

Significance
Torsin activity critically depends on accessory cofactors that activate their ATPase activity by a poorly understood mechanism. This study establishes the mechanistic framework for Torsin activation, which relies on a complementation of the fragmentary Torsin active site. Given these unusual properties and the fact that Torsin activation is dysregulated in the congenital movement disorder primary dystonia, our results suggest that pharmacological manipulation of Torsin activation may present a novel therapeutic opportunity.
Keywords: DYT1 dystonia, nuclear envelope, ATPase, Torsin
Abstract
Torsins are membrane-associated ATPases whose activity is dependent on two activating cofactors, lamina-associated polypeptide 1 (LAP1) and luminal domain-like LAP1 (LULL1). The mechanism by which these cofactors regulate Torsin activity has so far remained elusive. In this study, we identify a conserved domain in these activators that is predicted to adopt a fold resembling an AAA+ (ATPase associated with a variety of cellular activities) domain. Within these domains, a strictly conserved Arg residue present in both activating cofactors, but notably missing in Torsins, aligns with a key catalytic Arg found in AAA+ proteins. We demonstrate that cofactors and Torsins associate to form heterooligomeric assemblies with a defined Torsin–activator interface. In this arrangement, the highly conserved Arg residue present in either cofactor comes into close proximity with the nucleotide bound in the neighboring Torsin subunit. Because this invariant Arg is strictly required to stimulate Torsin ATPase activity but is dispensable for Torsin binding, we propose that LAP1 and LULL1 regulate Torsin ATPase activity through an active site complementation mechanism.
Torsin ATPases belong to the AAA+ (ATPase associated with a variety of cellular activities) superfamily of ATPases (1) but are unusual in that they lack conserved catalytic residues that are typically found in related ATPases (2, 3). Accordingly, Torsins do not display ATPase activity unless they are engaged by their regulatory cofactors lamina-associated polypeptide 1 (LAP1) or luminal domain-like LAP1 (LULL1) (4), which are type II transmembrane proteins residing in the nuclear envelope and endoplasmic reticulum (ER) (5, 6). This property stands in sharp contrast to the behavior of closely related Clp/Hsp100 ATPases, which display considerable basal ATPase activities that are moderately stimulated by their substrates (7, 8). Although a number of Clp/Hsp100 proteins use distinct cofactors that confer substrate specificity to the typically hexameric ATPase ring, they operate via similar principles (9). Substrates ultimately engage the pore at the center of the oligomeric assembly. This narrow annulus is defined by pore loops that emanate from each subunit and harbor a conserved aromatic residue that defines the center of the pore (10). The energy of ATP hydrolysis is invested in threading the substrate through this axial channel, and the substrate is unfolded in the process (11).
Much less is known about the mode of action of Torsin ATPases, although it is clear that regulation of Torsin activity is of vital importance in an organismal context. A mutation in TorsinA (TorA) causing the movement disorder primary dystonia (12) renders TorA unresponsive to its binding partners LAP1 and LULL1 (4), and a homozygous “knock-in” of this disease allele in mice causes a lethal phenotype, as does a TorA KO (13). A conditional deletion of TorA from the CNS in mice accurately replicates the symptoms of primary dystonia (14). In addition, LAP1-deficient mice display early perinatal lethality (15), again corroborating the physiological significance of regulation and dysregulation of Torsin activity.
Given the potency with which LAP1 and LULL1 induce ATP hydrolysis by Torsins, we suggested previously that LAP1 and LULL1 act as activating cofactors (4). However, the mechanism by which LAP1 and LULL1 induce Torsin ATPase activity has remained unclear.
In this study, we report a previously undetected predicted structural similarity between LAP1 and LULL1 activators and the AAA+ domain fold. Our modeling led to the identification of a conserved Arg residue in these activators that is necessary for ATPase activity in all known AAA+ ATPases but is missing from the Torsin family. This Arg is positioned at the activator–Torsin interface that we validate via site-specific cross-linking, and is vital for both LAP1- and LULL1-stimulated Torsin activity. We thus propose that LAP1 and LULL1 integrate into the Torsin ring to produce the biologically active ATPase machine.
Results
Structural Model for the Torsin–Cofactor Assembly.
The strict dependency of Torsin ATPase activity on LAP1 or LULL1 (4) prompted us to scrutinize these cofactors from a structural perspective with the aim of gaining insight into the underlying mechanism. We started our analysis by using HHpred (16) to search for likely structural homologs of LAP1 and LULL1. For LULL1, HHpred revealed homologies to several membrane fusion proteins in the N-terminal (cytosolic) domain (Dataset S1), the significance of which shall be addressed elsewhere. Surprisingly, the bulk of the LULL1 luminal domain that is necessary and sufficient for ATPase stimulation (4) is predicted to adopt a fold that is similar to the AAA+ domains of a plethora of AAA+ proteins (Dataset S1). This unanticipated structural homology was also found in LAP1 (Dataset S2), which is expected because the luminal domains of LAP1 and LULL1 share 60% sequence identity. This compelling homology between the luminal domains of LAP1/LULL1 and AAA+ ATPases on a secondary structure level (25 and 18 AAA+ domains were detected as homologous to LAP1 and LULL1, respectively) has escaped previous scrutiny, presumably because the defining sequence elements required for ATP binding and hydrolysis found in all AAA+ ATPases (1) are not conserved in either cofactor. Accordingly, a BLAST search (17) did not detect a significant homology to AAA+ ATPases due to limited sequence identity. Consistent with the lack of conservation in elements required for nucleotide binding, LAP1 or LULL1 alone does not display ATPase activity (4). Together, these findings underscore the usefulness of bioinformatics tools, such as HHpred, in revealing structural homologies between proteins that lack obvious sequence identity.
We next exploited the predicted structural similarity between LAP1/LULL1 and AAA+ modules and subjected the luminal domains of LAP1 and LULL1 to molecular modeling using an unbiased approach (Methods). The resulting structural models for LAP1 and LULL1 encompass the entire luminal domain of either cofactor (amino acids 356–583 and 236–470, respectively). Both models are indeed highly similar to the N-terminal alpha/beta-fold [or RecA fold (18)] of the AAA+ domain but do not feature a C-terminal four-helix bundle that is usually present in AAA+ domains (19) (Fig. 1 A and B).
Fig. 1.

LAP1 and LULL1 luminal domains are predicted to adopt an AAA+-like fold. (A) Predicted structural model of LAP1’s luminal domain using Phyre2. (B) As in A, but for LULL1. (C) Oligomeric model of TorA (green and yellow) using a least-squares superposition of alpha-carbons (Coot) onto ClpC’s hexameric structure (Protein Data Bank ID code 3PXI) bound to adenylylimidodiphosphate (AMPPNP). A least-squares superposition of LULL1LD (magenta) onto a TorA monomer is shown. AMPPNP is colored by element. The arrowhead indicates TorA’s C terminus. (D, Left) Proposed mixed-ring assembly of LAP1/LULL1 (blue) with TorA (green). CNDs, cytoplasmic/nuclear domains; PNS, perinuclear space; LDs, luminal domains. (D, Right) Space-filling structural model of the LAP1/LULL1-TorA heterodimer. Regions not modeled are shown as dashed lines, membrane domains are shown as blocks, and the CND is shown as an ellipse.
Given the striking structural similarity of LAP1 and LULL1 to AAA+ domains, one could envision that TorA and these cofactors can form mixed ring assemblies in which the luminal domains of LAP1/LULL1 assume a position that is equivalent to a regular AAA+ subunit. Based on structural superimpositions of molecular models of TorA and LULL1 onto the hexameric structure of the related AAA+ ATPase ClpC (20), such an arrangement appears feasible (Fig. 1 C and D). Because LAP1 and LULL1 are transmembrane proteins and Torsins are anchored to the ER/perinuclear space (PNS)-luminal side of the membrane, we can envision a heterooligomeric assembly of the type depicted in Fig. 1D. In this theoretical assembly, the luminal domains of LAP1 (or LULL1) would form a mixed ring with Torsin subunits in apposition to the membrane, whereas the nuclear and cytosolic domains of LAP1 and LULL1 would face the nucleus or cytosol, respectively (Fig. 1D).
We next investigated whether this proposed assembly is supported by the ratios of Torsin and activator molecules found in vivo. Copy numbers of TorA, TorsinB (TorB), LAP1, and LULL1 were approximated in three human cell types via quantitative immunoblotting (Methods, Table 1, and Fig. S1). The ratios obtained by dividing the sum of TorA and TorB molecules by the sum of the LAP1 and LULL1 molecules are 0.55 for HEK 293T cells, 1.4 for HeLa cells, and 0.82 for fibroblasts. Bearing in mind the limited accuracy of immunoblotting, as well as differences in gene dosage stemming from aneuploidy frequently observed in cell lines, we conclude that TorA, TorB, and their activators are present in approximately equimolar ratios.
Table 1.
Cellular copy numbers for TorA, TorB, LULL1, and LAP1
| Human cell line | Copies of TorA | Copies of TorB | Copies of LULL1 | Copies of LAP1 |
| HEK 293T | 1.5 ± 0.2 × 105 | 5.0 ± 0.6 × 104 | 1.9 ± 0.2 × 105 | 1.7 ± 0.6 × 105 |
| HeLa | 2.9 ± 0.5 × 105 | 7.8 ± 0.9 × 104 | 1.1 ± 0.06 × 105 | 1.6 ± 0.5 × 105 |
| Foreskin fibroblast | 9.2 ± 0.4 × 105 | 3.2 ± 0.6 × 105 | 9.4 ± 0.6 × 105 | 5.8 ± 0.9 × 105 |
TorA and Its Activators Form Mixed Oligomeric Assemblies in a 1:1 Ratio.
The interaction of Torsin with its cofactors is highly dynamic but strongly stabilized by a Walker B mutation (E171Q) that arrests TorA in the ATP-bound state (3, 4, 21). We therefore coexpressed TorA lacking its hydrophobic domain [maltose-binding protein (MBP)-TorA E171Q/Δ49] and the luminal domains of LAP1 or LULL1 in bacteria, and purified the resulting complexes in the presence of ATP to maintain a stable stoichiometry [Methods and Fig. S2 (equivalent WT complexes display enzymatic activity)]. Purified complexes were subjected to size exclusion chromatography as the final purification step, and the resulting fractions were resolved by SDS/PAGE. In both cases, TorA and cofactor coeluted at a position consistent with the expected mass of a heterodimer (Fig. 2 A and B). The observed ∼1:1 ratio of MBP-TorA E171Q/Δ49 and either cofactor seen in SDS/PAGE analysis (Fig. 2A) corroborates our interpretation of a heterodimeric complex.
Fig. 2.

TorA and LAP1 or LULL1 form a 1:1 stoichiometric complex. (A) Complex of MBP-tagged TorA Δ49EQ and LAP1LD or LULL1LD was analyzed by size exclusion chromatography. Elution positions of size markers are indicated by arrows. Elution fractions were subjected to SDS/PAGE, followed by colloidal blue staining. EQ, TorA E171Q Walker B mutant. (B) Size exclusion chromatography-purified complexes were incubated at room temperature for 10 min in the absence or presence of 0.1% (wt/vol) glutaraldehyde (GA) and resolved by gradient SDS/PAGE (6–9%), followed by silver staining.
Because low-affinity interactions can be disrupted by size exclusion chromatography, we additionally investigated the oligomeric state of the complexes (corresponding to the peak fraction in Fig. 2A) using a cross-linking approach. We used gentle glutaraldehyde cross-linking conditions that were previously used to establish the oligomeric state of the related AAA+ ATPase ClpB (22), which was independently confirmed by structural means (23). Following cross-linking for a duration that was just sufficient to deplete free cofactor and Torsin (t = 10 min), two major species were observed in a 6–9% gradient gel (Fig. 2B): a smaller species with an apparent mass of 80–120 kDa and a second species of ∼300–350 kDa (Fig. 2B and Fig. S3). The former corresponds to the heterodimer also observed via size exclusion chromatography (Fig. 2A). The 300- to 350-kDa species is consistent with the assembly of a mixed ring in which three Torsin-activator heterodimers associate to form a trimer of heterodimers, assuming that the Torsin/cofactor ratio is 1:1 as suggested above.
Confirmation of the Cofactor–TorA Interface by Site-Specific Cross-Linking.
We next focused on validating our proposal that LAP1 and LULL1 bind to TorA in a manner equivalent to a regular AAA+ subunit (Fig. 1C). As we will discuss in detail later, it is this nucleotide–proximal activator interface that is key for Torsin activation, irrespective of the precise stoichiometry of the higher order assembly. Importantly, our proposal is consistent with our previous finding that residues at the extreme C terminus of TorB are required for cofactor interaction, membrane remodeling, and ATPase stimulation (24). According to our model, these residues constitute the C-terminal alpha-helix (denoted by an arrowhead in Fig. 1C, Right) that contributes substantially to this LULL1–TorA (or LAP1–TorA) interface (Fig. 1C), explaining why nonconservative mutations or C-terminal deletions strongly impair cofactor binding and, consequently, ATPase induction. However, these previous data in isolation would also be consistent with a more indirect effect. We therefore set out to define the predicted cofactor–TorA interface using a site-specific cross-linking approach.
To this end, we coexpressed TorA (Δ51/E171Q), together with LAP1 luminal domain (LAP1LD) in Escherichia coli, and installed the UV cross-linkable amino acid analog p-benzoyl-l-phenylalanine (BPA) (25) at one of the three adjacent positions on TorA’s C-terminal alpha-helix that are expected to contribute to the interface (Fig. 3 A and B). We additionally engineered equivalent Torsin variants carrying the disease-causing single-glutamate deletion (ΔE302/E303 or ΔE). This mutation was previously shown to disrupt cofactor binding with a concomitant loss of ATPase activity (3, 4, 21). The Torsin–BPA/LAP1 complexes were partially purified in the presence of ATP by a single purification step. All BPA derivatives and cofactors were present in comparable quantities (Fig. S4). We then exposed one aliquot of each sample to UV light, isolated TorA by immunoprecipitation, and resolved the resulting complexes by SDS/PAGE. Cross-linking products were detected by immunoblotting. Spontaneous UV cross-linking was negligible, as judged by the absence of a cross-linking product in our negative control lacking BPA (Fig. 3C). A major UV-dependent cross-linking product of the expected molecular mass of 57 kDa was observed for all BPA derivatives, with slightly varying efficacies (Fig. 3C). Minor higher molecular mass cross-linking products were also observed, presumably arising from Torsin–Torsin–cofactor cross-linked assemblies. Notably, cross-linking products were absent or profoundly reduced in all cases in the presence of the additional ΔE mutation. Next, we treated an aliquot of each eluate with calf intestinal phosphatase (CIP) to remove ATP, allowing us to test whether the cross-linking efficacy mirrors the expected nucleotide dependency. Indeed, readdition of ATP strongly increased cross-linking efficacy relative to the aliquot exposed to CIP, whereas addition of ADP had no effect (Fig. 3D). We further showed that this equivalent Torsin–activator interface exists with LULL1 as well (Fig. 3E), consistent with our previous data for LULL1 and TorB (24).
Fig. 3.

Site-specific cross-linking between TorA and LAP1/LULL1. (A) Predicted interface between LAP1 (blue) and TorA (green). Cross-linker pBPA (orange) was installed on TorA at three sites (D327BPA, Y328BPA, and Y329BPA) at the predicted interface with LAP1. (B) Zoomed-in view of TorA and LAP1 interface. TorA D327BPA, Y328BPA, and Y329BPA are shown in orange. (C) HIS-LAP1LD and TorA-Δ51 constructs were copurified from E. coli in 1 mM ATP. Copurified complexes were incubated at 30 °C for 5 min, cooled to 20 °C, and UV-irradiated for 15 min. Cross-linked species were immunoprecipitated with a TorA antibody, resolved by SDS/PAGE, and analyzed via immunoblotting using HIS antibody to detect LAP1. Arrows indicate the Ig heavy chain, and arrowheads indicate the TorA-LAP1 cross-linked species. Note that the order of loading (±UV) was inadvertently reversed for Y329BPA. IP, immunoprecipitate. (D) Copurified HIS-LAP1LD and TorA-Δ51 constructs were treated with CIP overnight at 4 °C to remove nucleotide. ATP or ADP (10 mM) was added back to the cross-linking reaction. Cross-linked species were analyzed as described in C. (E) As in C, but for HIS-LULL1LD.
All in all, these cross-linking experiments faithfully recapitulate the previously established nucleotide dependency of the cofactor interaction, as well as its disruption by the ΔE mutation (3, 4, 21). Notably, similar results were obtained when BPA was installed instead on the activator side of the interface for both LAP1 and LULL1 in the context of full-length TorA (Fig. S5). Thus, the proposed activator interface is supported by several independent cross-linking experiments (Fig. 3 C–E and Fig. S5) and is in excellent agreement with all previously published mutagenesis and activity data (24).
Molecular Modeling Reveals the Presence of an Arginine Finger in LAP1 and LULL1.
Given that Torsins are outliers of the AAA+ family in that they lack nucleotide-proximal arginine (Arg) residues (3) and the similarity of Torsins to small GTPases in terms of their biochemical behavior (4, 26), it is a natural hypothesis to propose that this missing Arg may be located on the activating cofactor. Indeed, according to our model, both LAP1 and LULL1 project an Arg residue (LAP1 R563 and LULL1 R449) into the activator interface in close proximity to the gamma-phosphate of the nucleotide bound to the neighboring TorA subunit (Fig. 4B). This positioning is comparable to the Arg side chain that is known to act as the Arg finger in ClpB and ClpC (20, 27). This position is equivalent to the designated P1 Arg according to the nomenclature suggested by Ogura et al. (28), which is precisely the position where Torsins lack an Arg residue typically found in AAA ATPases. Somewhat unexpectedly, we found that TorA also has a nucleotide-proximal Arg (R260) in the vicinity of the expected Arg finger position. However, because this transacting Arg residue cannot properly function in a homooligomeric Torsin assembly, this Arg is not required for Torsin’s ATPase activity (Fig. S6).
Fig. 4.
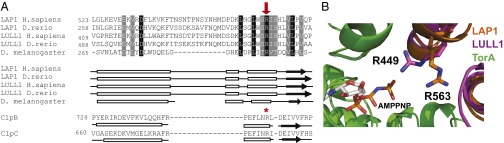
Conserved Arg residue in LAP1 and LULL1 aligns with the Arg finger of other AAA+ proteins. (A) Alignment of LAP1 and LULL1 amino acid sequences to ClpB and ClpC using predicted structural homology. LAP1 and LULL1 sequences from Homo sapiens (NP_056417 and NP_001186189), Danio rerio (NP_001017552 and XP_001339602), and Drosophila melanogaster (NP_649149) were aligned to each other using ClustalW and formatted using BoxShade; black boxes indicate strict amino acid agreement, and gray boxes indicate similar amino acid agreement. Predicted secondary structures were generated and aligned to Thermus thermophilus ClpB and Bacillus subtilis ClpC using HHPred; boxes represent helices, arrows indicate sheets, and lines indicate random coils. A conserved Arg from LAP1 (H. sapiens R563) and LULL1 (H. sapiens R449) aligns with ClpB’s and ClpC’s AAA-2 Arg finger (R747 and R704, respectively) and is indicated with a red arrow and a red star. (B) Zoomed-in view of LAP1 and LULL1’s Arg fingers in proximity to AMPPNP-bound TorA (green). LAP1 R563 (orange) and LULL1 R449 (magenta) are shown.
If the transactivator Arg residues are indeed critical for LAP1/LULL1’s ability to stimulate Torsins, we would expect them to be strictly conserved between species. Indeed, sequence alignments, including LAP1/LULL1 representatives, ranging from mammals to invertebrates confirm that this Arg residue is among the very few residues that are absolutely conserved in both cofactors (Fig. 4A and Fig. S7). We therefore propose that LAP1 R563 and LULL1 R449 complement the Torsin ATPase active site upon association.
Proposed Arginine Finger Residues in LAP1 and LULL1 Are Not Required for Torsin Binding.
To test our prediction, we made a series of mutants in which the proposed Arg finger was mutated to alanine or glutamate, resulting in the derivatives LAP1 R563A and R563E and LULL1 R449A and R449E, respectively. To exclude the possibility that the mutation of a surface-exposed charged residue perturbs Torsin binding, we first assessed whether these mutant derivatives are still functional in binding assays. To this end, we cotransfected TorA and the respective HA-tagged luminal domains and their mutant derivatives in 293T cells. We limited our analysis to HA-tagged variants of the luminal domains of LAP1/LULL1 and the hydrolysis-deficient TorA trap mutant TorA E171Q because LAP1 and LULL1 preferentially interact with TorA in its ATP-bound conformation and the luminal domains are necessary and sufficient for this interaction (3, 4, 21). Mild detergent extracts were prepared 24 h posttransfection and subjected to immunoprecipitation using anti-HA. The resulting immunoprecipitates and input controls were then analyzed by SDS/PAGE and subsequent immunoblotting. All constructs were expressed to comparable levels in these transfections (Fig. 5A), suggesting that the mutations did not perturb the structure to a degree that it would make them susceptible to proteolysis. As expected, TorA E171Q efficiently precipitated with the luminal domain of either LAP1 or LULL1 (Fig. 5A), indicative of a strong interaction. Most importantly, the mutant LAP1 and LULL1 proteins bound TorA E171Q to the same extent as their WT counterparts (Fig. 5A).
Fig. 5.

Mutations of LAP1 and LULL1 Arg residues do not affect Torsin binding. (A) LAP1 and LULL1 Arg mutants interact with TorA in vivo. The 293T cells were transfected with TorA E171Q and the indicated LD constructs, lysed with Nonidet P-40 (Roche), and immunoprecipitated using an HA antibody. Input controls and immunoprecipitates were resolved by SDS/PAGE and blotted with the indicated antibodies. IB, immunoblot. (B) LDs of LAP1 or LULL1 were incubated at 30 °C in the absence or presence of equal molar TorA E171Q and 2 mM ATP. Protein complex formation was analyzed by size exclusion chromatography. Elution positions of size markers are indicated by arrows. Elution fractions were subjected to immunoblotting using the indicated antibodies. (C) Identical analysis for LAP1LD R563A or LULL1LD R449A. (D) Identical analysis for LAP1LD R563E or LULL1LD R449E.
To exclude confounding effects by other binding partners that may be present in a cellular context, we confirmed these results in our previously established in vitro binding assay (4). In brief, this assay relies on size exclusion chromatography to monitor the shift of LAP1/LULL1 from its free monomeric form to the TorA E171Q-associated form, which elutes at an earlier position corresponding to an increase in the apparent molecular mass of the TorA E171Q–cofactor complex relative to the free cofactor. As a positive control, we subjected LULL1 and LAP1 to the column, either alone or in the presence of TorA E171Q and ATP. A profound shift to the higher molecular mass was observed in the presence of TorA E171Q, consistent with our previous observations (4) (Fig. 5B). We next tested the Arg mutant derivatives LAP1LD R563A and R563E and LULL1LD R449A and R449E. LULL1LD R449A eluted somewhat earlier in its free form compared with LULL1LD; however, in the additional presence of TorA E171Q, it was shifted to an earlier elution position, as was observed for its WT counterpart (compare Fig. 5 B and C). The other LAP1 and LULL1 mutant derivatives followed the pattern previously seen for their WT derivatives in that they were efficiently shifted to the high molecular mass fractions upon addition of TorA E171Q and ATP, relative to their free forms (Fig. 5 C and D). We conclude that all Arg mutants bind to TorA E171Q with an affinity that is highly similar or identical to their WT counterparts.
Conserved Arg Residue in LAP1 and LULL1 Is Essential for ATPase Induction.
Having ruled out major effects on Torsin binding, we next investigated the effect of the cofactor Arg mutations on ATP hydrolysis (Fig. 6). TorA WT was incubated either alone or with an equimolar concentration of cofactor, and ATPase activity was measured under steady-state conditions using a malachite green assay. As expected, ATPase activity was negligible in the absence of cofactors. ATPase activity was strongly increased in the presence of LAP1LD or LULL1LD, with the latter being the more potent activator (Fig. 6 A and B). At equivalent concentrations, the potency of ATPase stimulation by LAP1LD R563A and LULL1LD R449A was severely reduced (Fig. 6 A–D). The stimulatory effect of the cofactors was reduced to background levels when their putative Arg fingers were mutated to a glutamate (LAP1LD R563E and LULL1LD R449E) (Fig. 6 A–D). We conclude that the potency of ATPase induction is a function of the identity of the side chain at a position that is equivalent to the transacting Arg finger in AAA+ ATPases.
Fig. 6.

LAP1 and LULL1 Arg mutants fail to stimulate TorA’s ATPase activity. (A and B) TorA ATP hydrolysis rates in the presence of WT or Arg mutant cofactors. ATP hydrolysis rates were measured as a function of Pi-production over time. TorA (3 μM) was incubated with ATP (2 mM) at 37 °C, either alone or with 3 μM indicated WT or mutant cofactor, and Pi-production was measured at various time points using a malachite green assay. Data were fit with a linear regression in Prism (GraphPad) to yield the ATP hydrolysis rate. (C and D) Rate constants were obtained by dividing the ATP hydrolysis rate by the TorA concentration. (E and F) ATP single-turnover kinetics of TorA in the presence of WT or Arg mutant cofactor. Each data point represents the mean of three independent experiments. Error bars indicate SD. (G) Rate constants were obtained by fitting the data from E and F with a single exponential decay function in Prism. TorA basal ATP hydrolysis could not be fit to a single exponential decay function in a statistically significant manner. (H) LAP1 R563 and LULL1 R449 are required to stimulate TorB’s ATPase activity. TorB (3 μM) was incubated with ATP (2 mM), either alone or with 3 μM WT or Arg mutant cofactor. ATP hydrolysis was measured as a function of Pi-production after 60 min using a malachite green assay.
Arg Finger Is Involved in Nucleotide Hydrolysis.
We next determined if the profound effect on cofactor-stimulated TorA ATPase activity by mutation of the LAP1 or LULL1 Arg finger could be attributed to changes in the efficiency of ATP hydrolysis. To this end, we isolated a complex of TorA with alpha-32P ATP specifically to measure the hydrolysis step under single-turnover conditions (4). As described previously, ATP hydrolysis by TorA was negligible unless it was stimulated by LAP1 or LULL1 (4) (Fig. 6 E–G). All of the Arg mutants, at concentrations identical to their WT counterparts, displayed a markedly reduced potency as ATPase activators (Fig. 6 E–G). As expected, this effect was more pronounced for the Arg-to-glutamate mutants, LAP1LD R563E and LULL1LD R449E, than for the Ala variants, LAP1LD R563A and LULL1LD R449A (Fig. 6 E–G). These measurements indicate that ATP hydrolysis by TorA is strongly compromised if the Arg corresponding to the Arg finger in structurally characterized AAA+ ATPases is mutated in these Torsin activators.
Arg-Based Activation Mechanism Is Conserved.
Finally, we determined if the TorA activation mechanism is conserved in other representatives of the Torsin family. To this end, we measured the ATPase activity of another member of the Torsin family, namely TorB, under steady-state conditions and determined the effect of the Arg mutations. As described previously, TorB behaves like TorA in that ATPase activity strictly depends on LAP1 and LULL1 (4, 24) (Fig. 6H). As with TorA, the potency of ATPase stimulation of TorB is strongly reduced for all four Arg mutants relative to their WT counterparts, and a more severe effect is imposed by the glutamate mutations relative to the Ala mutations (Fig. 6H). Thus, the regulatory mechanism revealed in this study is conserved within the Torsin family.
Discussion
Deciphering the mechanism of Torsin regulation is a critical step toward a molecular understanding of Torsin function and its associated pathologies. Having previously established that Torsins do not display ATPase activity unless they encounter their binding partners LAP1 or LULL1 (4), one major question concerning the regulation of Torsin activity remained: How is the ATPase activation achieved on a molecular level? The kinetic properties seen for ATPase induction by LAP1 and LULL1 (4) (Fig. 6) are quite distinct from those kinetic properties seen in related AAA+ ATPases. Instead, the biochemical behavior of Torsin ATPases is more similar to the biochemical behavior of small GTPases: GTP hydrolysis is barely detectable in the absence of accessory cofactors [GTPase-activating proteins (GAPs)] but is induced by orders of magnitude upon addition of the cofactor that engages its cognate GTPase in its nucleotide triphosphate-bound form (26). These properties apply to Torsin and LAP1/LULL1 as well (4) (Fig. 6). LAP1/LULL1 accelerates the hydrolysis step by over two orders of magnitude at physiological temperature (Fig. 6 E and F).
Due to the apparent functional similarity between GAPs and LAP1/LULL1, we speculated that LAP1 and LULL1 function as cofactors of Torsin ATPases. We therefore used structural predictions to identify elements in LAP1 and LULL1 that could perform this function. This strategy revealed a striking structural homology to AAA+ domains (Fig. 1 A and B), even without conservation of sequence motifs that are usually present in AAA+ family proteins (1). Importantly, one key structural element that is conserved in both cofactors is an Arg residue (Fig. 4A and Fig. S7), which is positioned analogously to the P1 Arg finger in related ATPases (28). It is well established that GAPs operate by an active site complementation mechanism in which binding of the GAP to the GTPase contributes an Arg finger that catalytically stabilizes negative charges in the transition state of the GTP hydrolysis reaction (26). Here, we propose a similar, although not identical, mechanism for LAP1 and LULL1 stimulation of Torsin. The fact that Torsins lack a conserved Arg residue (3) that is otherwise highly conserved in AAA+ ATPases (1, 28) supports our proposal. Furthermore, the magnitude of the decrease in Torsin activity seen upon mutation of these Arg residues in LAP1 and LULL1 (for steady-state conditions: 10-fold for LULL1 R449A, 95-fold for LULL1 R449E, 17-fold for LAP1 R563A and 55-fold for LAP1 R563A) (Fig. 6 C and D) is within the range of analogous mutations in related AAA+ ATPases harboring a canonical Arg finger (22, 29, 30). Moreover, these Arg residues are strictly conserved in LAP1 and LULL1 homologs throughout evolution (Fig. 4A and Fig. S7). Final confirmation that these residues are functioning via this mechanism must come from a highly resolved structure of LAP1 and/or LULL1 in complex with TorA, ideally in the presence of a transition state analog. It is noteworthy that the effect imposed by Arg mutations is less pronounced under single-turnover conditions relative to steady-state measurements (compare Fig. 6 A–D and E–G), indicating that there must be an additional role for the Arg other than accelerating hydrolysis. In fact, the role of nucleotide-proximal Arg residues in AAA+ ATPases is more diverse than in GTPases (28), and might serve additional functions (e.g., intersubunit communication) (31). We therefore propose that the term “Arg trigger” might be appropriate in this case, to clearly distinguish between the Arg fingers of GTPases (26).
Although the precise in vivo stoichiometry of the Torsin–cofactor complex remains to be defined, a mixed ring assembly with a 1:1 stoichiometry suggested by our cross-linking data (Fig. 2) is biologically feasible based on the cellular copy numbers of Torsins and cofactors (Table 1). However, it is very clear that this assembly will be significantly more dynamic if not arrested by the Walker B mutation and cross-linking reagents. Various but not mutually exclusive symmetries and stoichiometries are feasible from a structural perspective. Importantly, however, these assemblies would all be reliant on the transactivation mechanism that was borne out by this study, and must therefore feature the activator interface identified by us.
Given that the disease-causing glutamate deletion in TorA (12) maps to the C-terminal four-helix bundle that contributes to the cofactor–Torsin interface (4) (Figs. 1C and 3A and Fig. S5E), it seems reasonable to propose that the dysfunction of this mutant is caused, at least in part, by a misalignment of the activator interface. The severe reduction of interface cross-links (Fig. 3 and Fig. S5), as well as the established reduction of the cofactor affinity (3, 4, 21), are consistent with this interpretation.
The juxtaposition of active and degenerated AAA+ domains borne out by our study is not without precedent: the bacterial clamp loader complex similarly operates by an active site complementation mechanism, in which an Arg residue is contributed by a neighboring, degenerated AAA+ subunit (32), and alternating subunits of active and inactive AAA+ modules are found in one polypeptide chain in dynein (33).
Apart from providing a structural rationale for Torsin activation, our experimental validation of the activator interface between cofactors and TorA (24) (Fig. 3 and Fig. S5) has important implications for the assembly of the Torsin–cofactor complex in vivo: We now envision LAP1 and LULL1 as integral parts of the Torsin machinery rather than serving as peripherally associated cofactors, creating important ramifications for how Torsins function. As transmembrane proteins that are part of the core Torsin machinery, LAP1 and LULL1 could act as crankshafts to transduce force across the membrane. Additionally, LAP1 and LULL1 may themselves be subject to regulatory events (e.g., posttranslational modifications) or be controlled by association of cytosolic/nuclear interaction partners. Binding of these interaction partners to LAP1 or LULL1 would then induce conformational changes propagated across the membrane to regulate Torsin activity on the luminal side. Finally, these cofactors might additionally serve to recruit substrates to the assembly, as we have speculated previously (4). It will therefore be important to determine the structure of the complex using full-length subunits to position the cytosolic and nuclear domains of LULL1 and LAP1 while establishing the structural relationship of the complex to the membrane.
Methods
Alignments and Structural Modeling.
LAP1 and LULL1 sequences were aligned using ClustalW (34) and formatted using BoxShade. LAP1 and LULL1 structural homology to other proteins was determined using HHpred (35). LAP1 and LULL1 luminal domains were modeled using Phyre2 (36). A Phyre2-generated structural model of TorA (residues 58–332) was superimposed on to the ClpC hexameric assembly (Protein Data Bank ID code 3PXI) using a least-squares superposition of alpha-carbons in Coot. Images were created in Ribbons.
Constructs for Mammalian Cell Expression and Transient Transfections.
Arg mutant constructs were cloned via quick-change mutagenesis from the LAP1LD and LULL1LD constructs previously described (25). The 293T cells were grown and transfected as previously described (25).
Antibodies, Immunoprecipitation, and Immunoblotting.
Antibodies, immunoprecipitation, and immunoblotting were used and performed as previously described (4). Anti-His antibody was purchased from Roche.
Cloning, Bacterial Coexpression, and Purification of TorA Complexes with LAP1 and LULL1 Luminal Domains.
Truncated version (Δ49) of MBP-tagged TorA E171Q and the luminal domains of LULL1 (LULL1LD or LULL1236–470) and LAP1 (LAP1LD or LAP1356–583) were cloned via PCR into the pRSFDuet-1 vector (Novagen) for expression in Origami2(DE3)pLysS cells (Novagen). Origami cells transformed with pRSFDuet constructs were grown at 30 °C to an OD600 of 1.0, and protein expression was induced at 18 °C overnight. Cells were harvested and resuspended in binding buffer [20 mM Hepes, 150 mM NaCl, 5 mM MgCl2, 5 mM KCl, 2 mM ATP, 10 mM imidazole (pH 7.5)] supplemented with complete protease inhibitors/DNase (Roche) and lysed in a French Pressure Cell (Thermo Scientific) at 1,000 psi. After clearing the lysate of cellular debris by centrifugation at 20,000 × g for 20 min, the supernatant was incubated with 0.5 mL of Ni-nitrilotriacetic (NTA) agarose (Qiagen) for 2 h. After removal of unbound material by washing with 20 column volumes of binding buffer, His-tagged LAP1LD/LULLLD in complex with TorA E171Q was eluted with elution buffer [30 mM Hepes, 75 mM NaCl, 5 mM MgCl2, 0.5 mM ATP, 500 mM imidazole (pH 7.5)]. TorA complexes were applied to a Superdex 200 Prep Grade column (GE Healthcare Life Sciences).
UV Cross-Linking.
For crosslinking experiments with co-expressed proteins, TorAΔ51 and LAP1LD or LULL1LD were expressed in BL21 Star (DE3) cells (Life Technologies) and co-purified by Ni-NTA purification in 2 mM ATP. Cross-linker pBPA was incorporated into TorA-Δ51, LAP1LD, or LULL1LD at the indicated amino acid positions, as previously described (25). Cross-linking was performed by incubating reactions at 30 °C for 5 min, cooling to 20 °C, and UV-irradiating for 15 min. Samples were boiled in 1% SDS and spun down at full speed, and the supernatant was transferred to a new tube. The final volume was increased with NET buffer (50 mM Tris/HCl, pH 7.5, 75 mM NaCl, 5 mM MgCl2, 0.5% NP-40) so that the final SDS concentration was 0.1%. The samples were immunoprecipitated with anti-TorA or anti-LAP1 antibodies overnight at 4 °C. Immunoprecipitates were washed three times with NET buffer plus 0.1% SDS, eluted with SDS reducing running buffer, and subjected to SDS/PAGE followed by Western blotting to detect the cross-linked product.
Quantitative Immunoblotting and Determination of Cellular Copy Numbers for Torsins, LAP1, and LULL1.
Human Torsins, LAP1LD, and LULL1LD were expressed and purified as previously described (4). Purified Torsins were treated with PNGase F (New England Biolabs). Extracts of 293T, HeLa, and Human Foreskin Fibroblast cells were prepared by pelleting cells from 90% to 100% confluent plates, suspending the pelleted cells in DPBS (Gibco), and counting live cell numbers by staining with trypan blue (Gibco). Cell pellets were washed in ice-cold PBS before lysis in 1% SDS in PBS (100 μL/mL of cells). After incubating lysates at 99 °C for 5 min and addition of benzonase, protein concentrations were measured with a Pierce BCA Assay (Thermo Scientific) and samples were diluted to ∼2 mg/mL total protein. After preparation in reducing SDS loading buffer, a standardized extract amount for each cell type was loaded onto a 12% polyacrylamide gel along with a range of 1–8 ng of the purified standard as measured by the Pierce BCA Assay. Following electrophoresis in SDS and transfer to PVDF membranes, blots were probed with homemade antibodies to each protein and rabbit HRP (SouthernBiotech), and were reacted with SuperSignal West Femto ECL Reagent (Thermo Scientific). Blots were scanned using a ChemiDoc imaging system (BioRad), and band intensity analysis, as well as background subtraction, was carried out using ImageLab software (BioRad). Molecule numbers were calculated by correlating protein mass detected in each lane of cellular extract to a mass within the linear range of the standard curve for purified protein on the same blot. The molecule number, calculated from the mass detected for each extract lane, was divided by the cell number loaded to give the copy number.
Glutaraldehyde Cross-Linking.
Cross-linking was carried out, as described previously (22), using 30 mM Hepes (pH 7.5), 75 mM NaCl, 5 mM MgCl2, and 2 mM ATP as cross-linking buffer and 2 μM TorA–cofactor complex in each reaction. Purified BRCA2 (breast cancer type 2 susceptibility protein) (37), used as a size standard, was a kind gift from Ryan Jensen (Yale School of Medicine).
Analytical Gel Filtration.
Analytical gel filtration was performed as described previously (25), except that 7 μM LAP1LD, LULL1LD, or their mutants was incubated with an equal concentration of TorA E171Q and 2 mM ATP at 30 °C for 5 min before gel filtrations. Fractions were collected and analyzed by SDS/PAGE and immunoblotting.
Cloning, Bacterial Expression, and Purification of LAP1LD and LULL1LD.
Arg mutants were cloned via quick-change mutagenesis. Proteins were purified as previously described (4). After PreScission Protease (GE Healthcare Life Sciences) digestion, LULL1LD R449A was incubated with ATP-agarose beads preequilibrated with gel filtration buffer for 1 h at 4 °C to remove residual contaminants.
Cloning, Expression, and Purification for Human Torsins In Insect Cells.
Human Torsins were cloned, expressed, and purified as previously described (25).
ATPase Activity Assay.
Steady-state ATPase activity assays were performed as previously described (4), with the following changes. For steady-state rates, 3 μM LAP1 or LULL1 protein was used with 3 μM Torsin protein, and activity was measured at the time points of 0, 5, 15, 30, and 60 min. Single-turnover assays were performed as previously described (4), except 45 μM TorA was used to form TorA–ATP complexes and single-turnover reactions were performed at 37 °C with 10 μM of LAP1 or LULL1.
Note Added in Proof.
While this manuscript was under review, Sosa et al. (38) reported structural evidence for the Torsin–cofactor ring assembly that is in excellent agreement with the molecular modeling and biochemical evidence presented in our study.
Supplementary Material
Acknowledgments
We thank members of the C.S. laboratory for critical reading of the manuscript. This work was funded by the Ellison Medical Foundation (Grant AG-NS-0662-10); NIH Grants DP2 OD008624-01 and P01 GM022778; and the Steitz Center for Structural Biology, Gwangju Institute of Science and Technology, Republic of Korea. R.S.H.B. is supported by Grant 5T32GM007223-39. A.R.C. is supported by a National Science Foundation Graduate Research Fellowship.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415271111/-/DCSupplemental.
References
- 1.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9(1):27–43. [PubMed] [Google Scholar]
- 2.Nagy M, Wu HC, Liu Z, Kedzierska-Mieszkowska S, Zolkiewski M. Walker-A threonine couples nucleotide occupancy with the chaperone activity of the AAA+ ATPase ClpB. Protein Sci. 2009;18(2):287–293. doi: 10.1002/pro.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu L, Millen L, Mendoza JL, Thomas PJ. A unique redox-sensing sensor II motif in TorsinA plays a critical role in nucleotide and partner binding. J Biol Chem. 2010;285(48):37271–37280. doi: 10.1074/jbc.M110.123471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao C, Brown RS, Chase AR, Eisele MR, Schlieker C. Regulation of Torsin ATPases by LAP1 and LULL1. Proc Natl Acad Sci USA. 2013;110(17):E1545–E1554. doi: 10.1073/pnas.1300676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol. 2005;168(6):855–862. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foisner R, Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 1993;73(7):1267–1279. doi: 10.1016/0092-8674(93)90355-t. [DOI] [PubMed] [Google Scholar]
- 7.Schlieker C, et al. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11(7):607–615. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 8.Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT. Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J. 2001;20(12):3092–3100. doi: 10.1093/emboj/20.12.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, et al. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure. 2001;9(2):177–184. doi: 10.1016/s0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 11.Weber-Ban EU, Reid BG, Miranker AD, Horwich AL. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature. 1999;401(6748):90–93. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- 12.Ozelius LJ, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17(1):40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 13.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48(6):923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest. 2014;124(7):3080–3092. doi: 10.1172/JCI72830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim CE, Perez A, Perkins G, Ellisman MH, Dauer WT. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc Natl Acad Sci USA. 2010;107(21):9861–9866. doi: 10.1073/pnas.0912877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerth VE, Vize PD. A Java tool for dynamic web-based 3D visualization of anatomy and overlapping gene or protein expression patterns. Bioinformatics. 2005;21(7):1278–1279. doi: 10.1093/bioinformatics/bti120. [DOI] [PubMed] [Google Scholar]
- 17.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Story RM, Weber IT, Steitz TA. The structure of the E. coli recA protein monomer and polymer. Nature. 1992;355(6358):318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 19.Hanson PI, Whiteheart SW. AAA+ proteins: Have engine, will work. Nat Rev Mol Cell Biol. 2005;6(7):519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 20.Wang F, et al. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 2011;471(7338):331–335. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- 21.Naismith TV, Dalal S, Hanson PI. Interaction of torsinA with its major binding partners is impaired by the dystonia-associated DeltaGAG deletion. J Biol Chem. 2009;284(41):27866–27874. doi: 10.1074/jbc.M109.020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mogk A, et al. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J Biol Chem. 2003;278(20):17615–17624. doi: 10.1074/jbc.M209686200. [DOI] [PubMed] [Google Scholar]
- 23.Carroni M, et al. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife. 2014;3:e02481. doi: 10.7554/eLife.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose AE, Zhao C, Turner EM, Steyer AM, Schlieker C. Arresting a Torsin ATPase reshapes the endoplasmic reticulum. J Biol Chem. 2014;289(1):552–564. doi: 10.1074/jbc.M113.515791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395(2):361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 26.Scheffzek K, Ahmadian MR, Wittinghofer A. GTPase-activating proteins: Helping hands to complement an active site. Trends Biochem Sci. 1998;23(7):257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- 27.Biter AB, Lee S, Sung N, Tsai FT. Structural basis for intersubunit signaling in a protein disaggregating machine. Proc Natl Acad Sci USA. 2012;109(31):12515–12520. doi: 10.1073/pnas.1207040109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 2004;146(1-2):106–112. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Karata K, Inagawa T, Wilkinson AJ, Tatsuta T, Ogura T. Dissecting the role of a conserved motif (the second region of homology) in the AAA family of ATPases. Site-directed mutagenesis of the ATP-dependent protease FtsH. J Biol Chem. 1999;274(37):26225–26232. doi: 10.1074/jbc.274.37.26225. [DOI] [PubMed] [Google Scholar]
- 30.Rombel I, et al. MgATP binding and hydrolysis determinants of NtrC, a bacterial enhancer-binding protein. J Bacteriol. 1999;181(15):4628–4638. doi: 10.1128/jb.181.15.4628-4638.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Augustin S, et al. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol Cell. 2009;35(5):574–585. doi: 10.1016/j.molcel.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Donnell M, Kuriyan J. Clamp loaders and replication initiation. Curr Opin Struct Biol. 2006;16(1):35–41. doi: 10.1016/j.sbi.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol. 2013;14(11):713–726. doi: 10.1038/nrm3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McWilliam H, et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41(web server issue):W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 36.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc. 2009;4(3):363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 37.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467(7316):678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sosa BA, et al. How lamina-associated polypeptide 1 (LAP1) activates Torsin. eLife. 2014;3:03239. doi: 10.7554/eLife.03239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.