14 is concentrated at excitatory synapses (33) suggests that PIKfyve is localized to excitatory synapses as well. Current antibodies are not suitable for immunofluorescence of endogenous PIKfyve. To test PIKfyve localization directly, neurons were cotransfected with HA-tagged PIKfyve and mCherry. Neurons were then stained for HA as well as vesicular glutamate transporter 1 (vGLUT1), the latter of which is a marker for excitatory presynaptic terminals. HA-PIKfyve puncta were found throughout the cell body and dendrites. To quantify the number of synapses with HA-PIKfyve nearby, the number of vGLUT1 puncta that overlap with HA-PIKfyve was counted in the first 55-μm segment of the dendrite. HA-PIKfyve puncta were detected at 39.6 ± 4.7% of the putative synapses (Fig. 1C and Fig. S2). Thus, PIKfyve is well positioned to regulate excitatory synapse strength.
Increasing PIKfyve Activity Causes Synaptic Depression.
The involvement of PIKfyve and other components of the PI(3,5)P2 synthesis complex in regulating synapse strength suggests that PI(3,5)P2 functions as a negative regulator of synaptic efficacy. Hence, we tested whether increasing PIKfyve activity has the opposite effect on synapse strength. A common approach for increasing the levels of specific metabolites in cells is to overexpress the enzyme required for their synthesis. In yeast, overexpression of Fab1, the homolog of mammalian PIKfyve, is insufficient to increase PI(3,5)P2 levels (15). To test the effect of overexpressing PIKfyve in mammalian cells, we generated two doxycycline-inducible stable cell lines using the Flp-In T-Rex-293 System (Invitrogen) with 3× FLAG-Citrine-PIKfyve or 3× FLAG for a control. This strategy was necessary to titrate the amount of expression. By Western blot, induction for 24 h yielded a robust increase in expression of 3× FLAG-Citrine-PIKfyve to ∼16-fold above the level of endogenous PIKfyve (Fig. 1D). Despite this strong overexpression, PI(3,5)P2 levels did not change (Fig. 1E). Thus, similar to overexpression of yeast Fab1, overexpression of PIKfyve in mammalian cells is insufficient to increase PI(3,5)P2 levels.
Dominant mutations in yeast Fab1 have been identified that cause a several-fold elevation in PI(3,5)P2 (6). Two of these mutants, Fab1-E1822K,N1832Y and Fab1-E1822V,F1833L,T2250, are mutated in conserved residues (Fig. S3), which suggests that dominant-active mammalian PIKfyve mutants can be generated. Indeed, we found that induction of PIKfyveKYA (E1620K, N1630Y, S2068A) mutant protein to levels fourfold above endogenous PIKfyve increased PI(3,5)P2 approximately fourfold (Fig. 1E). PIKfyve is required for most of the PI5P pools as well (10, 21, 24, 27), likely because PI(3,5)P2 is the major precursor to PI5P (10). We found a twofold increase in PI5P levels in PIKfyveKYA-expressing cells (Fig. 1E), which is consistent with a precursor–product relationship. Unexpectedly, we found that the levels of PI(3,4,5)P3 are reduced by ∼25% in PIKfyveKYA-expressing cells (Fig. 1E). Although there is currently no known connection between PI(3,5)P2 synthesis and PI(3,4,5)P3, potential links have not been tested directly.
To test the impact of increasing PIKfyve activity on synapse function, postnatal rat cultured hippocampal neurons were transfected with Citrine-PIKfyveKYA or WT Citrine-PIKfyve, and the amplitude of mEPSCs in transfected neurons was compared with untransfected neighbors. Whereas overexpression of Citrine-PIKfyve in neurons does not have an impact on synapse function (Fig. 1 F and G), the amplitude of mEPSCs recorded in Citrine-PIKfyveKYA–expressing neurons is significantly reduced (Fig. 1 F and G). These results suggest that elevated levels of PI(3,5)P2 negatively regulate postsynaptic strength. Although a number of neurological diseases have been linked to decreased PI(3,5)P2 synthesis and synaptic dysfunction, it is possible that mutations that activate PIKfyve may also contribute.
Neural Activity Dynamically Regulates PIP Levels.
PIPs play essential roles in cellular events important for Hebbian and homeostatic forms of synaptic plasticity. For example, PI(4,5)P2 regulates endocytosis, exocytosis, actin dynamics, and ion channel function (36–43). PI(3,4,5)P3 clusters membrane proteins both presynaptically (44) and postsynaptically (45), and postsynaptic strength is diminished by increased turnover of PI(3,4,5)P3 by the lipid 3-phosphatase, PTEN (46). These highly dynamic processes strongly imply that PIP metabolism is regulated by neural activity.
Detection of PIP species in cultured neurons.
To assess activity-dependent changes in PIP levels in neurons, we adapted methods designed for fibroblasts to cultured rat hippocampal neurons. Neuronal cultures were metabolically labeled with myo-[2-3H]-inositol for 24 h. Cells were precipitated by perchloric acid, and the resultant precipitate was treated with weak base to deacylate lipids. Following deacylation, water-soluble glycerol-inositol polyphosphates were separated by chromatography on an ion exchange column. In cultured hippocampal neurons at 21 days in vitro, peaks representing all seven PIP species were observed, including PI(3,5)P2 and PI5P (Table S1). We do not report the levels of PI(3,4)P2, which were observed in some samples but were often below the level of detection (<0.01% of PI). Compared with mouse embryonic fibroblasts (10), neurons have fivefold more PI4P, threefold more PI(4,5)P2, slightly more PI(3,4,5)P3, and half the levels of PI(3,5)P2 (Table S1). The high levels of PI4P and PI(4,5)P2 are likely due to the extensive plasma membrane, endoplasmic reticulum (ER), and abundance of somatic and dendritic Golgi in neurons.
Induction of chemical LTD dynamically regulates PIP levels.
To examine how PIP levels change during synaptic plasticity, we measured PI(3,5)P2 and other PIP levels during induction of NMDAR-dependent chemical LTD (cLTD) using an established NMDAR-cLTD induction protocol (47, 48). We analyzed five time points during the stimulus (30 s, 1 min, 2 min, 3 min, and 5 min), and the levels of PIPs were compared with sister cultures without stimulation (0 min) (Fig. 2A). We found dynamic changes in the levels of multiple PIPs (Fig. 2B). Two of the most abundant PIPs, PI4P and PI(4,5)P2, as well as PI5P, did not show consistent changes during cLTD stimulation. On the other hand, we found that the cLTD stimulus induced significant changes in three other PIPs. PI(3,4,5)P3 levels increased ∼50% within 30 s. In the first 3 min, PI(3,5)P2 rose rapidly and then returned to baseline levels within 5 min. Consistent with a transient elevation in PI(3,5)P2 levels, PI3P levels were initially stable and then dropped by ∼20%. Note that although the cLTD stimulus is associated with a rapid increase in both PI(3,4,5)P3 and PI(3,5)P2, we found that expression of Citrine-PIKfyveKYA, which also causes synaptic weakening, is associated with a reduction in PI(3,4,5)P3 and elevation in PI(3,5,)P2 levels (Fig. 1E). Thus, induction of cLTD involves dynamic changes in multiple PIPs. The transient increase in PI(3,5)P2 during induction of cLTD suggests that PIKfyve activity may be increased during the stimulus.
Fig. 2.
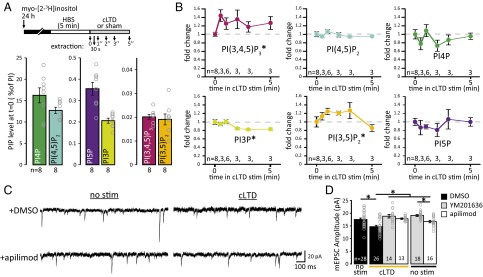
PIKfyve activity is required for NMDAR-dependent cLTD. (A) Schematic of experimental design. Samples were collected during cLTD induction (20 μM NMDA, 1 μM glycine, 0.2 mM Mg2+) at 30 s, 1 min, 2 min, 3 min, or 5 min. Mean (±SEM) PIP levels at time point 0. (B) Mean (±SEM) levels for each PIP normalized to 0 min. The induction of cLTD evokes a dynamic change in the levels of three PIPs. The distributions of each PIP level during the stimulation were compared with baseline values (0 min) using a two-sample Kolmogorov–Smirnov test (K-S test). PI(3,5)P2 levels transiently rise between 30 s and 3 min (K-S test: D = 0.55, P = 0.026). PI(3,4,5)P3 levels remain elevated during cLTD stimulus (K-S test: D = 0.65, P = 0.0045). The levels of PI3P decrease after 2 min of the cLTD stimulus (K-S test: D = 0.75, P = 0.0037). For PI4P, PI(4,5)P2, and PI5P, levels during the stimulus are from the same continuous distribution as at baseline. (C) Representative recording of mEPSCs. After cLTD or sham stimulation, neurons were incubated with reserved conditioned media without PIKfyve inhibitors for 30 min at 37 °C. (D) Mean (±SEM) mEPSC amplitude after stimulation in the presence or absence of 1 μM apilimod or 2 μM YM201636. The amplitude decreased after induction of cLTD, which was blocked by incubation with 1 μM apilimod or 2 μM YM201636 [control: 17.52 ± 0.55 pA, cLTD: 14.72 ± 0.53 pA, cLTD + YM201636: 18.83 ± 0.71 pA, cLTD + apilimod: 17.86 ± 0.30 pA; 7.5 min of YM201636: 19.11 ± 0.37 pA, 7.5 min of apilimod: 16.72 ± 0.48 pA; one-way ANOVA: F(5,109) = 9.77, P = 9.8 × 10−8]. *P < 0.05. stim, stimulus.
PIKfyve inhibition blocks induction of cLTD.
To test for a causal role for PIKfyve activity during induction of cLTD, we acutely inhibited PIKfyve kinase activity. One of the most widely used PIKfyve inhibitors is YM201636 (35). Recently, the small molecule apilimod was identified as a potent PIKfyve inhibitor (49). The potential for an off-target effect of YM201636 on Akt signaling (50) prompted us to examine the levels of PI(3,4,5)P3, a positive regulator of Akt, following PIKfyve inhibition with 1.6 μM YM201636 (Fig. S4 A and B). We also analyzed the change in levels of each PIP in response to 1 μM apilimod (Fig. S4 C and D). Note that a time course of changes in PIP levels following YM201636 inhibition of PIKfyve was reported previously (10), but the PI(3,4,5)P3 levels were not included. Consistent with both molecules acting as PIKfyve inhibitors, the level of the PIKfyve product PI(3,5)P2 rapidly decreases within minutes of treatment with YM201636 or apilimod (Fig. S4 B and D). Due to the interconvertibility of PIPs, in addition to a reduction in PI(3,5)P2, PIKfyve inhibition is expected to result in a concomitant accumulation of the PIKfyve substrate PI3P and reduction in PI5P, a product of PI(3,5)P2 (Fig. S4 B and D). Notably, 1.6 μM YM201636, but not 1 μM apilimod, produced a 50% decrease in the level of PI(3,4,5)P3 within 5 min of treatment.
To determine whether PIKfyve activity is required during cLTD, neurons were treated with 2 μM YM201636 or 1 μM apilimod for 2.5 min before and during the 5-min cLTD stimulus (7.5 min total). Neurons were then returned to media without PIKfyve inhibitors for an additional 30 min before electrophysiological recordings. As expected, the cLTD stimulus decreases mEPSC amplitude in the absence of PIKfyve inhibitors (Fig. 2 C and D). By contrast, the presence of apilimod or YM201636 during the 5-min cLTD stimulus prevented induction of enduring synaptic depression (Fig. 2 C and D). Thus, during induction of cLTD, PIKfyve activity is necessary for sustained synaptic weakening.
PIP levels are regulated by prolonged changes in neural activity.
To identify PIP species that dynamically respond to changes in network activity, we compared PIP levels in spontaneously active neuronal cultures with neuronal cultures treated with either 2 μM TTX for 10 min (to block action potentials) or 50 μM bicuculline for 1 min (to enhance firing by removing inhibitory tone) (Fig. 3A). Comparison of each PIP level with the levels in control samples from basally active neural cultures demonstrates that these acute manipulations in activity do not have a significant impact on PIP levels.
Fig. 3.

PI(3,5)P2 synthesis accompanies homeostatic synaptic weakening following prolonged hyperactivity. (A) Schematic of experimental design. Neurons were metabolically labeled with [3H]-inositol for 24 h. The media were replaced with Hepes-buffered saline (HBS). After 5 min, HBS was replaced with HBS + 2 μM TTX for 10 min or 50 μM bicuculline for 1 min and lipids were extracted. For each experiment, unstimulated controls (5 min of HBS) were collected and the mean (±SEM) PIP level after incubation with TTX or bicuculline is expressed as a percentage of the control (5 min of HBS). (B) Schematic of experimental design. Neurons were metabolically labeled with [3H]-inositol for 24 h in the presence of DMSO control, 2 μM TTX, or 50 μM bicuculline. The mean (±SEM) percent change in PIP levels relative to control from neurons incubated with 2 μM TTX (n = 5) or 50 μM bicuculline (n = 4) for 24 h is shown. The level of PI4P decreased after 24 h of TTX [percent change: −21.26 ± 1.77%; one-way ANOVA: F(2,11) = 15.2, P = 0.0007]. The level of PI(4,5)P2 decreased after 24 h of TTX [percent change: −15.34 ± 2.45%; one-way ANOVA: F(2,11) = 6.54, P = 0.014]. The level of PI(3,5)P2 increased after 24 h of bicuculline [percent change: 60.49 ± 16.05%; one-way ANOVA, F(2,11) = 7.13, P = 0.01]. (C) Representative images of neurons expressing the PI(3,5)P2 reporter, mCherry-ML1N*2, and GFP. (D) Mean (±SEM) ratio of dendritic mCherry-ML1N*2 fluorescence to soma mCherry-ML1N*2 fluorescence. The dendritic-to-soma ratio of mCherry-ML1N*2 intensity increased after 24 h of bicuculline [control: 1.0 ± 0.04, 24 h of bicuculline: 1.30 ± 0.06; t test: t(63) = 4.12, P = 0.0001]. *P < 0.05.
Chronic suppression or elevation in network activity is known to engage homeostatic synaptic control mechanisms that strengthen or weaken synapses, respectively. This ability to adapt to persistent changes in network activity through compensatory changes in synapse function is important for neurons to maintain levels of activity in a stable range (51). Notably, the levels of PI(3,5)P2 significantly increased after 24 h of bicuculline-induced hyperactivation (Fig. 3B). In contrast, following 24 h of activity suppression with TTX, the levels of both PI4P and PI(4,5)P2 decreased (Fig. 3B).
To test for changes in PI(3,5)P2 independently and determine whether there are specific subcellular locations where its synthesis occurs during homeostatic downscaling, we monitored a fluorescent probe for PI(3,5)P2 under periods of normal or hyperactive network activity. The reporter (mCherry-ML1N*2) is mCherry fused to a tandem duplication of the PI(3,5)P2 binding domain of the cytosolic N-terminal polybasic domain (ML1N) of transient receptor potential mucolipin1 (TRPML1) and has been validated previously (52). We found that relative to conditions of basal neural activity, chronic hyperactivity induced by bicuculline produced a robust increase in dendritic reporter intensity (Fig. 3 C and D). To normalize for differences in expression levels, we analyzed the average intensity of mCherry-ML1N*2 in the dendrite relative to the soma and found network hyperactivation increased this ratio. These data are consistent with the increased PI(3,5)P2 levels detected by HPLC following homeostatic synaptic weakening (Fig. 3B) and suggest that a component of the rise in PI(3,5)P2 level is due to new synthesis in dendrites during synaptic depression. The changes in PI4P and PI(4,5)P2 are potentially interesting as well, and analysis of these species in future studies may shed light on mechanisms governing homeostatic synaptic strengthening.
PIKfyve Activity Is Required for Homeostatic Synaptic Weakening.
The observations that PI(3,5)P2 levels are elevated after prolonged hyperactivation with bicuculline and, conversely, that basal synapse strength is elevated in PI(3,5)P2-deficient neurons (33) suggest that PIKfyve activity may be important for homeostatic synaptic weakening. We first tested whether Vac14−/− on a C57BL/6J background shares the elevation in synapse strength observed in a “mixed” background (strain 129 and C57BL/6J) (33) and found that the amplitude of mEPSCs is increased in Vac14−/− relative to WT neurons in a C57BL/6J background (Fig. 4 A and B). Then, we induced prolonged increases or decreases in the level of neural activity and monitored synaptic strength by measuring the amplitude of mEPSCs. To test whether homeostatic plasticity was altered in Vac14−/− neurons, we incubated neurons with 2 μM TTX or 50 μM bicuculline for 24 h. As expected, WT neurons exhibited an increase in mEPSC amplitude following chronic activity deprivation with TTX. Vac14−/− neurons, despite larger basal mEPSC amplitudes, exhibited homeostatic synaptic strengthening that was indistinguishable from WT neurons (Fig. 4 A and C). On the other hand, whereas chronically increasing network activity induced a significant decrease in mEPSC amplitude in WT neurons, Vac14−/− neurons failed to downscale synaptic strength homeostatically (Fig. 4 A and C). Together, these data suggest that Vac14 is required for homeostatic downscaling but not for homeostatic upscaling, which fits with multiple studies that suggest these two processes are controlled by independent mechanisms (reviewed in refs. 53–58).
Fig. 4.

PIKfyve activity is required for synaptic depression. (A) Representative example recordings from WT and Vac14−/− mouse hippocampal cultured neurons treated for 24 h with vehicle, 2 μM TTX, or 50 μM bicuculline (Bic). (B) Mean (±SEM) mEPSC amplitude. The amplitude of Vac14−/− mEPSCs is increased relative to WT [WT: 15.16 ± 0.39 pA, Vac14−/−: 19.12 ± 0.88 pA; t(43) = 31.04, P = 4.45 × 10−31]. (C) Cumulative distribution frequency of mEPSC amplitude from WT (Left) and Vac14−/− (Right) neurons treated for 24 h with vehicle control (black line), 2 μM TTX (purple line), or 50 μM Bic (green line). In WT neurons, the distribution of mEPSC amplitude is left-shifted following 24 h of Bic and right-shifted following 24 h of TTX. Although Vac14−/− neurons normally scale-up mEPSC amplitude in response to 24 h of TTX, they fail to scale-down. All six distributions were compared using the one-way Kruskal–Wallis ANOVA [χ2(5, n = 4,016) = 411.56, P = 9.6 × 10−87] and the results of Tukey–Kramer post hoc test reported here: WT + Bic is different from every group. WT + TTX is different from WT. Vac14−/− + TTX is different from every group. WT control is different from Vac14−/− control. (Insets) Mean (±SEM) percent change in mEPSC amplitude relative to unstimulated controls of the corresponding genotype. In WT neurons, 24 h of treatment with TTX increases mEPSC amplitude and 24 h of treatment with Bic decreases mEPSC amplitude [WT, percent change from control (n = 24): +TTX: 28.79 ± 6.99% (n = 7), +Bic: −11.11 ± 2.97% (n = 23); one-way ANOVA: F(2,51) = 21.41, P = 1.8 × 10−7]. In Vac14−/− neurons, 24 h of treatment with TTX increases mEPSC amplitude but 24 h of treatment with Bic does not decrease mEPSC amplitude [Vac14−/−, percent change from control (n = 20): +TTX: 24.28 ± 8.99% (n = 7), +Bic: 13.72 ± 6.03% (n = 20); one-way ANOVA: F(2,45) = 4.06, P = 0.02]. *P < 0.05.
Acute PIKfyve Inhibition Reverses Established Homeostatic Changes in Synaptic Strength.
Unlike cLTD induction, homeostatic downscaling is associated with a persistent increase in PI(3,5)P2 levels (Fig. 3 B and D) during a time at which compensatory synaptic adaptations have already been established (59–63). Given that Vac14−/− neurons fail to express homeostatic synaptic downscaling in response to hyperexcitation (Fig. 4C) and that elevation of PIKfyve activity leads to reductions in excitatory synaptic strength (Fig. 1 F and G), we hypothesized that the changes in PI(3,5)P2 following chronic hyperactivity play a direct role in maintaining synaptic depression. In agreement with this hypothesis, we found that knocking down PIKfyve using shRNA similarly prevents the weakening of synapses following 24 h of treatment with bicuculline (Fig. 5 A and B). If PIKfyve activity is required for maintaining synaptic depression, then acute blockade of PI(3,5)P2 synthesis may be sufficient to reverse homeostatic downscaling rapidly after it has been established. To test this prediction, we acutely blocked PI(3,5)P2 production with 1 μM apilimod or 2 μM YM201636 in cultured rat hippocampal neurons for 1 h after a 23-h period of basal (control) or elevated (bicuculline) activity. Under control conditions, neither 1 h of apilimod nor 1 h of YM201636 significantly increased mEPSC amplitude, although there was a trend toward an increase with apilimod (Fig. S5). Thus, brief inhibition of PIKfyve activity may predispose synapses toward an increase in function. Notably, when neurons were treated for the previous 23 h with bicuculline to induce homeostatic synaptic weakening, PIKfyve inhibition by either apilimod or YM201636 completely reversed downscaling of mEPSC amplitude within 1 h (Fig. 5 C and D). Together, these data suggest that the maintenance of synaptic adaptations induced by network hyperactivity requires PIKfyve activity.
Fig. 5.

Loss of PI(3,5)P2 synthesis reverses homeostatic synaptic depression. (A) Representative recordings of mEPSCs from mouse cultured hippocampal neurons infected with scrambled or PIKfyve shRNA for 1 wk and then treated with 50 μM Bic for 24 h. (B) Cumulative distribution frequency of mEPSC amplitude is scaled-down after 24 h of Bic treatment in scrambled (scram) but not PIKfyve shRNA-expressing neurons [one-way Kruskal–Wallis ANOVA: χ2(3, n = 2,486) = 331.7963, P = 1.3 × 10−71; Tukey–Kramer post hoc results: scrambled + 24 h of Bic is different from each group, and PIKfyve-shRNA and PIKfyve-shRNA + 24 h of Bic are different from scrambled but not different from each other]. (Inset) Mean percent change (±SEM) in mEPSC amplitude relative to untreated neurons. Treatment with 50 μM Bic for 24 h decreases mEPSC amplitude in scrambled but not PIKfyve-shRNA–expressing neurons [scrambled + Bic: −21.50 ± 4.57% change from scrambled, PIKfyve-shRNA + Bic: −4.33 ± 6.11% change from PIKfyve-shRNA; one-way ANOVA, F(3,79) = 5.17, P = 0.0026]. (C) Representative recordings of mEPSCs from rat cultured hippocampal neurons treated or untreated with 50 μM Bic for 24 h. After 23 h, the PIKfyve inhibitor apilimod or YM201636 was added for 1 h. (D) Mean (±SEM) mEPSC amplitude. Treatment with 50 μM Bic for 24 h decreases mEPSC amplitude. Application of 1 μM apilimod or 2 μM YM201636 for 1 h does not affect the amplitude of mEPSCs in control neurons not treated with Bic. Application of 1 μM apilimod or 2 μM YM201636 for 1 h in Bic-treated neurons restores mEPSC amplitude to control levels [control + DMSO: 16.15 ± 0.49 pA, 24 h of Bic + DMSO: 12.58 ± 0.50 pA, 24 h of Bic + YM201636: 14.80 ± 0.41 pA, 24 h of Bic + apilimod: 17.53 ± 0.73 pA; one-way ANOVA: F(3,51) = 13.9, P = 9.6 × 10−7]. (E) Representative images of sGluA2 and PSD-95 staining with or without 50 μM Bic for 24 h. Neurons were live-labeled with an aminoterminal antibody for the GluA2 subunit. After permeabilization, the cells were stained for PSD-95. Intensity is presented in the “fire” lookup table color scheme. (F) Mean (±SEM) sGluA2 intensity quantified in the first 50 μm of dendrite relative to control. Treatment with Bic for 24 h decreased the abundance of sGluA2, which was restored to control levels within 1 h by PIKfyve inhibition with either 1 μM apilimod or 2 μM YM201636 [control: 100 ± 2.29%, 24 h of Bic: 83.71 ± 1.97, 24 h of Bic + 1 h of YM201636: 95.41 ± 2.27%, 24 h of Bic + 1 h of apilimod: 96.94 ± 2.68%, one-way ANOVA: F(3,224) = 11.16, P = 7.5 × 10−7]. *P < 0.05.
AMPARs are the predominant mediator of fast neurotransmission in the CNS, and AMPAR trafficking is critical for many enduring forms of synaptic plasticity (64, 65). Although no direct interaction has been demonstrated between PI(3,5)P2 and AMPARs, multiple groups have found that AMPAR trafficking is sensitive to PIKfyve activity (32, 33, 66). Because bidirectional regulation of AMPAR surface expression is a signature of homeostatic synaptic plasticity, where prolonged hyperactivity reduces surface levels of AMPAR subunits GluA1 and GluA2 (59, 63, 67–70), we tested whether PIKfyve inhibition following induction of homeostatic synaptic depression has an impact on surface AMPAR levels. Surface GluA2 (sGluA2) subunits were detected with an amino-terminal antibody under nonpermeabilization conditions, followed by permeabilization and detection of the excitatory synaptic scaffolding molecule postsynaptic density 95 (PSD-95). Consistent with previous reports, sGluA2 expression is significantly reduced during homeostatic downscaling induced by 24 h of bicuculline treatment (Fig. 5 E and F). We also found that this period of hyperactivity did not have an impact on the total levels of PSD-95 (Fig. S6 A and B), consistent with previous results from hippocampal neurons (63, but see ref. 71). Notably, sGluA2 expression in bicuculline-treated neurons was restored to baseline levels by PIKfyve inhibition for 1 h with either YM201636 or apilimod (Fig. 5 E and F). Although 1 h of apilimod treatment reversed sGluA2 expression during network hyperactivation (Fig. 5 E and F), the same course of apilimod treatment did not alter sGluA2 expression under basal conditions (Fig. S6 C and D). However, 1 h of YM201636 treatment was effective in increasing sGluA2 in control neurons (Fig. S6 C and D), suggesting that 1 h of PIKfyve inhibition likely favors enhanced surface expression of AMPARs even under basal levels of activity. Nevertheless, the reversal of synaptic downscaling by both PIKfyve inhibitors suggests that the increase in PI(3,5)P2 levels during network hyperactivation plays a direct role in maintaining weakened synaptic strength via effects on surface AMPAR dynamics.
AMPAR Trafficking at the Plasma Membrane Is Sensitive to PIKfyve Activity.
PI(3,5)P2 likely regulates multiple endosomal functions via interactions with specific downstream protein effectors. However, the diversity of membranes on which PI(3,5)P2 is synthesized and the identity of most PI(3,5)P2 binding proteins are largely unknown. To test whether PIKfyve associates with membranes involved in AMPAR trafficking, we cotransfected neurons with HA-PIKfyve and GFP and measured the degree of colocalization of HA-PIKfyve puncta in the first 55 μm of dendrites with a marker of early endosomes, early endosome antigen 1 (EEA1). We observed punctate HA-PIKfyve in the soma and dendrites of cultured pyramidal neurons (Fig. S7A). HA-PIKfyve puncta were observed both inside (Fig. S7B) and at the base (Fig. 6A) of dendritic spines, and 44.7 ± 1.6% HA-PIKfyve puncta colocalize with EEA1 (Fig. S7C). The proportion of HA-PIKfyve puncta that colocalize with EGFP-LAMP1, a late endosome/lysosomal marker, is lower (28.7 ± 2.6%; Fig. S7 D and E); thus, PIKfyve in dendrites is found with both early and late endosomes/lysosomes.
Fig. 6.
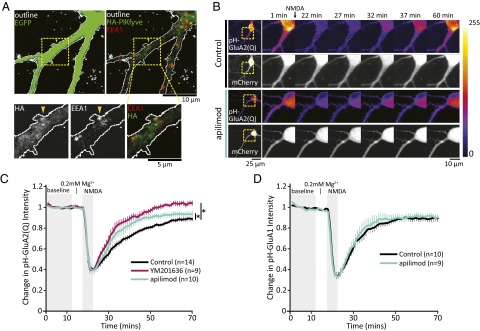
PIKfyve activity regulates AMPAR trafficking. (A) Representative images of neurons transfected with EGFP and HA-PIKfyve, and stained to mark early endosomes (EEA1). The dashed box indicates the enlarged regions below. The yellow arrowheads indicate an example of HA-PIKfyve colocalizing with EEA1 at the base of a dendritic spine. (Top Left) Z-projected images of a neuron expressing EGFP (green, outlined in white). (Top Right) Merged 0.41-μm slice of HA-PIKfyve (green) and EEA1 (red). (B) Representative images of neurons cotransfected with pH-GluA2(Q) and mCherry. Neurons were incubated with DMSO or PIKfyve inhibitors for 1 h before live confocal imaging. During imaging, all solutions were continuously perfused at 32 °C. (C) Mean (±SEM) intensity of pH-GluA2(Q) relative to the mean intensity of the first 10 min of imaging. Once a stable baseline was obtained, HBS (0.2 mM Mg2+) was washed for 5 min, followed by 5 min of stimulation with NMDA (20 μM NMDA, 10 μM glycine, 0.2 mM Mg2+), which quenches the fluorescence of pH-GluA2(Q). After NMDA stimulation, normal HBS was continuously perfused for the remainder of the experiment. PIKfyve inhibition by either 1 μM apilimod or 2 μM YM201636 enhances the rate of pH-GluA2(Q) fluorescence recovery [Kruskal–Wallis ANOVA: χ2(2, 2,146) = 137.27, P = 1.6 × 10−30]. (D) Mean (±SEM) intensity of pH-GluA1 relative to the mean intensity of the first 10 min of imaging. The fluorescence of pH-GluA1 is strongly quenched by NMDA stimulation. Brief PIKfyve inhibition does not have an impact on the rate of recovery. *P < 0.05.
Following endocytosis, AMPARs are known to traverse through EEA1-positive endosomes, after which receptors may recycle back to the plasma membrane or remain internalized. Localization of HA-PIKfyve to early endosomal compartments suggested that this subcellular compartment may be where PI(3,5)P2 synthesis has a direct impact on AMPAR trafficking. Thus, we tested whether PIKfyve activity is required for constitutive and regulated AMPAR trafficking using exogenously expressed AMPAR subunits tagged with the pH-sensitive GFP variant pHluorin, pH-GluA1, or pH-GluA2(Q). pHluorin is fluorescent at neutral pH and is quenched in acidic environments. Thus, changes in the abundance of surface-exposed pH-GluAs can be detected as a change in fluorescence intensity. Note that we used pH-GluA2(Q) to minimize contamination from the ER (72).
Initially, we tested the effect of acute PIKfyve inhibition on steady-state fluorescence and found no change compared with baseline intensity. To widen the scope of this analysis, neurons were treated for 1 h with YM201636 or apilimod and then stimulated with 20 μM NMDA to drive endocytosis of AMPAR in the continuous presence of inhibitors. Of note, this NMDA stimulation does not lead to persistent loss of AMPARs, as in the case of cLTD, but rather drives a transient loss of surface receptors through endocytosis that is restored over time by recycling back to the plasma membrane. Indeed, although 5 min of NMDA stimulation strongly decreased pHluorin fluorescence by ∼60%, surface fluorescence slowly reappeared over the next 45 min due to recycling of AMPARs back to the cell surface (Fig. 6B). We found that PIKfyve inhibition specifically enhanced the recovery rate of pH-GluA2(Q) fluorescence (Fig. 6 B and C) yet had no detectable impact on the dynamics of pH-GluA1 (Fig. 6D and Fig. S8). Subunit-specific trafficking of AMPAR has been observed previously in a variety of contexts (67, 73–77). Of particular relevance, coordinate loss of sGluA1 and sGluA2 during slow homeostatic downscaling requires subunit-specific trafficking events controlled by the GluA2 subunit (78, 79). Thus, our results are consistent with the idea that PIKfyve activity has a selective impact on the GluA2 subunit to control the surface expression of AMPARs.
To determine whether the effects of PIKfyve inhibition on pH-GluA2(Q) dynamics reflect a role for PIKfyve in native AMPAR trafficking, we measured surface abundance of endogenous GluA2 using immunocytochemistry. Using NMDA stimulation to drive endocytosis of AMPAR, we determined the ratio of surface GluA2 to internal GluA2 using an aminoterminal antibody without or with permeabilization, respectively. In control neurons, NMDA stimulation for 5 min reduced the ratio of surface GluA2 to internal GluA2, but pretreatment with apilimod for 1 h blocked this NMDA-induced decrease (Fig. 7 A and B). Thus, PIKfyve activity may contribute to GluA2 endocytosis. Alternatively, given that this NMDA regimen induces only a transient loss of surface AMPARs, these effects could reflect a situation where inhibition of PIKfyve results in a rapid return of internalized receptors to the surface.
Fig. 7.

PIKfyve inhibition blocks activity-dependent trafficking of GluA2. (A) Representative images of neurons stained for surface or internal GluA2. (B) Ratio (±SEM) of surface GluA2 to internal GluA2. The sGluA2/inGluA2 ratio is reduced by 5 min of NMDA stimulation (20 μM NMDA, 10 μM glycine, 0.2 mM Mg2+) in control neurons. Incubation with 1 μM apilimod blocks this decrease [relative to average baseline; baseline: 1.0 ± 0.02; 5 min of NMDA: 0.86 ± 0.03, 1 h of apilimod: 1.0 ± 0.02, 1 h of apilimod + 5 min of NMDA: 1.07 ± 0.03; one-way ANOVA, F(3,101) = 14.68, P = 5.26 × 10−8]. (C) Schematic of experimental design and representative images of sGluA2 staining in dendrites. After 5 min of NMDA stimulation, PIKfyve was inhibited by 1 μM apilimod for 10 min or 30 min before staining for sGluA2. (D) Mean (±SEM) sGluA2 puncta integrated density (mean intensity × puncta size) relative to unstimulated baseline. After 10 min, GluA2 is decreased in control but not in apilimod-treated neurons [NMDA + 10 min: 78.43 ± 5.46%, NMDA + 30 min: 95.47 ± 7.62%, NMDA + 10 min with apilimod: 100 ± 6.47%, NMDA + 30 min with apilimod: 98.84 ± 6.47%; Kruskal–Wallis ANOVA: χ2 (4,154) = 12.1, P = 0.017]. *P < 0.05.
To determine whether PIKfyve activity has additional roles in GluA2 trafficking beyond the acute response to NMDA stimulation, we limited PIKfyve inhibition to time points after NMDAR activation. Neurons were first stimulated with NMDA for 5 min without PIKfyve inhibition and then incubated with 1 μM apilimod for 10 or 30 min. In control neurons, the intensity of sGluA2 after NMDA stimulation compared with unstimulated sister cultures remained decreased at the 10-min time point but returned to basal levels by 30 min (Fig. 7 C and D). By contrast, inhibition of PIKfyve after NMDA stimulation resulted in the rapid return of sGluA2 to baseline levels within 10 min and sGluA2 expression remained stable at the 30-min time point. Together, these results suggest that a target of PIKfyve activity is the GluA2 subunit of AMPARs and that PIKfyve inhibition has an impact on multiple AMPAR trafficking steps, which collectively result in enrichment of AMPARs on the cell surface. Thus, PIKfyve activity is important for GluA2 trafficking during NMDA stimulation and then continues to play a critical role in the subsequent membrane trafficking of AMPARs during the recycling of internalized receptor pools.
Discussion
PIPs play crucial roles in membrane trafficking and cell signaling in eukaryotic cells. Despite their clear biological significance, the pathways that are regulated by these lipids are largely unknown. This lack of knowledge is especially true in neurons, where activity-dependent regulation of PIPs is poorly understood. In this work, we determined that PIKfyve lipid kinase activity plays a regulatory role at synapses and that PI(3,5)P2 is a downstream effector of neural activity (Fig. S9). PIKfyve activity drives synaptic depression and is critical for two distinct forms of synaptic plasticity: cLTD and homeostatic synaptic weakening. Moreover, dynamic regulation of PIPs occurs in response to acute and chronic changes in activity. Changes in levels of PI(3,5)P2 often result in parallel changes in PI5P (10). Thus, it is notable that PI5P levels were stable during both cLTD and homeostatic weakening. Therefore, the impact on synaptic receptors due to loss of PIKfyve activity is likely due to the loss of PI(3,5)P2. Although future studies are required to delineate the precise mechanism by which PIKfyve is activated by neural activity, these results suggest that this process occurs close to synapses.
We found that prolonged hyperactivation of neural network activity leads to a sustained increased in PI(3,5)P2 levels (∼1.6-fold). This result raises the possibility that neural activity alters PI(3,5)P2 metabolism; however, little is known about the upstream activators of PIKfyve even in lower eukaryotes, such as yeast. The degree of elevation in neurons is similar to increases found in mammalian cells via induction by EGF [1.5-fold (80)], IL-2 [1.75-fold (81)], and insulin [twofold (30)]. Interestingly, in neurons, insulin stimulation causes synaptic weakening (73), raising the possibility that enhanced PI(3,5)P2 synthesis contributes to this effect. Another potential link to neuronal activity is that PIKfyve interacts with some L-type calcium channels (32), suggesting that PIKfyve may be well positioned to respond to calcium influx. Interactions with other members of the PIKfyve protein complex may also be important for activation of PIKfyve at synapses. For example, Vac14 has a motif that interacts with PDZ domains (82), which are highly abundant protein interaction domains found in many scaffolding proteins in the postsynaptic density.
The pleiotropic effects caused by defects in PI(3,5)P2 synthesis (3) suggest that multiple pathways are regulated by PI(3,5)P2. A complete understanding of PI(3,5)P2 signaling requires identification of the downstream protein effectors. However, the motifs that interact specifically with PI(3,5)P2 are not known, thus limiting the utility of bioinformatic approaches. In neurons with chronically reduced PIKfyve activity, stimulus-dependent endocytosis of AMPARs is impaired (33). Here, we found that brief PIKfyve inhibition during NMDA stimulation blocks internalization of GluA2 and that PIKfyve inhibition immediately following NMDA stimulation hastens the recovery of surface GluA2 levels after internalization. The GluA2 subunit plays an instructive role in homeostatic plasticity induced by chronic changes in activity (67, 69, 78, 79, 83, 84), thereby providing a potential link between PI(3,5)P2 synthesis during sustained activity changes and the regulation of AMPAR expression at the cell surface. Our results are consistent with up-regulation of PI(3,5)P2 levels driving AMPAR internalization and PI(3,5)P2 acting by delaying or diverting internalized receptors from recycling back to the plasma membrane. Impairment of AMPAR trafficking toward the late endosome favors receptor recycling and attenuates synaptic depression (48). Therefore, PIKfyve blockade may also cause defects in the late endosome/lysosome that result in accumulation of GluA2 on the plasma membrane. In metazoans, PIKfyve is found on early endosomes, late endosomes, and lysosomes (3). This diverse localization strongly suggests that PIKfyve regulates multiple trafficking steps.
Removal of synaptic AMPARs involves lateral diffusion (reviewed in ref. 85), constitutive cycling, regulated endocytosis and exocytosis, and intracellular trafficking (reviewed in refs. 64, 65, 86). Although both NMDAR-cLTD and downscaling result in a decrease in AMPARs, different signaling cascades are thought to mediate these forms of synaptic depression. Thus, PIKfyve activity may be a point of convergence in these mechanisms. In addition to the finding that maintenance of homeostatic synaptic weakening requires PIKfyve activity, we found that disruption of PI(3,5)P2 synthesis results in a specific loss of the ability to downscale, but not to upscale, synapse strength. This unidirectional role is consistent with a growing literature suggesting that distinct mechanisms underlie homeostatic synaptic strengthening and weakening (reviewed in refs. 53, 58). Candidate pathways that may regulate PIKfyve or may be regulated by PIKfyve are mechanisms known to be important for homeostatic synaptic weakening (reviewed in refs. 53, 58, 87). These include PSD-95, Polo-like kinase 2 (Plk2), cyclin-dependent kinase 5 (CDK5), and L-type voltage-gated calcium channels.
The focus on PI(3,5)P2 metabolism in this study does not preclude roles for other PIPs in regulation of synapses. Indeed, there are crucial roles for PIPs in regulation of both basal synaptic strength and activity-dependent modifications to synaptic efficacy. For example, clathrin-mediated endocytosis of AMPARs, which is regulated by PI(4,5)P2 and the 5-phosphatase synaptojanin (43), are critical for internalization of AMPARs during synaptic depression (88). Dynamic changes in the metabolism of PI(4,5)P2 (88, 89) and PI(3,4,5)P3 (46, 90, 91) are critical for LTD and other forms of synaptic plasticity (reviewed in ref. 92). We found that PI(3,4,5)P3 levels are detected under basal conditions, which is consistent with the hypothesis that PI(3,4,5)P3 is required continuously for maintaining synaptic AMPARs (45) and AMPAR trafficking (93). Collectively, these results illustrate the complexity and importance of PIP signaling in neurons and raise the possibility that therapies targeted at PIP metabolism and signaling may be beneficial for treatment of neurological disorders characterized by aberrant synapse function.
Materials and Methods
All experiments with animals were performed in compliance with guidelines of the University Committee on the Use and Care of Animals of the University of Michigan and the NIH. Details of experimental procedures are provided in SI Materials and Methods. Cell culture, transient transfection, lentivirus shRNA knockdown, and measurement of PIPs (10), as well as electrophysiology experiments, immunocytochemistry, fluorescent recovery after NMDA stimulation, image quantification, and statistical analysis (33), were performed as described. Additional details, as well as details on the generation of the Flp-In cell lines generated from T-Rex 293 cells (Life Technologies) are presented in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Christian Althaus and Cindy Carruthers for their assistance in preparing neuronal cultures. We thank Dr. Roberto Malinow and Addgene for the SEP-GluA1 and SEP-GluA2(Q) plasmids. This work was supported by NIH Grants R01-NS064015 and R01-GM050403 (to L.S.W.) and NIH Grant RO1-MH085798 (to M.A.S.), and, in part, by the Protein Folding Diseases FastForward Initiative, University of Michigan. A.J.M. was supported, in part, by National Research Service Award F31NS07470 and the Rackham Predoctoral Fellowship award from the University of Michigan. B.S.S. was supported by a postdoctoral fellowship from the Jane Coffin Child Fund.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411117111/-/DCSupplemental.
References
- 1.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443(7112):651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 2.Balla T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93(3):1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: Low abundance, high significance. BioEssays. 2014;36(1):52–64. doi: 10.1002/bies.201300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow CY, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448(7149):68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duex JE, Nau JJ, Kauffman EJ, Weisman LS. Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell. 2006;5(4):723–731. doi: 10.1128/EC.5.4.723-731.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006;172(5):693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gary JD, et al. Regulation of Fab1 phosphatidylinositol 3-phosphate 5-kinase pathway by Vac7 protein and Fig4, a polyphosphoinositide phosphatase family member. Mol Biol Cell. 2002;13(4):1238–1251. doi: 10.1091/mbc.01-10-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudge SA, Anderson DM, Emr SD. Vacuole size control: Regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell. 2004;15(1):24–36. doi: 10.1091/mbc.E03-05-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikonomov OC, Sbrissa D, Fenner H, Shisheva A. PIKfyve-ArPIKfyve-Sac3 core complex: Contact sites and their consequence for Sac3 phosphatase activity and endocytic membrane homeostasis. J Biol Chem. 2009;284(51):35794–35806. doi: 10.1074/jbc.M109.037515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zolov SN, et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci USA. 2012;109(43):17472–17477. doi: 10.1073/pnas.1203106109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson G, et al. Distinctive genetic and clinical features of CMT4J: A severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain. 2011;134(Pt 7):1959–1971. doi: 10.1093/brain/awr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow CY, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84(1):85–88. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baulac S, et al. Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology. 2014;82(12):1068–1075. doi: 10.1212/WNL.0000000000000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campeau PM, et al. Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet. 2013;92(5):781–791. doi: 10.1016/j.ajhg.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gary JD, Wurmser AE, Bonangelino CJ, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol. 1998;143(1):65–79. doi: 10.1083/jcb.143.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto A, et al. Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol Biol Cell. 1995;6(5):525–539. doi: 10.1091/mbc.6.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooke FT, et al. The stress-activated phosphatidylinositol 3-phosphate 5-kinase Fab1p is essential for vacuole function in S. cerevisiae. Curr Biol. 1998;8(22):1219–1222. doi: 10.1016/s0960-9822(07)00513-1. [DOI] [PubMed] [Google Scholar]
- 18.Sbrissa D, Ikonomov OC, Shisheva A. PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. Effect of insulin. J Biol Chem. 1999;274(31):21589–21597. doi: 10.1074/jbc.274.31.21589. [DOI] [PubMed] [Google Scholar]
- 19.Bonangelino CJ, et al. Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol. 2002;156(6):1015–1028. doi: 10.1083/jcb.200201002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin N, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27(24):3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, et al. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci USA. 2007;104(44):17518–17523. doi: 10.1073/pnas.0702275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Botelho RJ, Efe JA, Teis D, Emr SD. Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig4 phosphoinositide phosphatase. Mol Biol Cell. 2008;19(10):4273–4286. doi: 10.1091/mbc.E08-04-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Lartigue J, et al. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10(7):883–893. doi: 10.1111/j.1600-0854.2009.00915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikonomov OC, et al. The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve-/- embryos but normality of PIKfyve+/- mice. J Biol Chem. 2011;286(15):13404–13413. doi: 10.1074/jbc.M111.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeffries TR, Dove SK, Michell RH, Parker PJ. PtdIns-specific MPR pathway association of a novel WD40 repeat protein, WIPI49. Mol Biol Cell. 2004;15(6):2652–2663. doi: 10.1091/mbc.E03-10-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morishita M, et al. Phosphatidylinositol 3-phosphate 5-kinase is required for the cellular response to nutritional starvation and mating pheromone signals in Schizosaccharomyces pombe. Genes Cells. 2002;7(2):199–215. doi: 10.1046/j.1356-9597.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- 27.Sbrissa D, Ikonomov OC, Filios C, Delvecchio K, Shisheva A. Functional dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P2 by means of the PIKfyve inhibitor YM201636. Am J Physiol Cell Physiol. 2012;303(4):C436–C446. doi: 10.1152/ajpcell.00105.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takasuga S, et al. Critical roles of type III phosphatidylinositol phosphate kinase in murine embryonic visceral endoderm and adult intestine. Proc Natl Acad Sci USA. 2013;110(5):1726–1731. doi: 10.1073/pnas.1213212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whiteford CC, Brearley CA, Ulug ET. Phosphatidylinositol 3,5-bisphosphate defines a novel PI 3-kinase pathway in resting mouse fibroblasts. Biochem J. 1997;323(Pt 3):597–601. doi: 10.1042/bj3230597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bridges D, et al. Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell. 2012;23(15):2955–2962. doi: 10.1091/mbc.E11-12-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dove SK, et al. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997;390(6656):187–192. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- 32.Tsuruta F, Green EM, Rousset M, Dolmetsch RE. PIKfyve regulates CaV1.2 degradation and prevents excitotoxic cell death. J Cell Biol. 2009;187(2):279–294. doi: 10.1083/jcb.200903028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, et al. Modulation of synaptic function by VAC14, a protein that regulates the phosphoinositides PI(3,5)P2 and PI(5)P. EMBO J. 2012;31(16):3442–3456. doi: 10.1038/emboj.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonangelino CJ, Catlett NL, Weisman LS. Vac7p, a novel vacuolar protein, is required for normal vacuole inheritance and morphology. Mol Cell Biol. 1997;17(12):6847–6858. doi: 10.1128/mcb.17.12.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jefferies HB, et al. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008;9(2):164–170. doi: 10.1038/sj.embor.7401155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McPherson PS, et al. A presynaptic inositol-5-phosphatase. Nature. 1996;379(6563):353–357. doi: 10.1038/379353a0. [DOI] [PubMed] [Google Scholar]
- 37.Cremona O, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99(2):179–188. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- 38.Haffner C, Di Paolo G, Rosenthal JA, de Camilli P. Direct interaction of the 170 kDa isoform of synaptojanin 1 with clathrin and with the clathrin adaptor AP-2. Curr Biol. 2000;10(8):471–474. doi: 10.1016/s0960-9822(00)00446-2. [DOI] [PubMed] [Google Scholar]
- 39.Wenk MR, et al. PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron. 2001;32(1):79–88. doi: 10.1016/s0896-6273(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 40.Kim WT, et al. Delayed reentry of recycling vesicles into the fusion-competent synaptic vesicle pool in synaptojanin 1 knockout mice. Proc Natl Acad Sci USA. 2002;99(26):17143–17148. doi: 10.1073/pnas.222657399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Paolo G, et al. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431(7007):415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- 42.Mani M, et al. The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron. 2007;56(6):1004–1018. doi: 10.1016/j.neuron.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong LW, De Camilli P. Regulation of postsynaptic AMPA responses by synaptojanin 1. Proc Natl Acad Sci USA. 2008;105(45):17561–17566. doi: 10.1073/pnas.0809221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khuong TM, et al. Synaptic PI(3,4,5)P3 is required for Syntaxin1A clustering and neurotransmitter release. Neuron. 2013;77(6):1097–1108. doi: 10.1016/j.neuron.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 45.Arendt KL, et al. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2010;13(1):36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jurado S, et al. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J. 2010;29(16):2827–2840. doi: 10.1038/emboj.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21(5):1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 48.Fernández-Monreal M, Brown TC, Royo M, Esteban JA. The balance between receptor recycling and trafficking toward lysosomes determines synaptic strength during long-term depression. J Neurosci. 2012;32(38):13200–13205. doi: 10.1523/JNEUROSCI.0061-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai X, et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem Biol. 2013;20(7):912–921. doi: 10.1016/j.chembiol.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikonomov OC, Sbrissa D, Shisheva A. YM201636, an inhibitor of retroviral budding and PIKfyve-catalyzed PtdIns(3,5)P2 synthesis, halts glucose entry by insulin in adipocytes. Biochem Biophys Res Commun. 2009;382(3):566–570. doi: 10.1016/j.bbrc.2009.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80(3):718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, et al. Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5-bisphosphate localization and dynamics. Proc Natl Acad Sci USA. 2013;110(52):21165–21170. doi: 10.1073/pnas.1311864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turrigiano G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor Perspect Biol. 2012;4(1):a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L, Lau AG, Sarti F. Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:3–12. doi: 10.1016/j.neuropharm.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pribiag H, Stellwagen D. Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology. 2014;78:13–22. doi: 10.1016/j.neuropharm.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Thalhammer A, Cingolani LA. Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:23–30. doi: 10.1016/j.neuropharm.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 57.Lee KFH, Soares C, Béïque JC. Tuning into diversity of homeostatic synaptic plasticity. Neuropharmacology. 2014;78:31–37. doi: 10.1016/j.neuropharm.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 58.Siddoway B, Hou H, Xia H. Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology. 2014;78:38–44. doi: 10.1016/j.neuropharm.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Brien RJ, et al. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21(5):1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 60.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391(6670):892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 61.Maffei A, Turrigiano GG. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci. 2008;28(17):4377–4384. doi: 10.1523/JNEUROSCI.5298-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu LM, Goda Y. Dendritic signaling and homeostatic adaptation. Curr Opin Neurobiol. 2009;19(3):327–335. doi: 10.1016/j.conb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 63.Jakawich SK, Neely RM, Djakovic SN, Patrick GN, Sutton MA. An essential postsynaptic role for the ubiquitin proteasome system in slow homeostatic synaptic plasticity in cultured hippocampal neurons. Neuroscience. 2010;171(4):1016–1031. doi: 10.1016/j.neuroscience.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22(3):461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henley JM, Wilkinson KA. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin Neurosci. 2013;15(1):11–27. doi: 10.31887/DCNS.2013.15.1/jhenley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seebohm G, et al. Identification of a novel signaling pathway and its relevance for GluA1 recycling. PLoS ONE. 2012;7(3):e33889. doi: 10.1371/journal.pone.0033889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cingolani LA, et al. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58(5):749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu JH, et al. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron. 2010;68(6):1128–1142. doi: 10.1016/j.neuron.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evers DM, et al. Plk2 attachment to NSF induces homeostatic removal of GluA2 during chronic overexcitation. Nat Neurosci. 2010;13(10):1199–1207. doi: 10.1038/nn.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee KJ, et al. Requirement for Plk2 in orchestrated ras and rap signaling, homeostatic structural plasticity, and memory. Neuron. 2011;69(5):957–973. doi: 10.1016/j.neuron.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun Q, Turrigiano GG. PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. J Neurosci. 2011;31(18):6800–6808. doi: 10.1523/JNEUROSCI.5616-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rathje M, et al. AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc Natl Acad Sci USA. 2013;110(35):14426–14431. doi: 10.1073/pnas.1312982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Man HY, et al. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25(3):649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 74.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105(3):331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 75.Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43(2):221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 76.Shepherd JD, et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci. 2011;31(6):2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29(20):6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68(3):512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsujita K, et al. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279(14):13817–13824. doi: 10.1074/jbc.M312294200. [DOI] [PubMed] [Google Scholar]
- 81.Jones DR, et al. The identification of phosphatidylinositol 3,5-bisphosphate in T-lymphocytes and its regulation by interleukin-2. J Biol Chem. 1999;274(26):18407–18413. doi: 10.1074/jbc.274.26.18407. [DOI] [PubMed] [Google Scholar]
- 82.Lemaire JF, McPherson PS. Binding of Vac14 to neuronal nitric oxide synthase: Characterisation of a new internal PDZ-recognition motif. FEBS Lett. 2006;580(30):6948–6954. doi: 10.1016/j.febslet.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 83.Lambo ME, Turrigiano GG. Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. J Neurosci. 2013;33(20):8810–8819. doi: 10.1523/JNEUROSCI.4502-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pozo K, et al. β3 integrin interacts directly with GluA2 AMPA receptor subunit and regulates AMPA receptor expression in hippocampal neurons. Proc Natl Acad Sci USA. 2012;109(4):1323–1328. doi: 10.1073/pnas.1113736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Opazo P, Sainlos M, Choquet D. Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol. 2012;22(3):453–460. doi: 10.1016/j.conb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 86.Esteban JA. Intracellular machinery for the transport of AMPA receptors. Br J Pharmacol. 2008;153(Suppl 1):S35–S43. doi: 10.1038/sj.bjp.0707525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vitureira N, Letellier M, Goda Y. Homeostatic synaptic plasticity: From single synapses to neural circuits. Curr Opin Neurobiol. 2012;22(3):516–521. doi: 10.1016/j.conb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Unoki T, et al. NMDA receptor-mediated PIP5K activation to produce PI(4,5)P2 is essential for AMPA receptor endocytosis during LTD. Neuron. 2012;73(1):135–148. doi: 10.1016/j.neuron.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 89.Horne EA, Dell’Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci. 2007;27(13):3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim JI, et al. PI3Kγ is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat Neurosci. 2011;14(11):1447–1454. doi: 10.1038/nn.2937. [DOI] [PubMed] [Google Scholar]
- 91.Takeuchi K, et al. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc Natl Acad Sci USA. 2013;110(12):4738–4743. doi: 10.1073/pnas.1222803110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Knafo S, Esteban JA. Common pathways for growth and for plasticity. Curr Opin Neurobiol. 2012;22(3):405–411. doi: 10.1016/j.conb.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 93.Qin Y, et al. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 2005;19(17):2000–2015. doi: 10.1101/gad.342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.