

Significance
Ischemia reperfusion (I/R)-induced tissue injury and inflammation encompasses a wide range of human disease, including stroke, hepatic and renal failure, and myocardial infarction. Generation of highly reactive oxygen and nitrogen species during I/R results in DNA damage that is subject to numerous DNA repair processes. Base excision repair (BER) initiated by various DNA glycosylases is critical for the repair of reactive oxygen and nitrogen species (RONS)-induced DNA damage. Our data describe a new paradigm wherein the Aag BER DNA glycosylase enzyme promotes, rather than prevents, tissue injury and inflammation in liver, brain, and kidney following I/R. This finding reveals a detrimental facet of DNA repair during inflammation and presents a novel target for controlling I/R-induced injury.
Keywords: DNA repair, base excision, Aag/Mpg DNA glycosylase, ischemia reperfusion, liver
Abstract
Inflammation is accompanied by the release of highly reactive oxygen and nitrogen species (RONS) that damage DNA, among other cellular molecules. Base excision repair (BER) is initiated by DNA glycosylases and is crucial in repairing RONS-induced DNA damage; the alkyladenine DNA glycosylase (Aag/Mpg) excises several DNA base lesions induced by the inflammation-associated RONS release that accompanies ischemia reperfusion (I/R). Using mouse I/R models we demonstrate that Aag−/− mice are significantly protected against, rather than sensitized to, I/R injury, and that such protection is observed across three different organs. Following I/R in liver, kidney, and brain, Aag−/− mice display decreased hepatocyte death, cerebral infarction, and renal injury relative to wild-type. We infer that in wild-type mice, Aag excises damaged DNA bases to generate potentially toxic abasic sites that in turn generate highly toxic DNA strand breaks that trigger poly(ADP-ribose) polymerase (Parp) hyperactivation, cellular bioenergetics failure, and necrosis; indeed, steady-state levels of abasic sites and nuclear PAR polymers were significantly more elevated in wild-type vs. Aag−/− liver after I/R. This increase in PAR polymers was accompanied by depletion of intracellular NAD and ATP levels plus the translocation and extracellular release of the high-mobility group box 1 (Hmgb1) nuclear protein, activating the sterile inflammatory response. We thus demonstrate the detrimental effects of Aag-initiated BER during I/R and sterile inflammation, and present a novel target for controlling I/R-induced injury.
Ischemia reperfusion (I/R)-induced tissue injury is one of the most common examples of acute, sterile inflammation-induced tissue damage. Health events that incur I/R include ischemic stroke, acute liver or kidney failure, myocardial infarction, various forms of circulatory shock, sickle cell disease, and organ transplantations. These conditions are frequently accompanied by profound morbidity and mortality worldwide (1). The need for effective approaches to manage patients with I/R-induced organ damage is highlighted by the fact that current treatment is primarily supportive care (1).
During I/R, a burst of reactive oxygen and nitrogen species (RONS) occurs within the first moments of reperfusion, and this burst is thought to be primarily responsible for collateral tissue damage (2). Furthermore, RONS are also generated during the preceding ischemia, despite the low oxygen tension (3). I/R alters cellular metabolism and redox state, and induces so-called “sterile inflammation” by activating the innate immune system. The activation of macrophages and recruitment of neutrophils to sites of I/R by intravascular danger signals results in the release of an arsenal of RONS capable of inducing DNA damage and lipid peroxidation, thus causing major collateral tissue damage. Base excision repair (BER), initiated by various DNA glycosylases, is critical for the repair of RONS-associated DNA damage, including oxidized, deaminated, and etheno (ε)-adducted DNA bases (4, 5). BER involves the orchestration of the following enzymatic steps: damaged bases are excised by DNA glycosylase, followed by cleavage of the DNA backbone at the resulting abasic site; DNA ends are trimmed to generate a 3′OH and 5′P; the gap is then filled by DNA polymerase and the remaining nick sealed by DNA ligase to complete BER (6, 7) (Fig. 1A). The murine alkyladenine DNA glycosylase (Aag; also known as Mpg) acts efficiently on 1,N6-ethenodeoxyadenosine (ε-A) lesions and deaminated adenosine (hypoxanthine, Hx) (8, 9), and with lower efficiency on 8-oxoguanine (8-oxoG) (10), all of which are induced directly or indirectly by RONS (11). Aag therefore seemed likely to provide resistance to I/R-induced toxicity. In support of this hypothesis, we previously found that Aag−/− mice suffer more inflammation-associated intestinal tissue damage and colon carcinogenesis than WT mice (5, 12); with this in mind, we set out to examine whether the Aag DNA glycosylase modulates I/R-induced tissue injury in three distinct tissue environments—namely liver, brain, and kidney. To our surprise, we find that Aag-initiated BER actually exacerbates, rather than attenuates, I/R tissue injury in all three organs.
Fig. 1.

Aag deficiency protects liver against I/R-induced tissue damage. (A) The BER pathway initiated by various DNA glycosylases consists of four key enzymatic steps: (i) glycosylase-mediated excision of a damaged base generating an abasic site; (ii) incision of the sugar-phosphate backbone at the abasic site by APE, yielding a 3′-OH adjacent to a 5′-dRP moiety; (iii) 5′-dRP removal and DNA synthesis by DNA polymerase β (Pol β); and (iv) sealing of the DNA nick by DNA ligase (LIG). (B) Serum ALT concentrations after 90 min of ischemia and 24 h of reperfusion (n = 3 in sham groups and n = 7–8 in I/R groups). (C) Representative images of necrotic areas 24 h after I/R in Aag−/− (n = 7) or WT livers (n = 8); liver sections (4 μm) were stained with H&E. (D) Mean necrotic area in ischemic liver samples and (E) serum LDH concentrations 24 h after reperfusion (n = 3 in sham groups and n = 7–8 in I/R groups); necrosis analyzed by ImageJ (necrotic areas indicated by green color). (F) Serum ALT in control mice (no surgery) or after 60 min of ischemia plus either 6- or 24-h reperfusion in WT and Aag−/− mice. Error bars represent the mean ± SEM of at least three independent experiments.
Results
Aag Deficiency Protects the Liver Against I/R-Induced Tissue Damage.
To address the role of Aag during I/R-associated tissue injury, we first focused on liver, comparing I/R-induced hepatic injury in WT vs. Aag−/− mice subjected to 90-min liver ischemia followed by 24-h reperfusion. To our surprise, serum alanine aminotransferase (ALT) levels, a canonical marker of hepatic injury, indicated that Aag deficiency actually attenuates the detrimental effects of I/R (Fig. 1B). In other words, Aag-initiated BER in WT liver appears to increase rather than decrease I/R-induced hepatic injury. Quantitative evaluation of hepatic necrosis and serum lactate dehydrogenase (LDH; another marker of tissue injury) corroborated this finding; Aag−/− mice subjected to I/R displayed significantly smaller areas of necrosis (Fig. 1 C and D) and lower LDH compared with WT mice (Fig. 1E).
Following I/R, histological assessment of livers from both genotypes showed centrilobular and midzonal necrosis as the prominent types of necrosis. However, WT liver had a higher total pathological score for necrosis and exhibited additional types of necrosis, including portal/periportal and subcapsular/capsular necrosis (Fig. S1 A–C). Reduced liver toxicity in Aag−/− vs. WT was also evident with shorter ischemic duration (60 min vs. 90 min of ischemia). ALT levels were raised significantly in all groups undergoing I/R at both 6 and 24 h of reperfusion, but WT mice demonstrated significantly higher serum ALT levels compared with Aag−/− mice (Fig. 1F).
Wild-Type Mice Accumulate More BER Intermediates and Fewer Oxidized DNA Base Lesions than Aag Null Mice.
DNA glycosylases generate potentially toxic abasic (AP) sites that can block replication and transcription (13, 14), and their cleavage by AP endonuclease generates highly toxic DNA single-strand breaks (15); indeed, until the final ligation step of BER, toxic lesions are present in DNA (Fig. 1A). Thus, if BER is initiated under conditions where subsequent processing of BER intermediates is limiting, this can result in cell death and tissue damage; under these circumstances, cells are said to have an imbalanced BER pathway (6). Having shown that Aag in WT mice stimulates more I/R-induced liver toxicity than that seen in Aag−/− mice, we determined whether this toxicity correlates with the accumulation of BER intermediates. At 24 h following I/R, WT liver DNA contained twice as many abasic sites as Aag−/− liver DNA (Fig. 2A). Aag activity in WT liver also showed a mild increase following 6 and 24 h of reperfusion, which was not accompanied by an increase in apurinic–apyrimidinic endonuclease (APE) activity; this could further contribute to imbalanced BER and the accumulation of AP sites in WT tissue (Fig. S2 A and B). Interestingly, of the damaged bases measured [εA, 1,N2 ethenodeoxyguanosine (εG) and 8-oxoG], only 8-oxoG accumulated to a higher level in DNA isolated from Aag−/− vs. WT livers (Fig. 2B), whereas there was no difference in the basal level of 8-oxoG in WT vs. Aag−/− mice (Fig. S2 C–E).
Fig. 2.

WT mice accumulate more BER intermediates and fewer 8-oxoG DNA lesions than Aag−/− mice. (A) The level of abasic sites (AP sites) 24 h after I/R treatment in liver DNA isolated from WT and Aag−/− mice. (B) εA, εG, and 8-oxoG base lesions in liver DNA isolated from WT and Aag−/− mice after ischemia followed by 6 h of reperfusion. Error bars represent the mean ± SEM of at least three independent experiments.
I/R-Induced Poly(ADP ribose) Polymerase Activation and Intracellular NAD and ATP Depletion Is More Extreme in WT vs. Aag−/− Liver.
Poly(ADP-ribose) polymerase 1 (Parp1), an abundant nuclear protein, is potently activated by DNA strand breaks and BER intermediates. Activation of Parp consumes NAD+, causing ATP depletion, which has been linked to DNA damage-induced programmed necrosis (16). Using a single-cell analysis approach (17), we evaluated nuclear poly(ADP-ribose) (PAR) formation in hepatocytes after I/R (Figs. S3 and S4). We focused on viable cells immediately adjacent to necrotic areas to capture dynamic cellular responses to I/R preceding cellular disintegration, and where immunostaining is technically more reliable. We quantitated both the average level and the distribution of Parp activation at single-cell resolution. PAR was present at a low level in hepatocytes of sham-treated WT animals, and this level clearly increased upon I/R (Fig. 3A). Hepatocyte PAR staining in sham-treated WT or Aag−/− mice showed a relatively low unimodal distribution; following I/R, nearly all WT hepatocytes showed increased nuclear PAR with a broad multimodal distribution, but only a subset of Aag−/− hepatocytes showed increased PAR, generating a bimodal distribution that was narrower than that of WT (Fig. 3B). Using a MatLab code to quantitate the average nuclear PAR signal intensity after I/R, we found that WT hepatocytes accumulate more nuclear PAR than Aag−/− hepatocytes following I/R injury (Fig. 3C), consistent with the accumulation of more BER intermediates upon initiation of BER by the Aag enzyme (Fig. 2A). The PAR polymer has been characterized as a signaling molecule that triggers cell death, making the PARP1 enzyme an innovative therapeutic target for the prevention of tissue injury (18). If PAR polymer formation plays a critical role in I/R-induced tissue damage, Parp1−/− mice may be expected to suffer less I/R-induced liver damage. We therefore compared I/R-induced hepatic injury (assessed by serum ALT levels) in Parp1−/− vs. WT mice; Parp1−/− mice showed significantly less hepatic injury compared with WT mice and, moreover, the lack of Aag in Parp1−/− mice (Parp1−/−Aag−/− genotype) conferred no additional protection against I/R-induced tissue damage, suggesting that Aag and Parp1 act in the same pathway (Fig. 3D).
Fig. 3.

Parylation is differentially induced by I/R in WT vs. Aag−/− liver. (A) Images of livers from WT sham-operated mice and mice that have undergone I/R (90 min, 24 h) are shown. Nuclei are in blue; PAR is in red. (B) Histograms of nuclear PAR average fluorescence intensity among individual cells. (n = 500 cells for each conditions in each histogram; gray shows sham and red indicates I/R conditions). (C) Quantification of PAR average nuclear intensity in the nuclei. (D) Serum ALT in WT, Parp1−/−, and Aag−/− Parp1−/− mice after I/R. (E) Total NAD content and (F) ATP level in liver from WT and Aag−/− mice. Error bars are mean ± SEM (n = 3 in sham and n = 5–6 in I/R groups).
Stimulated by DNA strand breaks, Parp1 consumes NAD+ to form PAR chains on various nuclear proteins, including histones, polymerases, transcription factors, and Parp1 itself. Hyperactivation of Parp1 leads to defective glycolysis or cellular NAD+ depletion, which consequently results in diminished cellular ATP, mitochondrial depolarization, and a bioenergetics collapse that ensures cell death (16, 19, 20). We assayed total NAD and ATP in hepatic tissue 24 h after I/R; total NAD and ATP were significantly decreased in WT mice 24 h after I/R, consistent with Parp1 hyperactivation, whereas Aag−/− mice only showed moderately decreased NAD and ATP levels that were not statistically significant (P > 0.05; Fig. 3 E and F).
Aag−/− Mice Are Significantly Protected Against I/R-Driven Sterile Inflammation.
Several studies have demonstrated that during reperfusion of ischemic tissue there is an activation of both the innate and adaptive immune responses, similar to that stimulated by microbial infections, despite no microbial involvement—hence the term “sterile inflammation” (21, 22). High-mobility group box 1 (Hmgb1), a highly abundant nonhistone nuclear protein, is now known to act as an extracellular damage-associated molecular pattern (DAMP) molecule that activates the innate immune system in response to excessive cell death (23). Hmgb1 activates immune cells through a variety receptors, including the receptor for advanced glycation end-products (RAGE), TLR2, and TLR4 (24). Past studies have also identified Hmgb1 as an interacting partner of BER enzymes, suggesting that it functions as modulator of BER following DNA damage (25). We first examined intracellular Hmgb1 expression and localization following I/R using quantitative immunofluorescence staining. Hmgb1 exhibited exclusively nuclear localization in liver tissue from sham-operated WT and Aag−/− mice (Fig. 4A). By 24 h after I/R, Hmgb1 was almost entirely cytoplasmic in cells immediately adjacent to necrotic liver tissue in WT mice, whereas in Aag−/− mice the protein was still largely nuclear in most cells (Fig. 4A). The mean Hmgb1 nuclear to cytoplasmic ratio (NCR) was assessed for at least 1,000 cells from each mouse; after I/R, the Hmgb1 NCR was significantly lower in WT mice vs. Aag−/− (Fig. 4B), whereas the initial NCR values did not differ between genotypes (NCR = 2.05 in WT sham and 1.95 in Aag−/−sham). Because mean Hmgb1 NCR values do not reflect the fact that the protein is cytoplasmic in some cells and nuclear in others, even within the same sample, especially for Aag−/− mice (Fig. 4A), we also compared the distributions of Hmgb1 NCR among cells using a cumulative distribution function (CDF). The results in Fig. 4C show significantly different NCR distributions between WT and Aag−/− livers (after I/R) with Aag−/− hepatocytes having higher NCR values across the entire distribution (∼1,000 cells). Parylation of nuclear Hmgb1 is thought to control its cytoplasmic translocation after cellular DNA damage (26). We therefore simultaneously monitored immunofluorescence for both PAR polymers and Hmgb1 in the same cells; after I/R, WT hepatocytes exhibited a much higher fraction of cells with both high PAR intensity and high Hmgb1 cytoplasmic intensity (35%) than did Aag−/− liver (2%) (Fig. S5).
Fig. 4.

Differential Hmgb1 translocation phenotypes between WT and Aag−/− mice upon I/R. (A) Images of livers from sham-operated mice and mice that have undergone I/R are shown. Nuclei are in blue; Hmgb1 is in red. Necrotic regions stain low for Hmgb1 as it is released from the cells. (B) Quantification of mean Hmgb1 nuclear-to-cytoplasmic ratio (NCR) (n = 3 mice each). (C) The cumulative distribution function (CDF) is plotted. The large value of the mean KS statistic, that quantifies the mean distance between WT and Aag−/− histograms, indicates that the difference in distributions is indeed significant. (D) Serum concentration of Hmgb1 24 h after I/R (n = 3 in sham and n = 5 in I/R groups). Error bars are mean ± SEM.
The extracellular release of Hmgb1 requires translocation of Hmgb1 from nucleus to cytoplasm before release into the extracellular space. High levels of extracellular HMGB1 can accumulate in patients with infectious or sterile inflammatory diseases (23, 27). We therefore measured serum levels of Hmgb1, 24 h after I/R, as an indicator of liver necrosis or active Hmgb1 release from hepatic cells, or both. Hmgb1 serum levels were significantly higher in WT mice vs. Aag−/− mice, which had levels similar to sham operated mice (Fig. 4D).
Extracellular Hmgb1 serves as a proinflammatory signal, inducing macrophage activation and promoting recruitment of inflammatory cells to damaged tissues (27). Upon tissue injury, the release of Hmgb1, along with other DAMPs, guides neutrophils and monocytes to sites of sterile inflammation. Neutrophils and monocytes in turn release a vast arsenal of hydrolytic, oxidative, and pore-forming molecules with the potential to cause profound collateral tissue destruction (28); such destruction signals for further recruitment of neutrophils and monocytes, eliciting even more tissue damage in a vicious cycle that is ultimately regulated by both pro- and antiinflammatory signals (29, 30). Histological assessment of livers after I/R shows lobular and portal inflammation in both WT and Aag−/− samples (Fig. S1B). To further characterize I/R-induced inflammation in the liver, we isolated nonparenchymal liver cells and, by immunophenotyping, assessed the presence of different immune cells. There was no difference in neutrophil or T-cell populations in the livers of WT and Aag−/− sham-treated mice (Fig. 5). Following I/R, livers from both genotypes exhibited increased numbers of neutrophils (CD11b+ Gr-1+), but the increase in Aag−/− livers was only one-third of that in WT liver, reflecting decreased tissue injury and a protected phenotype upon I/R (Fig. 5 A and B); there was no difference in CD4+ or CD8+ T cells in Aag−/− vs. WT liver (Fig. 5 C and D). To further explore the inflammatory response in Aag−/− vs. WT animals, we tested a panel of genes involved in tissue homeostasis, inflammation, and oxidative stress. The transcriptional expression pattern between sham-operated groups was not significantly different, suggesting no basal level disturbance in Aag−/− vs. WT animals following sham operation (Fig. S6). However, after I/R, the gene expression pattern reflected a reduced inflammatory response in Aag−/− livers compared with WT (Fig. S6). We observed significant differences in neutrophil trafficking factors, such as intercellular adhesion molecule 1 (ICAM-1), chemokine (C-X-C motif) ligand 2 (CXCL2), also known as macrophage inflammatory protein 2-alpha (MIP-2), and P-selectin. Additionally, CD68, a marker of macrophage lineage, heme oxygense-1 (HO-1) and neutrophil cytosolic factor 2 (Ncf2), a component of the phagocyte NADPH oxidase, showed significantly higher expressions in WT vs. Aag−/− mice 24 h after I/R (Fig. S6). We infer that the initial I/R-induced tissue damage is more severe in WT vs. Aag−/− liver tissue and that this results in enhanced recruitment of neutrophils that in turn generate more tissue damage in the WT mice.
Fig. 5.

Aag−/− mice are significantly protected against I/R-driven sterile inflammation (A) Representative flow cytometry plots of total nonparenchymal liver cells gated for CD11b+ Gr1+ cells. CD11b and Gr-1 together mark neutrophils and immature monocyte populations. (B) Percentage of CD11b+ Gr1+ cells among total hepatic nonparenchymal cells after 90 min of ischemia and 24 h of reperfusion, or after sham surgery, in WT and Aag−/− mice. (C) Percentage of CD4+ cells among total hepatic nonparenchymal cells after I/R or sham surgery. (D) Percentage of CD8+ cells among total hepatic nonparenchymal cells after I/R or sham surgery. NPCs, total hepatic nonparenchymal cells. Error bars represent the mean ± SEM of at least three independent experiments.
Aag-Initiated BER Contributes to I/R-Induced Tissue Injury in Other Organs.
Having established the role of Aag in I/R-mediated liver injury, we next determined whether Aag modulates I/R-mediated injury in other organs—namely, brain and kidney—for which I/R-induced damage is associated with significant morbidity and mortality (31, 32). Ischemic stroke is the third leading cause of death in industrialized countries and the most frequent cause of permanent disability in adults worldwide (31), and more than 16,000 kidney transplants, necessarily involving I/R, are performed annually in the United States (33).
We used middle cerebral artery occlusion (MCAO) to induce transient focal cerebral ischemia followed by reperfusion in mice (Fig. S7A). Fig. 6A shows WT and Aag−/− coronal brain sections (at 1-mm intervals) 24 h following 60-min MCAO. Multiple coronal slice levels showed smaller lesion areas in Aag−/− vs. WT brains (Fig. 6B). The cerebral infarct volume, assessed by computer-assisted volumetry, revealed that the mean volume of necrosis was reduced by almost half in Aag−/− vs. WT mice (Fig. 6C and Fig. S7B).
Fig. 6.
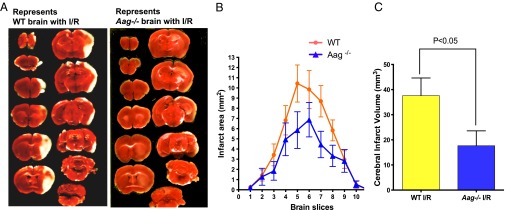
MCAO model of stroke demonstrates protective phenotypes against I/R in Aag−/− mice. (A) Representative triphenyltetrazolium chloride-stained brain slices 24 h after reperfusion. The white areas indicate regions of infarction. (B) Infarct areas evaluated by ImageJ analysis (n = 10 for WT; n = 11 for Aag−/−). (C) Indirect infarct volume is calculated as the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere; vertical bars represent ± SEM (n = 10 for WT; n = 11 for Aag−/−).
We also performed bilateral renal ischemia (30 min), harvesting blood and kidneys 24 h following reperfusion. Serum creatinine levels and blood urea nitrogen (BUN), two surrogate markers of renal function, indicated that WT mice sustained significantly greater loss of kidney function than Aag−/− mice (Fig. 7 A and B); this conclusion was supported by histological scores of kidney pathology (Fig. 7 C and D). Together these data indicate that Aag-initiated BER is an important contributor to I/R-mediated damage in multiple tissues.
Fig. 7.

Bilateral renal ischemia model shows protective phenotype against I/R in Aag−/− mice. (A) Serum creatinine levels in WT and Aag−/− mice after bilateral kidney I/R (30 min of ischemia and 24 h of reperfusion); n = 3 in sham groups and n = 5–6 in I/R groups. (B) BUN in WT and Aag−/− mice after bilateral kidney I/R; n = 3 in sham groups and n = 5–6 in I/R groups. Error bars represent mean ± SEM. (C) Representative sections of kidneys from WT or Aag−/− mice 24 h after reperfusion (H&E stained). (D) WT mice displayed augmented renal tissue injury presented by higher percent of high-power fields (%HPFs) that scored 5 in our pathological examination (*P < 0.05 in comparison with corresponding WT group, n = 5 in WT I/R and n = 6 in Aag−/− I/R).
Discussion
How BER modulates I/R-induced tissue damage and inflammation has not been fully elucidated. Absence of the Ung, Neil1, or Ogg1 DNA glycosylases (recognizing uracil, oxidized pyrimidines, and 8-oxoG) confers increased susceptibility to brain damage induced by ischemia or I/R (34–36), indicating that BER initiated by these enzymes protects against I/R-induced tissue damage. Though the exact mechanisms contributing to increased cell death in those studies remain to be determined, they clearly suggest that BER initiated by these glycosylases plays a protective role during tissue repair. In stark contrast, the study presented herein clearly shows that Aag-initiated BER promotes and exacerbates I/R-induced tissue damage, and that this unexpected phenotype pertains to three different tissues—namely, liver, brain, and kidney. In all three organs, I/R-induced tissue injury is markedly suppressed in the absence of Aag-initiated BER.
Exploring the mechanism of Aag-induced I/R-induced liver injury revealed more abasic sites in hepatocyte DNA, and higher PAR polymer levels (accompanied by greater depletion of NAD and ATP) in livers, from WT vs. Aag−/− mice. Evidence from several laboratories indicates that BER intermediates are often more cytotoxic than the initiating DNA base lesion, possibly because translesion DNA polymerases are capable of bypassing the unrepaired base lesions (37, 38), whereas strand breaks cannot be bypassed. Further, BER intermediates can induce Parp1 hyperactivation that in turn depletes NAD+/ATP pools, thus eliciting an energetic crisis and cell death (39). Though we previously reported that Aag-initiated BER promotes alkylation-induced tissue damage in specific neural and lymphoid tissues (40, 41), here we demonstrate the importance of this process in regulating I/R-induced tissue injury and sterile inflammation. Together, the decreased generation of BER intermediates and consequent decrease in the accumulation of PAR polymers likely preserves adequate NAD and ATP levels in Aag−/− livers; this is accompanied by lower Hmgb1 release from Aag−/− hepatocytes and attenuated post-I/R neutrophilic infiltration in injured Aag−/− livers compared with WT. Our data support the conclusion that Aag-initiated BER, in contrast to BER initiated by three other DNA glycosylases (34–36), acts upstream of Parp1 hyperactivation in I/R-induced tissue damage and sterile inflammation.
Aag has been shown to act on Uracil (an Ung substrate) and 8-oxoG (substrates for both Ogg1 and Neil1) (9, 10, 42). Thus, in contrast to most other DNA glycosylases that have a narrow substrate range, Aag catalyzes the excision of a broad range of modified bases; this is achieved by its unique active site structure that can both discriminate against normal DNA bases and yet accommodate a structurally diverse set of aberrant DNA bases (8, 9). This broad substrate specificity enables Aag to initiate BER at wide variety of DNA base lesions during pathological situations where many different kinds of DNA lesions are generated. When a panoply of damaged bases is induced, base excision by Aag, in contrast to that by Ung, Neil1, and Ogg1, may exceed the capacity for the downstream BER enzymes to complete repair, thus generating toxic BER intermediates and cell death. Consequently, when Ung, Ogg1, or Neil1 are deficient, their unrepaired substrates become subject to Aag-initiated BER such that in the Ung/Ogg1/Neil1-deficient mice, Aag has even more substrates to repair, causing increased I/R-mediated tissue damage (34–36).
Additionally, Aag is a monofunctional DNA glycosylase that creates AP sites for incision by APE-1, whose action generates 5′-deoxyribose-5-phosphate (5′-dRP) and 3′-hydroxyl (3′-OH) DNA ends; in contrast, the bifunctional DNA glycosylases Ogg1 and Neil1, after creating AP sites, can also incise the DNA backbone 3′ to the AP site, generating a 3′-deoxyribose moiety and a 5′-P. Thus, Neil1 and Ogg1 do not generate 5′dRPs that have been shown to be especially toxic (14). Indeed, it was shown that Parp1 has specific affinity to AP sites and 5′-dRP intermediates that are generated during BER initiated by monofunctional glycosylases (43, 44), and excessive Parp1 activation is known to elicit necrotic cell death (16, 18, 19). Therefore, repair by Aag, a monofunctional glycosylase with an unusually broad range of substrates, may generate excessive amounts of BER intermediates and, in particular, highly toxic dRP intermediates, possibly explaining why Aag-initiated BER is toxic and Ung/Ogg1/Neil1 initiated repair is not (34–36).
We previously observed that Aag−/− mice are more susceptible than WT mice to inflammation-associated colon cancer (12); this may at first appear to contradict the findings reported here. However, Aag-mediated cell death in the inflamed colon may serve to protect the colon against the accumulation of mutant cells, thus reducing carcinogenesis in the long term. This adaptation may be beneficial because cell death rids the tissue of potentially mutated cells, albeit at the cost of an acute but transient decline in tissue function. However, in disease models such as I/R-induced acute tissue injury and inflammation, extensive cell death may lead to tissue dysfunction or organ failure as demonstrated in this study. Additionally, though there are some similarities, it is likely that there are inherent differences between microbially induced chronic inflammation in the colon and acute sterile inflammation following I/R in the liver, brain, and kidney.
Among the DNA base adducts measured, only 8-oxoG increased in Aag−/− liver DNA, with no significant change in ε-DNA adducts. The low redox potential of guanine makes this base particularly vulnerable to oxidation (45), and 8-oxoG is readily generated during oxidative stress (46). Both Ogg1 and Aag can excise 8-oxoG, although Ogg1 is normally much more efficient (10, 47). However, it was recently shown that Ogg1 is a specific target of calpain I, a Ca2+-dependent protease activated during oxidative stress (48); further, Ogg1 is directly inhibited by nitric oxide during inflammation (49). Thus, Aag mediated 8-oxoG excision may assume a very important role during conditions of oxidative tissue damage. Indeed, increased levels of 8-oxoG after I/R in Aag−/− livers underscores the significance of Aag-mediated 8-oxoG excision during I/R-induced injury. That Aag is important for 8-oxoG excision during inflammation is further supported by our previous study in which Aag−/− mice accumulated higher levels of genomic 8-oxoG than WT during chronic inflammation in the colon (50). The lack of increased ε-base adducts in Aag−/− liver DNA after I/R could be due to their efficient repair by the Alkbh2 and Alkbh3 direct reversal enzymes (5, 51, 52) in this tissue, or could perhaps reflect differences in the types of DNA lesions induced during sterile inflammation in the liver vs. microbially induced inflammation in the colon. The removal of each different base lesion by Aag generates an AP site in DNA, and we cannot exclude the possibility that small changes in the excision of several ε-base lesions as well as hypoxanthine also contributes to the significantly increased level of AP sites in WT liver DNA after I/R.
Several other pathways may contribute to the observed phenotype in Aag−/− mice. For instance, mitochondria are well established as a critical player in several cell death pathways, and damage to mitochondrial genes has been linked to a number of diseases and aging. There is good evidence for BER in mitochondria, and we have shown that a small fraction of the human AAG protein localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (53). However, this interaction specifically inhibits AAG activity in the context of a single-stranded DNA, preventing the formation of abasic sites, which could potentially lead to formation of harmful DNA breaks (53). In another study, overexpression of the mitochondrially targeted AAG dramatically increased cancer cells’ sensitivity to an alkylating agent, presumably through an imbalanced BER pathway and accumulation of BER intermediates (54). Additionally, upon release from necrotic cells, mitochondrial DNA can activate innate immune cells through pattern recognition receptors (55). Thus, the possibility that Aag-initiated mitochondrial BER may also play a role in modulation of I/R-induced tissue damage warrants further study.
Additionally it was shown that some DNA repair-deficient mice induce a protective response that prevents further damage during stress response (56–58). Accordingly, a mouse model of Cockayne syndrome is less susceptible to renal I/R than WT mouse, possibly due to a reduced inflammatory response and improved insulin sensitivity (58). It thus seems possible that Aag−/− animals may have undergone an adaptive response that diminishes tissue injury and sterile inflammation in response to I/R. However, we did not observe a significant difference in the expression of multiple genes important for ROS production and inflammation between sham-operated Aag−/− and WT mice; in contrast, following I/R, we found significant differences between WT and Aag−/− livers for expression of a subset of inflammatory genes (e.g., CXCR2, ICAM-1, P-selectin) and genes important in oxidative stress [e.g., subunits of NADPH oxidase (Cyba, Ncf1, Ncf2) or HO-1] (Fig. S6). Though these changes are likely to be secondary to differences in I/R-induced cell death between WT and Aag−/− liver, we cannot rule out the possibility that the Aag−/− mouse may have developed a different threshold for activation of these inflammatory pathways.
How mechanisms regulating genome integrity modulate the innate immune response is critical for understanding the regulation of tissue homeostasis. It was previously shown that the activation of Parp1 by DNA alkylation damage induces parylation of Hmgb1 and its release from the nucleus (26). Such nuclear-to-cytosolic translocation of Hmgb1 serves to enable cellular release of this potent inflammatory mediator through either active secretion or passive release upon necrotic cell death (26). Hmgb1 also stimulates production of ssDNA breaks during BER, thus the cytoplasmic translocation of Hmgb1 can impact BER pathway in the nucleus by altering the nuclear Hmgb1 levels (25). Following release from hepatocytes, Hmgb1 signals danger to other cells such as macrophages or endothelial cells and induces neutrophil chemoattractants (e.g., CXCL1) and adhesion ligands (e.g., ICAM-1), both of which are indispensable for guiding immune cells to the site of injury (59). Accordingly, after I/R, WT mice showed higher hepatic transcriptional levels of ICAM-1, P-selectin, and CXCL2, and greater postischemic neutrophilic infiltrate than Aag−/− mice.
In summary, the data presented here reveal an important role for Aag-initiated BER in modulating tissue injury during I/R in liver, kidney, and brain. Indeed, exposure of a single organ to I/R can subsequently ignite inflammatory activation in other organs, eventually leading to multiorgan failure (1). Our results suggest that interindividual differences in BER activity could influence the pathological outcome of I/R. Though PARP inhibition is currently being explored for the amelioration of I/R injury, we argue that the inhibition of the upstream-acting AAG enzyme, eliminating the generation of toxic BER intermediates and the signal for PARP hyperactivation, may present a better target. However, long-term mutagenic effects of both approaches warrant future studies.
Materials and Methods
All animals were maintained in pathogen-free conditions at the Massachusetts Institute of Technology or Massachusetts General Hospital facilities. The Animal Care and Use Committee of both institutes approved related animal protocols, and the experiments were conducted in adherence with the National Institutes of Health Guidelines for the Use of Laboratory Animals (60). I/R-induced tissue-injury models were established. Multiple metrics of tissue injury; tissue and serum Hmgb1; levels of AP sites; DNA adducts; Aag and APE activities; PAR polymers; and ATP and total NAD in tissues were determined. Liver nonparenchymal cells were isolated and analyzed by flow cytometry analysis. For full procedures, please see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Catherine A. Moroski-Erkul and Lindsey W. Eichinger for technical help with mouse colonies during the study, the Swanson Biotechnology Center, the David H. Koch Institute for Integrative Cancer Research, and the Center for Environmental Health Sciences Bioanalytical Core Facility. This work was supported by National Institutes of Health Grants R01-CA055042, R01-CA149261, and P30-ES02109 and the Ellison Medical Foundation (to L.D.S.); National Institutes of Health Grant R01-NS055104 (to C.A.); and the Heitman Foundation and the Ellison Foundation (C.A.). L.D.S. is an American Cancer Society Research Professor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413582111/-/DCSupplemental.
References
- 1.Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brass CA, Narciso J, Gollan JL. Enhanced activity of the free radical producing enzyme xanthine oxidase in hypoxic rat liver. Regulation and pathophysiologic significance. J Clin Invest. 1991;87(2):424–431. doi: 10.1172/JCI115013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61(3):461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 4.Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. 2011;14(12):2491–2507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calvo JA, et al. DNA repair is indispensable for survival after acute inflammation. J Clin Invest. 2012;122(7):2680–2689. doi: 10.1172/JCI63338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12(2):104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobol RW, et al. Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses. J Biol Chem. 2003;278(41):39951–39959. doi: 10.1074/jbc.M306592200. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien PJ, Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem. 2004;279(11):9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 9.Lee CY, et al. Recognition and processing of a new repertoire of DNA substrates by human 3-methyladenine DNA glycosylase (AAG) Biochemistry. 2009;48(9):1850–1861. doi: 10.1021/bi8018898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bessho T, et al. Repair of 8-hydroxyguanine in DNA by mammalian N-methylpurine-DNA glycosylase. Proc Natl Acad Sci USA. 1993;90(19):8901–8904. doi: 10.1073/pnas.90.19.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nair J, Barbin A, Velic I, Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat Res. 1999;424(1-2):59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 12.Meira LB, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118(7):2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kothandapani A, et al. Novel role of base excision repair in mediating cisplatin cytotoxicity. J Biol Chem. 2011;286(16):14564–14574. doi: 10.1074/jbc.M111.225375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sobol RW, et al. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405(6788):807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, et al. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J Biol Chem. 2007;282(18):13532–13541. doi: 10.1074/jbc.M611295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo X, Kraus WL. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26(5):417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazumder A, Pesudo LQ, McRee S, Bathe M, Samson LD. Genome-wide single-cell-level screen for protein abundance and localization changes in response to DNA damage in S. cerevisiae. Nucleic Acids Res. 2013;41(20):9310–9324. doi: 10.1093/nar/gkt715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrabi SA, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103(48):18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25(5):259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Andrabi SA, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci USA. 2014;111(28):10209–14. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen GY, Nuñez G. Sterile inflammation: Sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 24.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Prasad R, Wilson SH. HMGB1: Roles in base excision repair and related function. Biochim Biophys Acta. 2010;1799(1-2):119–130. doi: 10.1016/j.bbagrm.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282(24):17845–17854. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orlova VV, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26(4):1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, et al. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest. 2012;122(11):3931–3942. doi: 10.1172/JCI63170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gandolfo MT, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76(7):717–729. doi: 10.1038/ki.2009.259. [DOI] [PubMed] [Google Scholar]
- 31.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 32.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matas AJ, et al. OPTN/SRTR 2012 Annual Data Report: Kidney. Am J Transplant. 2014;14(suppl 1):11–44. doi: 10.1111/ajt.12579. [DOI] [PubMed] [Google Scholar]
- 34.Endres M, et al. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113(12):1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canugovi C, et al. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc Natl Acad Sci USA. 2012;109(37):14948–53. doi: 10.1073/pnas.1204156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu D, et al. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J Cereb Blood Flow Metab. 2011;31(2):680–692. doi: 10.1038/jcbfm.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roos WP, et al. The translesion polymerase Rev3L in the tolerance of alkylating anticancer drugs. Mol Pharmacol. 2009;76(4):927–934. doi: 10.1124/mol.109.058131. [DOI] [PubMed] [Google Scholar]
- 38.Johnson RE, Yu SL, Prakash S, Prakash L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol Cell Biol. 2007;27(20):7198–7205. doi: 10.1128/MCB.01079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173(1):2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvo JA, et al. Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by Parp1. PLoS Genet. 2013;9(4):e1003413. doi: 10.1371/journal.pgen.1003413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meira LB, et al. Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice. Proc Natl Acad Sci USA. 2009;106(3):888–893. doi: 10.1073/pnas.0807030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5(4):a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavrik OI, et al. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276(27):25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 44.Khodyreva SN, et al. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1) Proc Natl Acad Sci USA. 2010;107(51):22090–22095. doi: 10.1073/pnas.1009182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol. 2006;19(4):491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 46.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447(7147):941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hazra TK, Hill JW, Izumi T, Mitra S. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog Nucleic Acid Res Mol Biol. 2001;68:193–205. doi: 10.1016/s0079-6603(01)68100-5. [DOI] [PubMed] [Google Scholar]
- 48.Hill JW, Hu JJ, Evans MK. OGG1 is degraded by calpain following oxidative stress and cisplatin exposure. DNA Repair (Amst) 2008;7(4):648–654. doi: 10.1016/j.dnarep.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaiswal M, LaRusso NF, Nishioka N, Nakabeppu Y, Gores GJ. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61(17):6388–6393. [PubMed] [Google Scholar]
- 50.Hofseth LJ, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112(12):1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mishina Y, Yang CG, He C. Direct repair of the exocyclic DNA adduct 1,N6-ethenoadenine by the DNA repair AlkB proteins. J Am Chem Soc. 2005;127(42):14594–14595. doi: 10.1021/ja055957m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ringvoll J, et al. AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA. Cancer Res. 2008;68(11):4142–4149. doi: 10.1158/0008-5472.CAN-08-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Loon B, Samson LD. Alkyladenine DNA glycosylase (AAG) localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (mtSSB) DNA Repair (Amst) 2013;12(3):177–187. doi: 10.1016/j.dnarep.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003;63(3):608–615. [PubMed] [Google Scholar]
- 55.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van de Ven M, et al. Extended longevity mechanisms in short-lived progeroid mice: Identification of a preservative stress response associated with successful aging. Mech Ageing Dev. 2007;128(1):58–63. doi: 10.1016/j.mad.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Pluijm I, et al. Impaired genome maintenance suppresses the growth hormone—insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2007;5(1):e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Susa D, et al. Congenital DNA repair deficiency results in protection against renal ischemia reperfusion injury in mice. Aging Cell. 2009;8(2):192–200. doi: 10.1111/j.1474-9726.2009.00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86(3):573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 60. Committee on Care and Use of Laboratory Animals (1985) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85-23.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.