

Significance
Transmembrane receptors often dimerize to facilitate signaling across the cell membrane. Here we show that the intrinsically disordered cytoplasmic domain of a disintegrin and metalloprotease (ADAM)10, an important transmembrane protease, can acquire structure and dimerize when it is adjoined to a transmembrane helix and thus placed near the membrane surface. This study is, to our knowledge, the first report of a transmembrane helix enabling dimerization of an adjacent intrinsically disordered sequence, which not only advances our understanding of the activation mechanism of ADAM protease, but also provides novel insights on the principles of membrane–protein interactions and their roles in transmembrane signaling.
Keywords: Intrinsic disorder, ADAM10, membrane protein dimerization, FRET
Abstract
Intrinsically disordered protein regions are widely distributed in the cytoplasmic domains of many transmembrane receptors. The cytoplasmic domain of a disintegrin and metalloprotease (ADAM)10, a transmembrane metalloprotease mediating ectodomain shedding of diverse membrane proteins, was recently suggested to mediate the homodimerization of ADAM10. Here we show that a recombinant cytoplasmic domain of ADAM10 (A10Cp) is unstructured as judged by its susceptibility to limited trypsin digestion and its circular dichroism spectrum. In comparison, recombinant transmembrane-cytoplasmic domain of ADAM10 (A10TmCp) reconstituted in dodecylphosphocholine (DPC) micelles exhibits much greater resistance to trypsin digestion, with its cytoplasmic domain taking on a significant ordered structure. FRET analysis demonstrates that, although A10Cp remains monomeric, A10TmCp forms a tight homodimer (Kd ∼ 7 nM) in DPC micelles. Phospholipid-conjugated A10Cp dose-dependently inhibits formation of A10TmCp homodimer, whereas A10Cp achieves only limited inhibition. Placing the transmembrane and cytoplasmic domains of ADAM10, but not the transmembrane domain alone, in their native orientation in the inner membrane of Escherichia coli produces specific and strong dimerization signal in the AraC-based transcriptional reporter assay. A chimeric construct containing the otherwise monomeric transmembrane domain of l-selectin and the cytoplasmic domain of ADAM10 produces a similar dimerization signal. Overall, these results demonstrate that a transmembrane domain imparts a stable structure to the adjacent and intrinsically disordered cytoplasmic domain of ADAM10 to form a homodimer in the membrane. This finding advances our understanding of the regulatory mechanism of ADAMs and has general implications for membrane–protein interactions in the process of transmembrane signaling.
Intrinsically disordered regions are prevalent in proteins (1, 2). Compared with the structured region, the intrinsically disordered region is flexible, which enables it to interact with a wider range of binding partners for diverse biological functions (3–5). The presence of relatively high net charge with a low content of bulky hydrophobic residues signifies an intrinsically disordered region (6, 7). For many transmembrane proteins, the cytoplasmic domain is intrinsically disordered, but it can adopt stable conformation from binding intracellular partners or on posttranslational modification (8, 9). However, little is known about how the membrane bilayer affects the intrinsic disorder of a nearby cytoplasmic domain of a membrane protein.
ADAM10, a member of a disintegrin and metalloprotease (ADAM) family, is expressed in diverse tissues and cells. ADAM10 proteolyzes a broad range of membrane protein substrates in a process often known as ectodomain shedding (10). ADAM10 plays a critical role in cell proliferation and tissue development, as its deficiency in mouse embryo leads to prenatal lethality during the early development period (11–13). ADAM10 is composed of an N-terminal signal sequence followed by a prodomain, a metalloprotease domain, a disintegrin domain, a cysteine-rich region, an EGF-like repeat, a transmembrane helix, and a cytoplasmic region. The proteolytic activity of ADAM10 is tightly regulated by multiple factors. External stimulations such as ionophore and phorbol ester can modulate the activity of ADAM10 (14–16), likely through an inside-out mechanism in which its cytoplasmic region is involved. Recently, it was suggested that ADAM10 forms a homodimer in the cell membrane as does ADAM17, a key feature in the proposed regulation mechanism of ADAM activation (17). Because ADAM10 with a truncated cytoplasmic domain could not form a homodimer, the cytoplasmic domain was suggested to be important for dimerization (17). However, the cytoplasmic domain of ADAM10 appears disordered and does not contain any known dimerization sequence motifs.
In the current study, we report the characterization of transmembrane-cytoplasmic domains of ADAM10. We found that, although the isolated cytoplasmic domain of ADAM10 is intrinsically disordered, the inclusion of a neighboring transmembrane domain imparts an ordered structure to it and enables its dimerization.
Results
The Intrinsically Disordered Cytoplasmic Domain of ADAM10 Takes on Ordered Conformation in the Presence of Its Adjacent Transmembrane Domain.
The cytoplasmic domain of ADAM10 is enriched with proline and positively charged residues (Fig. S1), which are signatures of intrinsic disorder (2). To assess the intrinsic disorder of the ADAM10 cytoplasmic domain, the sequence of human ADAM10 transmembrane and cytoplasmic domains was analyzed using SPINE-D, a program that predicts disordered regions based on a single neural network (18). In the program, each residue is assigned a score that denotes the probability of being disordered. Except for the juxtamembrane region (residues Lys697–His702), most residues in the cytoplasmic domain were assigned a disorder score >0.5, suggesting that they are highly disordered (Fig. 1). In contrast, most residues in the transmembrane domain were assigned a disorder score <0.5, consistent with the expectation of ordered α-helical conformation.
Fig. 1.
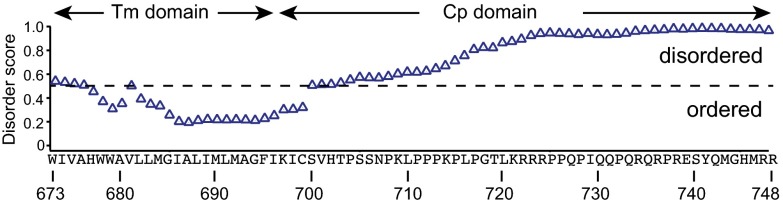
Prediction of intrinsic disorder in the transmembrane (Tm) and cytoplasmic (Cp) domains of ADAM10 by the SPINE-D program. The protein sequence is shown at the bottom with marked residue numbers. A score of >0.5 (above the dashed line) indicates disorder for the residue.
Consistent with the assessment by SPINE-D, recombinant cytoplasmic domain of human ADAM10 (A10Cp-6H, containing residues Lys697–Arg748 and a C-terminal hexahistidine tag) in the dodecylphosphocholine (DPC) micellar solution (10 mM DPC, 20 mM Tris·HCl, 100 mM NaCl, and 1 mM DTT, pH 8.0) contained little stable secondary structure as indicated by its circular dichroism (CD) spectrum (Fig. 2A and Fig. S1). Neither removal of the hexahistidine tag from A10Cp-6H (i.e., A10Cp) nor DPC from the solvent significantly altered the CD spectrum (Fig. S2). However, recombinant transmembrane-cytoplasmic domains of human ADAM10 (A10TmCp-6H, containing residues Leu666–Arg748 and a C-terminal hexahistidine tag) reconstituted in the same DPC micelles contained significant amount of α-helical structure (Fig. 2A and Fig. S1). Deconvolution of its CD spectrum (α helix: 87%, β strand: 1%, coil: 12%) suggested that most residues in A10TmCp-6H adopt the α-helical conformation (19). Because the transmembrane domain constitutes only 30% of the residues in A10TmCp-6H, significant amount of helical structure should be present in the cytoplasmic domain.
Fig. 2.

The intrinsically disordered cytoplasmic domain of ADAM10 takes on stable helical conformation in the presence of its adjacent transmembrane domain. (A) CD spectra of A10Cp-6H (brown) and A10TmCp-6H (red) in 10 mM DPC, 20 mM Tris·HCl, 100 mM NaCl, and 1 mM DTT, pH 8.0 at 20 °C. (B) HPLC traces of A10Cp-6H (brown) and A10TmCp-6H (red) before and after (black) 1-h trypsin digestion at 37 °C.
The susceptibility to trypsin digestion is widely used to confirm the lack of stable structure in a protein (20, 21). There are multiple trypsin cleavage sites in the cytoplasmic domain of ADAM10. Limited trypsin digestion of A10Cp-6H or A10TmCp-6H in DPC micelles was carried out. Subsequent HPLC analysis of the digestion products indicated that, whereas less than 20% of A10TmCp-6H was cleaved after 1 h of trypsin digestion, A10Cp-6H was completely cleaved (Fig. 2B). Removal of DPC from the solution did not improve the resistance of A10Cp-6H to trypsin digestion (Fig. S3). A similar difference in trypsin digestion resistance was observed when both proteins were reconstituted in or mixed with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposomes (Fig. S3). Collectively, these results indicate that the cytoplasmic domain in A10TmCp-6H is more stable than the same sequence in A10Cp-6H. In other words, the cytoplasmic domain of ADAM10 is intrinsically disordered, but in the presence of its neighboring transmembrane domain, it takes on an ordered conformation.
Recombinant ADAM10 Transmembrane-Cytoplasmic Domain, but Not ADAM10 Cytoplasmic Domain, Forms a Strong Homodimer in DPC Micelles.
We next analyzed the homo-association of A10Cp-6H and A10TmCp-6H in DPC micellar solutions using FRET. Trinitrilotriacetic acid (tri-NTA) was synthesized (22) and conjugated to fluorescein (FS) and tetramethylrhodamine (TMR) as fluorescent donor and acceptor, respectively (Fig. 3A and Fig. S4). The fluorophore-tri-NTA bound the hexahistidine tag in A10Cp-6H or A10TmCp-6H by nanomolar-affinity chelation to Ni2+ ions (Fig. 3B and Fig. S5). The dissociation constants for the fluorophore/protein pairs needed were measured by fluorescence anisotropy and listed in Table 1. Little FRET was observed between 100 nM FS-tri-NTA and TMR-tri-NTA that were equally mixed in the DPC micellar solution (Fig. S6A). Adding 200 nM A10TmCp-6H to the fluorophore mixture, which induced binding of all fluorophore-tri-NTA to the protein, resulted in significant quenching of FS fluorescence emission and concurrent increase of TMR fluorescence (Fig. 3 C and E). The FRET was abolished by addition of EDTA, which dissociated fluorophores from A10TmCp-6H (Figs. S5 and S6). In comparison, adding 200 nM A10Cp-6H did not induce quenching of FS fluorescence (Fig. 3D). These results indicate that A10TmCp-6H, but not A10Cp-6H, forms homo-oligomers in DPC micelles. It is noteworthy that the protein/detergent molar ratio in this experiment was 1/50,000 (200 nM protein/10 mM DPC). It is much lower than 1/200–1/1,000, a range typically used in the studies of homo-association of transmembrane helical peptides (23–26), suggesting that homo-association of A10TmCp-6H is not mediated by its transmembrane domain.
Fig. 3.

A10TmCp-6H, but not A10Cp-6H, forms an oligomer in DPC micelles. (A) Illustration of the FRET experiment. Fluorophore (FS, TMR)-conjugated tri-NTA noncovalently associates with the hexahistidine (6H) tag in the target protein. When the protein self-associates, FRET will occur between the bound FS and TMR. (B) Binding of A10TmCp-6H to FS-tri-NTA dissolved in the DPC micellar solution containing 1 µM NiSO4 monitored by FS fluorescence anisotropy. The binding curve was fitted to a hyperbolic binding equation with a binding affinity of 8 ± 2 nM (Table 1). A10TmCp-6H induced little change in FS anisotropy in the presence of 10 mM EDTA (filled diamond), indicating that FS-tri-NTA bound specifically to the 6H sequence in A10TmCp-6H. (C and D) Fluorescence emission spectra of 10 nM FS-tri-NTA (green), 100 nM TMR-tri-NTA (pink), and the mixture of both in complex with 200 nM A10TmCp-6H (red) or A10Cp-6H (brown) in DPC micelles. (E) Comparison of fluorescence quenching for the noted proteins that were added to the mixture of FS- and TMR-tri-NTA.
Table 1.
Binding affinity of tri-NTA to A10Cp and A10TmCp in 10 mM DPC, 20 mM Tris·HCl, 100 mM NaCl, 1 mM DTT, and 1 µM NiSO4, pH 8.0
| Fluorophore | Protein | KNTA (nM) |
| FS-tri-NTA | A10Cp-6H | 18 ± 2 |
| TMR-tri-NTA | A10Cp-6H | 16 ± 3 |
| FS-tri-NTA | A10TmCp-6H | 8 ± 2 |
| TMR-tri-NTA | A10TmCp-6H | 12 ± 4 |
To determine the oligomeric state of A10TmCp-6H, quenching of FS fluorescence was measured as a function of the mole ratio of TMR-tri-NTA/FS-tri-NTA. The quenching should increase with the acceptor/donor mole ratio, the extent of which is dependent on the degree of protein association (24, 27). A linear increase indicates a monomer-dimer association (27). In this study, the concentrations of A10TmCp-6H and FS-tri-NTA were kept constant at 200 and 100 nM, respectively. The total tri-NTA concentration was also kept constant at 200 nM by adding unconjugated tri-NTA along with TMR-tri-NTA. Fig. 4A shows that quenching of the FS fluorescence increased linearly with the TMR/FS mole ratio, indicating that A10TmCp-6H forms a homodimer. Consistently, the quenching of FS fluorescence reached the maximum when the TMR/FS ratio reached 1, and it did not change further at higher ratios (Fig. 4A).
Fig. 4.

Homodimerization of A10TmCp-6H in the DPC micelle. (A) Plot of FS fluorescence quenching (F/F0) as a function of the molar ratio of TMR-tri-NTA of FS-tri-NTA. F/F0 values expected for a dimer (black dashed line), a trimer (green), and a tetramer (pink) are calculated according to (24, 27). (B) Plot of FRET efficiency (E) as a function of A10TmCp-6H concentration. A mixture of A10TmCp-6H/FS-tri-NTA/TMR-tri-NTA at a molar ratio of 2/1/1 was serially diluted and the FRET efficiency at each dilution measured. The plot was fitted to a linked equilibria of A10TmCp-6H dimerization and its binding to tri-NTA fluorophores (dashed line), with a dimerization constant of 7.2 ± 2.0 nM.
To determine the dimerization constant of A10TmCp-6H (Kd), the FRET efficiency (E) was recorded as a function of A10TmCp-6H concentration (1–200 nM), whereas the molar ratio of A10TmCp-6H/FS-tri-NTA/TMR-tri-NTA was kept at 2/1/1. As shown in Fig. 4B, E increased with the A10TmCp-6H concentration but leveled off at higher concentrations (∼200 nM). Because only when both FS and TMR fluorophores were bound to the A10TmCp-6H dimer did FRET occur, equations were derived to describe the dependence of E on the linked equilibria of A10TmCp-6H dimerization and its association with fluorophores (see SI Materials and Methods for details). To simplify the equation, the dissociation constant for the binding of fluorophore-tri-NTA with A10TmCp-6H (KNTA) was considered as a constant of 10 nM for both FS and TMR fluorophores (Table 1). The plot of E vs. A10TmCp-6H concentration was fitted globally to the following equation:
where x is the concentration of A10TmCp-6H and also that of total tri-NTA, Eint is the intrinsic FRET efficiency in this system, and A is a constant used to denote the distribution of FS-tri-NTA in all of the complexes. The best fit produced a Kd of 7.2 ± 2.0 nM and EintA of 0.47 ± 0.02 (Fig. 4B). Therefore, A10TmCp-6H formed a strong homodimer in the DPC micellar solution.
The Transmembrane-Cytoplasmic Domain but Not the Transmembrane Domain of ADAM10 Self-Associates in a Cell Membrane.
The AraC-based transcriptional reporter assay (AraTM) (28) was used to test whether the transmembrane and cytoplasmic domains of ADAM10 self-associate in a cell membrane. In this assay, transmembrane and cytoplasmic domains of the target protein are fused N-terminally to maltose binding protein (MBP) to enable its native placement in the inner membrane of Escherichia coli. The C terminus of the chimera is fused to the AraC transcriptional factor, the dimerization of which activates the PBAD promoter and induces expression of eGFP in the engineered bacteria. The eGFP fluorescence can be quantitated directly from culture and is correlated with the extent of dimerization of inserted transmembrane and cytoplasmic domains in the membrane (28). The MBP-TmCp-AraC constructs containing the ADAM10 transmembrane domain (a10Tm), the ADAM10 transmembrane and cytoplasmic domains (a10TmCp), the l-selectin transmembrane and cytoplasmic domains (lselTmCp), and a chimeric l-selectin transmembrane and ADAM10 cytoplasmic domains (lselTm-a10Cp) were transformed into the AraC-deficient E. coli strain SB1676 (Fig. 5A). Each chimeric protein was inserted into the inner membrane of E. coli with the native topology, and each was expressed at a level comparable with that of the positive control construct (containing a tightly dimerizing integrin αIIb transmembrane sequence; Fig. 5B and C). The GFP fluorescence intensity in transformed bacteria was recorded and normalized with the cell density as previously described (28). Comparison of the GFP fluorescence intensities indicated that, although MBP-a10Tm-AraC exhibited little dimerization in the membrane, MBP-a10TmCp-AraC dimerized as strongly as the positive control (Fig. 5C and Fig. S7). Furthermore, coexpression of MBP-a10TmCp-AraC with MBP-a10TmCp-DNAraC, a dominant negative construct with a loss-of-function mutation in AraC (29), produced significantly lowered GFP fluorescence, confirming the specificity of a10TmCp-mediated dimerization (Fig. 5D and Fig. S7).
Fig. 5.

The cytoplasmic domain of ADAM10 dimerizes in a cell membrane. (A) Illustration of MBP-AraC constructs used in the study. Construct a10TmCp-DN contains the same ADAM10 residues as in a10TmCp plus a loss-of-function mutation in the AraC domain (AraC*). (B) MalE complementation to test the topology of chimeric proteins. The pMal-p2 and pMal-c2 plasmids express MBP protein in the periplasm and cytoplasm of E. coli, respectively. The positive control construct (pos. contr.) expresses the MBP-AraC chimeric protein containing a tightly dimerizing integrin αIIb transmembrane sequence (28). (C and D) Ratios of GFP fluorescence intensity vs. OD600 for each construct are compared with the background (bkgd) and positive control. In the background sample, E. coli was not transformed with the MBP-AraC–expressing vector. (Upper) Comparable expression levels of chimeric MBP-AraC proteins probed by Western blot using the anti-MBP antibody.
Consistent with recent reports that the transmembrane and cytoplasmic domains of l-selectin or full-length l-selectin are monomeric in membrane conditions (30, 31), expression of MBP-lselTmCp-AraC induced little GFP fluorescence in the AraTM assay (Fig. 5C). In comparison, expression of MBP-lselTm-a10Cp-AraC produced as strong a GFP fluorescence as the positive control or MBP-A10TmCp-AraC. These results indicate that the cytoplasmic domain of ADAM10, when placed next to an unrelated transmembrane domain, can mediate strong dimerization of the host protein.
Phospholipid-Conjugated A10Cp, but Not Unconjugated A10Cp, Inhibits Dimerization of A10TmCp in DPC Micelles.
To test whether the proximity to a membrane surface is sufficient for the ADAM10 cytoplasmic domain to adopt ordered structure and to form homodimer, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidomethyl)cyclohexane-carboxamide] was conjugated to Cys699 of A10Cp and A10Cp-6H to produce DiPal-A10Cp and DiPal-A10Cp-6H, respectively (Fig. S8). Neither conjugated protein was water soluble. Appending the hexahistidine tag did not affect the structure of the ADAM10 cytoplasmic domain because little difference was noted between the CD spectra of DiPal-A10Cp and DiPal-A10Cp-6H in DPC micelles (Fig. 6A and Fig. S9). With a single minimum at 207 nm, both CD spectra were significantly different from that of A10Cp, suggesting that the ADAM10 cytoplasmic domain in DiPal-A10Cp-6H took on a different conformation than the disordered one adopted by A10Cp-6H (Fig. 6A). They were also different from that of A10TmCp in DPC micelles. Additional feature of this conformation could not be ascertained from the CD spectra, partly because the contribution from the chiral atoms in the lipid moiety was unclear. After 1 h of trypsin digestion, all of DiPal-A10Cp-6H reconstituted in DPC micelles was cleaved, but several intermediate digestion products were still present (Fig. S9B). Overall, these results indicate that attachment of a phospholipid molecule imparted some ordered structure to the ADAM10 cytoplasmic domain, although the structure is not as stable as that in A10TmCp.
Fig. 6.

DiPal-A10Cp, but not A10Cp, forms an oligomer in DPC micelles and inhibits dimerization of A10TmCp-6H. (A) CD spectra of DiPal-A10Cp (blue) and A10Cp (brown) in the DPC micellar solution (10 mM DPC, 20 mM Tris·HCl, 100 mM NaCl, and 1 mM DTT, pH 8.0) at 20 °C. (B) Fluorescence emission spectra of 10 nM FS-tri-NTA (green), 100 nM TMR-tri-NTA (pink), and the mixture of both in complex with 200 nM DiPal-A10Cp-6H (blue) in the DPC micellar solution. (C) Plots of FS fluorescence quenching (F/F0) vs. the molar ratio of added protein to A10TmCp-6H in DPC. Stock of DiPal-A10Cp (blue), A10Cp (brown) or lysozyme (black, as control) was added individually to the mixture of A10TmCp-6H with tri-NTA fluorophores, and the FS fluorescence intensities were measured as described in SI Materials and Methods.
Under the same condition as described above for A10TmCp-6H and A10Cp-6H, FRET was clearly observed for 200 nM DiPal-A10Cp-6H in 10 mM DPC (Fig. 6B). The F/F0 value was 0.85 ± 0.03, close to that observed for A10TmCp-6H (0.75 ± 0.02) and distinct from that for A10Cp-6H (1.03 ± 0.02) (Fig. 3E). Considering the protein/DPC molar ratio of 1/50,000 in the experiment and the usual aggregation number of 50–60 for the DPC micelle (32), the average number of DiPal-A10Cp-6H molecules per DPC micelle is significantly lower than 1/900. By comparison, in our earlier study of tje recombinant l-selectin transmembrane-cytoplasmic domain at a protein/DPC molar ratio of 1/500, no fluorescence quenching (F/F0 ∼1.0) was observed between covalently conjugated FS-selectin and TMR-selectin (30). Therefore, the FRET observed for DiPal-A10Cp-6H indicates that it forms an oligomer in DPC micelles, although the extent of homo-association is less than that of A10TmCp-6H.
To test whether dimerization of A10TmCp is mediated by its cytoplasmic domain, A10Cp, DiPal-A10Cp, or lysozyme (as control) was added, in varying doses, to 200 nM A10TmCp-6H in the DPC micellar solution that also contained appropriate tri-NTA fluorophores. None of the added peptides contained the hexahistidine tag; thus, their effects on the observed quenching of FS fluorescence were attributed to their abilities to inhibit dimerization of A10TmCp-6H. Compared with addition of lysozyme that caused no change, DiPal-A10Cp dose-dependently reduced quenching in apparently two stages (Fig. 6C). Complete inhibition of fluorescence quenching, and thus A10TmCp-6H dimerization, was achieved when DiPal-A10Cp was added to a molar ratio of 12. In comparison, addition of A10Cp to molar ratios of 10–20 failed to inhibit dimerization of A10TmCp-6H (Fig. 6C). It is noteworthy that addition of A10Cp at a lower molar ratio (2–5) reduced quenching to a similar extent as DiPal-A10Cp, but the underlying mechanism was not clear. Overall, these results indicate that the dimerization of A10TmCp is mediated by its cytoplasmic domain.
Discussion
It was reported recently that ADAM17 and ADAM10, both of which are important members of the ADAM protease family, form homodimers on the cell surface (17). Deletion of the cytoplasmic domain significantly reduced the presence of ADAM10 dimer. ADAM17-B, the ADAM17 variant without its cytoplasmic domain, is a monomer; however, appending the ADAM10 cytoplasmic domain to ADAM17-B confers its ability to form a homodimer (17). In the current study, we elucidated the molecular mechanism underlying the cytoplasmic domain-mediated dimerization of ADAM10. A10Cp, representing the cytoplasmic domain of ADAM10 in isolation, is intrinsically disordered and monomeric (Figs. 2 and 3). This result is not surprising because it is enriched with prolines and polar residues (2). However, when adjoined to the neighboring transmembrane domain (i.e., within its native context), the cytoplasmic domain of ADAM10 in A10TmCp-6H takes on an ordered conformation and A10TmCp-6H forms a strong homodimer (Figs. 3 and 4). Furthermore, the transmembrane domain of ADAM10 does not dimerize in a cell membrane, and fusing the cytoplasmic domain of ADAM10 to an unrelated monomeric transmembrane sequence from l-selectin induces dimerization of the chimera (Fig. 5). Finally, dimerization of A10TmCp-6H is inhibited by DiPal-A10Cp, in which the cytoplasmic domain of ADAM10 is attached to a phospholipid molecule and thus located at the membrane surface (Fig. 6), demonstrating that the dimerization sequence motif resides entirely in the cytoplasmic domain of ADAM10.
Understanding the molecular basis for ADAM10 dimerization will facilitate characterization of the dimerization of closely related ADAM17 and elucidation of the ADAM activation mechanism. It was recently reported that TIMP-3 preferably bound ADAM17 dimer to its monomer form and thus inhibited the activity of ADAM17 in a dimeric form (17). Phosphorylation of residue Thr735 in the cytoplasmic domain of ADAM17 by activated p38 MAPK was considered a crucial step for activation of ADAM17 induced by anisomycin or IL-1β (33). However, it was also recently reported that the cytoplasmic domain of ADAM17 or its phosphorylation at Thr735 was not required for ADAM17 activation (34). In this study, we established, to our knowledge, the first system that quantitatively measures the dimerization of an ADAM cytoplasmic domain. Applying this system to ascertain the effect of Thr735 phosphorylation, as well as phosphorylation at other sites in the cytoplasmic domain (35), on the dimerization of ADAM17 cytoplasmic domain may help to reconcile the above observations. Comparison of the difference between ADAM10 and ADAM17 dimerization may also provide insights on the mechanism underlying differential activation of these two sheddases (14, 15, 36, 37).
What is unique and intriguing about our finding is that dimerization of the cytoplasmic domain of ADAM10 requires attachment to a transmembrane domain, yet the attached transmembrane domain does not directly mediate dimerization. MBP-a10Tm-AraC produced little dimerization signal in the AraTM assay, but MBP-a10TmCp-AraC produced as strong a dimerization signal as MBP-lselTm-a10Cp-AraC (Fig. 5C). Consistently, dimerization of A10TmCp-6H could be completely inhibited by DiPal-A10Cp that does not contain a transmembrane domain (Fig. 6). It appears that a role of the transmembrane domain is to anchor the cytoplasmic domain of ADAM10 in proximity to a membrane surface. What such proximity effect entails is not clear, because A10Cp is disordered in the presence of DPC micelles (Fig. S2B). It is possible that only by placing the cytoplasmic domain of ADAM10 in proximity to the membrane surface does it interact with the surface and take on a more structured conformation. This possibility is supported by our observation that DiPal-A10Cp is more structured than A10Cp and it self-associates in DPC micelles (Fig. 6). However, the ADAM10 cytoplasmic domain in DiPal-A10Cp is less structured and less resistant to trypsin digestion than that in A10TmCp, which is consistent with a lower FRET observed for the former. The comparison of DiPal-A10Cp and A10TmCp suggests an additional role of an adjoining transmembrane domain in imparting the ordered structure to the ADAM10 cytoplasmic domain.
Cytoplasmic domains of many transmembrane receptors, especially the juxtamembrane region in the cytoplasmic domain, have been reported to associate with the membrane surface. For instance, the juxtamembrane region in the transmembrane-juxtamembrane fragment of EGFR associates with the anionic membrane surface and assumes a nonhelical structure, and the membrane association can be reversed by the competing binding of calmodulin (38). Similarly, the juxtamembrane region in a transmembrane-cytoplasmic fragment of l-selectin associates with the anionic membrane surface, but it cannot be competed off the surface by calmodulin binding (39, 40). In addition, the cytoplasmic domains of CD3ε, CD3ζ, and PECAM-1 bind the membrane surface, shielding the tyrosine residues therein from phosphorylation (41–43). In contrast to these reports in which the membrane association impedes the biological function of the cytoplasmic domain, the cytoplasmic domain of ADAM10 acquires its ordered conformation and function (i.e., dimerization) when placed in proximity to the membrane surface. This feature is akin to the membrane-dependent acquisition of a stable structure by the Nogo-66 domain (44), although the cytoplasmic domain of ADAM10 is shorter than Nogo-66 and shares little sequence homology with the latter. Understanding the biophysical and energetic basis for the membrane-enabled dimerization of ADAM10 cytoplasmic domain will provide important insights on the relationship of the protein–lipid interaction and transmembrane signaling.
To assess the interaction between a cytoplasmic domain with intracellular binding proteins, it is a widely adopted practice to fuse the target cytoplasmic sequence to GST for pull-down studies. The absence of the target binding protein in the pull-down as detected by Western blot is interpreted as the lack of interaction between the said cytoplasmic domain and the protein in the cell. In this study, the clear difference between A10Cp and A10TmCp indicates that an isolated cytoplasmic domain without its neighboring transmembrane domain may not retain its endogenous binding function. Therefore, caution should be exercised regarding the interpretation of negative pull-down results of GST-cytoplasmic fusion proteins.
Materials and Methods
Detailed methods can be found in SI Materials and Methods.
Materials.
Recombinant proteins A10Cp, A10TmCp-6H, and their derivatives were produced following previously published protocols (40, 45). Synthesis of tri-NTA was carried out as previously described (22) and its identity was confirmed by MS.
Characterization of ADAM10 Fragments.
A10TmCp-6H was reconstituted in DPC micelles and POPC liposomes as detailed in SI Materials and Methods. CD spectra were collected on a JASCO J810 instrument as described previously (39). For limited trypsin digestion, the protein was mixed with trypsin to a mass ratio of 20/1 and incubated at 37 °C for 1 h, followed by LC-MS analysis using a reverse phase C4 column. The change in fluorescence anisotropy during the titration of ADAM10-derived fragments was monitored using a polarizer-equipped PTI QuantaMaster spectrometer, and the binding isotherms were fitted to a hyberbolic equation as described previously (25, 39).
FRET Measurements.
For steady-state FRET, FS-tri-NTA and TMR-tri-NTA were added separately or together in the DPC micellar solution (10 mM DPC, 20 mM Tris·HCl, 100 mM NaCl, and 1 mM DTT, pH 8.0) containing 1 µM NiSO4 to the final concentration of 10 and 100 nM, respectively. The protein stocks were diluted to the same DPC solution containing the fluorophores to the final concentration of 200 nM. Fluorescence emission spectra of these samples were recorded with excitation at 494 nm. To determine the association state of A10TmCp-6H, unlabeled tri-NTA and TMR-tri-NTA, both of which were prepared in the same DPC micellar solution and had a stock concentration of 10 µM, were added in various combinations to a solution that contained 200 nM A10TmCp-6H and 100 nM FS-tri-NTA. The overall tri-NTA concentration was kept constant at 200 nM. Quenching of FS emission intensity in each sample was recorded and plotted vs. the TMR/FS mole ratio as described previously (24, 27). To determine the Kd of the A10TmCp-6H dimer, 200 nM A10TmCp-6H, 100 nM FS-tri-NTA, and 100 nM TMR-tri-NTA in the DPC micellar solution were serially diluted while the FS fluorescence intensity was recorded. The solution containing the same fluorophores but not the protein was used as the nonquenching control. Data fitting of the plot of FRET efficiency vs. A10TmCp-6H concentration to estimate Kd are described in detail in SI Materials and Methods. For inhibition of dimerization, peptides without the hexahistidine tag were mixed with A10TmCp-6H in the DPC micellar solution containing appropriate fluorophores to various molar ratios. After incubation, FS emission intensity was measured, and the extent of fluorescence quenching was calculated as described in SI Materials and Methods.
AraTM Assay.
Expression plasmids encoding various MBP-TmCp-AraC proteins were cloned largely as described previously (28, 30). The a10Tm construct contained ADAM10 residues Leu666–Lys697, a10TmCp contained ADAM10 residues Leu666–Arg748, lselTmCp contained l-selectin residues Pro295–Tyr334, and lselTm-a10Cp contained l-selectin residues Pro295–Ala317 followed by ADAM10 residues Lys697–Arg748. Each plasmid was cotransformed with pAraGFP plasmid into E. coli strain SB1676, which were selected with 100 µg/mL ampicillin and 50 µg/mL kanamycin (28). For the DN-AraTM test, vectors encoding the AraC and DNAraC proteins were mixed equally and transformed into E. coli (29). The topology of the fusion protein was checked by its expression in E. coli MM39 cells on maltose-only growth media (28). The expression level of the MBP-TmCp-AraC protein was measured by Western blot using an anti-MBP monoclonal antibody (Sigma) as described previously (25). Measurement of GFP fluorescence in the bacterial culture was performed largely as described (28), and the results were plotted as the ratio of fluorescence emission at 530 nm to optical density at 600 nm.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health Grant GM084175.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409354111/-/DCSupplemental.
References
- 1.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 2.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 3.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580(8):2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Alonso LG, et al. High-risk (HPV16) human papillomavirus E7 oncoprotein is highly stable and extended, with conformational transitions that could explain its multiple cellular binding partners. Biochemistry. 2002;41(33):10510–10518. doi: 10.1021/bi025579n. [DOI] [PubMed] [Google Scholar]
- 5.Kami K, Chidgey M, Dafforn T, Overduin M. The desmoglein-specific cytoplasmic region is intrinsically disordered in solution and interacts with multiple desmosomal protein partners. J Mol Biol. 2009;386(2):531–543. doi: 10.1016/j.jmb.2008.12.054. [DOI] [PubMed] [Google Scholar]
- 6.Romero P, et al. Sequence complexity of disordered protein. Proteins. 2001;42(1):38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 7.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins. 2003;52(4):573–584. doi: 10.1002/prot.10437. [DOI] [PubMed] [Google Scholar]
- 8.Devarakonda S, et al. Disorder-to-order transition underlies the structural basis for the assembly of a transcriptionally active PGC-1α/ERRγ complex. Proc Natl Acad Sci USA. 2011;108(46):18678–18683. doi: 10.1073/pnas.1113813108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glaves JP, Trieber CA, Ceholski DK, Stokes DL, Young HS. Phosphorylation and mutation of phospholamban alter physical interactions with the sarcoplasmic reticulum calcium pump. J Mol Biol. 2011;405(3):707–723. doi: 10.1016/j.jmb.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arribas J, Borroto A. Protein ectodomain shedding. Chem Rev. 2002;102(12):4627–4638. doi: 10.1021/cr010202t. [DOI] [PubMed] [Google Scholar]
- 11.Reiss K, et al. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24(4):742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prinzen C, et al. Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice. BMC Genomics. 2009;10:66. doi: 10.1186/1471-2164-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber S, et al. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development. 2011;138(3):495–505. doi: 10.1242/dev.055210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanderson MP, et al. ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J Biol Chem. 2005;280(3):1826–1837. doi: 10.1074/jbc.M408804200. [DOI] [PubMed] [Google Scholar]
- 15.Horiuchi K, et al. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18(1):176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allinson TM, et al. The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. Eur J Biochem. 2004;271(12):2539–2547. doi: 10.1111/j.1432-1033.2004.04184.x. [DOI] [PubMed] [Google Scholar]
- 17.Xu P, Liu J, Sakaki-Yumoto M, Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci Signal. 2012;5(222):ra34. doi: 10.1126/scisignal.2002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang T, et al. SPINE-D: Accurate prediction of short and long disordered regions by a single neural-network based method. J Biomol Struct Dyn. 2012;29(4):799–813. doi: 10.1080/073911012010525022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2011;80:374–381. doi: 10.1002/prot.23188. [DOI] [PubMed] [Google Scholar]
- 20.Noda Y, Fujiwara K, Yamamoto K, Fukuno T, Segawa S. Specificity of trypsin digestion and conformational flexibility at different sites of unfolded lysozyme. Biopolymers. 1994;34(2):217–226. doi: 10.1002/bip.360340208. [DOI] [PubMed] [Google Scholar]
- 21.Coulter-Mackie MB, Lian Q. Partial trypsin digestion as an indicator of mis-folding of mutant alanine:glyoxylate aminotransferase and chaperone effects of specific ligands. Study of a spectrum of missense mutants. Mol Genet Metab. 2008;94(3):368–374. doi: 10.1016/j.ymgme.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Huang Z, Park JI, Watson DS, Hwang P, Szoka FC., Jr Facile synthesis of multivalent nitrilotriacetic acid (NTA) and NTA conjugates for analytical and drug delivery applications. Bioconjug Chem. 2006;17(6):1592–1600. doi: 10.1021/bc0602228. [DOI] [PubMed] [Google Scholar]
- 23.Zhou FX, Merianos HJ, Brunger AT, Engelman DM. Polar residues drive association of polyleucine transmembrane helices. Proc Natl Acad Sci USA. 2001;98(5):2250–2255. doi: 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7(2):161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 25.Luo SZ, Li R. Specific heteromeric association of four transmembrane peptides derived from platelet glycoprotein Ib-IX complex. J Mol Biol. 2008;382(2):448–457. doi: 10.1016/j.jmb.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You M, Li E, Wimley WC, Hristova K. Forster resonance energy transfer in liposomes: Measurements of transmembrane helix dimerization in the native bilayer environment. Anal Biochem. 2005;340(1):154–164. doi: 10.1016/j.ab.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adair BD, Engelman DM. Glycophorin A helical transmembrane domains dimerize in phospholipid bilayers: A resonance energy transfer study. Biochemistry. 1994;33(18):5539–5544. doi: 10.1021/bi00184a024. [DOI] [PubMed] [Google Scholar]
- 28.Su PC, Berger BW. Identifying key juxtamembrane interactions in cell membranes using AraC-based transcriptional reporter assay (AraTM) J Biol Chem. 2012;287(37):31515–31526. doi: 10.1074/jbc.M112.396895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su PC, Berger BW. A novel assay for assessing juxtamembrane and transmembrane domain interactions important for receptor heterodimerization. J Mol Biol. 2013;425(22):4652–4658. doi: 10.1016/j.jmb.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasan S, Deng W, Li R. L-selectin transmembrane and cytoplasmic domains are monomeric in membranes. Biochim Biophys Acta. 2011;1808(6):1709–1715. doi: 10.1016/j.bbamem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, et al. P-selectin glycoprotein ligand-1 forms dimeric interactions with E-selectin but monomeric interactions with L-selectin on cell surfaces. PLoS ONE. 2013;8(2):e57202. doi: 10.1371/journal.pone.0057202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lauterwein J, Bösch C, Brown LR, Wüthrich K. Physicochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim Biophys Acta. 1979;556(2):244–264. doi: 10.1016/0005-2736(79)90046-4. [DOI] [PubMed] [Google Scholar]
- 33.Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell. 2010;37(4):551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall KC, Blobel CP. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLoS ONE. 2012;7(2):e31600. doi: 10.1371/journal.pone.0031600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan H, Turck CW, Derynck R. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. J Biol Chem. 2003;278(20):18617–18627. doi: 10.1074/jbc.M300331200. [DOI] [PubMed] [Google Scholar]
- 36.Nagano O, et al. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J Cell Biol. 2004;165(6):893–902. doi: 10.1083/jcb.200310024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blobel CP. ADAMs: Key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6(1):32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 38.Sato T, Pallavi P, Golebiewska U, McLaughlin S, Smith SO. Structure of the membrane reconstituted transmembrane-juxtamembrane peptide EGFR(622-660) and its interaction with Ca2+/calmodulin. Biochemistry. 2006;45(42):12704–12714. doi: 10.1021/bi061264m. [DOI] [PubMed] [Google Scholar]
- 39.Deng W, Srinivasan S, Zheng X, Putkey JA, Li R. Interaction of calmodulin with L-selectin at the membrane interface: Implication on the regulation of L-selectin shedding. J Mol Biol. 2011;411(1):220–233. doi: 10.1016/j.jmb.2011.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng W, Cho S, Li R. FERM domain of moesin desorbs the basic-rich cytoplasmic domain of l-selectin from the anionic membrane surface. J Mol Biol. 2013;425(18):3549–3562. doi: 10.1016/j.jmb.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu C, et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135(4):702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7(11):1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- 43.Paddock C, et al. Residues within a lipid-associated segment of the PECAM-1 cytoplasmic domain are susceptible to inducible, sequential phosphorylation. Blood. 2011;117(22):6012–6023. doi: 10.1182/blood-2010-11-317867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vasudevan SV, Schulz J, Zhou C, Cocco MJ. Protein folding at the membrane interface, the structure of Nogo-66 requires interactions with a phosphocholine surface. Proc Natl Acad Sci USA. 2010;107(15):6847–6851. doi: 10.1073/pnas.0911817107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo S-Z, et al. Glycoprotein Ibalpha forms disulfide bonds with 2 glycoprotein Ibbeta subunits in the resting platelet. Blood. 2007;109(2):603–609. doi: 10.1182/blood-2006-05-024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.