

Abstract
To understand the mechanism of fiber development and pigmentation formation, the mRNAs of two cotton lines were sequenced: line Z128 (light brown fiber) was a selected mutant from line Z263 (dark brown fiber). The primary walls of the fiber cell in both Z263 and Z128 contain pigments; more pigments were laid in the lumen of the fiber cell in Z263 compared with that in Z128. However, Z263 contained less cellulose than Z128. A total of 71,895 unigenes were generated: 13,278 (20.26%) unigenes were defined as differentially expressed genes (DEGs) by comparing the library of Z128 with that of Z263; 5,345 (8.16%) unigenes were up-regulated and 7,933 (12.10%) unigenes were down-regulated. qRT-PCR and comparative transcriptional analysis demonstrated that the pigmentation formation in brown cotton fiber was possibly the consequence of an interaction between oxidized tannins and glycosylated anthocyanins. Furthermore, our results showed the pigmentation related genes not only regulated the fiber color but also influenced the fiber quality at the fiber elongation stage (10 DPA). The highly expressed flavonoid gene in the fiber elongation stage could be related to the fiber quality. DEGs analyses also revealed that transcript levels of some fiber development genes (Ca2+/CaM, reactive oxygen, ethylene and sucrose phosphate synthase) varied dramatically between these two cotton lines.
Introduction
Upland cotton (Gossypium hirsutum L.) is the largest natural fiber producer of the plants. In recent years, interest in naturally colored cotton has grown because it may reduce pollution, making it preferable to white fiber which requires a dyeing process [1]–[3]. However, its commercial application is very limited due to the lack of fiber color diversity and low fiber quality [2]. Limited brown (different color depth) and green fiber lines, among other varieties, have been used in the textile industry. A previous study demonstrated that there was a significant negative correlation between the degree of fiber color and lint percentage and fiber quality traits in cotton [4]. Therefore, subsequent studies should focus on improving the fiber quality and revealing the underlying mechanisms for pigmentation formation in naturally colored cotton.
Early genetic analysis suggested that the brown color of cotton fiber was controlled by one incompletely dominant major gene [5]. Furthermore, gene expression analysis and dimethylaminocinnaldehyde staining showed that tannins could be the key chemical responsible for the brown color in cotton fiber [6]. Subsequent chemical research indicated that the brown pigment in cotton fiber might be the chinone compound oxidated from condensed tannins, and the accumulation period of condensed tannins was from 10 DPA-25 DPA [7], [8]. However, the molecular mechanism that underlies pigmentation in colored cotton fiber is still unknown.
With the development of next generation sequencing technology, RNA-seq provides a powerful tool to rebuild our knowledge of transcriptomics. By directly sequencing and assembling the mRNA, the whole transcriptome could be de novo reconstructed precisely and efficiently [9], aligned with public databases for function annotation and the critical genes could be assessed using pathway classification. In addition, gene expression can be measured and the number of transcripts can be obtained if the appropriate level of sequencing was performed. The application of RNA-seq technology has been used successfully for various species [10]–[14].
Lines Z263 and Z128, two brown fiber inbred lines with dark and light brown fiber respectively, both derived from a cross between white and brown fiber cotton. To reveal the whole transcriptome landscape of the natural colored cotton fiber, and to understand the molecular mechanism of pigment formation, the transcriptomes of these two lines at the whole early developmental stage (0 dpa–20 dpa) were sequenced using RNA-seq technology and analyzed.
Materials and Methods
1. Plant material
Line Z263 (Gossypium hirsutum L.), with dark brown fiber, was selected from a cross between white cotton (Zhong 6331) and brown fiber cotton (Crd). Line Z128 (G. hirsutum L.) was a selected mutant from Z263. They were planted in an experimental field at the Institute of Cotton Research, Chinese Academy of Agricultural Sciences (ICR, CAAS) under normal agronomic management conditions. The bolls at the day of anthesis (0 day post anthesis/DPA), 5 DPA, 15 DPA and 20 DPA were harvested and stored in an ice box, then the ovules of 0 DPA and fibers of other stages were dissected and separated on ice as fast as possible, and then stored at −80°C immediately.
2. Fiber quality measurement, fiber microstructure detection and cellulose test
To test the fiber quality of Z263 and Z128, the following measurements were included: upper half mean length (mm), uniformity index (%), micronaire, fiber elongation (%), fiber strength, 15 g lint samples of each line were analyzed using USTER HVI 1000 (USTER Technologies, Inc., Uster, Switzerland). The fiber microstructure detection and cellulose test were performed according to Ru et al [15].
3. RNA extraction and cDNA library construction
Total RNAs were extracted from each sample using the CTAB method described by Wan and Wilkins [16], with minor modifications to increase the yield. All RNA quality and quantity were measured by 1.0% agarose gel and an ultraviolet spectrophotometer. Four stages of RNAs (0 DPA, 5 DPA, 15 DPA and 20 DPA) with the same concentration and quality from each line were mixed together as one mixed library. The two libraries of Z263 and Z128 were constructed using the method described by Xia et al. [17].
4. RNA-seq and sequence de novo assembly
The sequencing of two libraries of Z263 and Z128 were performed on HiSeq 2000 (illumina) by the Beijing Genomics Institute (BGI) (Shenzhen, Guangdong, China). The raw reads, transformed from images, were first processed by removing adaptors and redundant fragments to generate clean reads. The clean reads de novo assembly was carried out using the short reads assembling program-SOAPdenovo [18]. Clean reads with a certain length of overlap were first combined to form “contigs”. Then the clean reads were mapped back to the contigs; it was possible to detect contigs from the same transcript, as well as the distances between them, by paired-end reads. Next, SOAPdenovo connected the contigs into “scaffolds”; “N” was used to present unknown sequences. Paired-end reads were used again to fill the gaps between scaffolds, longer sequences which could not extend at either end were assembled and defined as “unigenes”. In this study, the unigenes from the two libraries were further processed by sequence splicing and redundancy removal with the sequence cluster program-TGICL [19] to acquire non-redundant unigenes that were as long as possible.
5. Unigene functional annotation and functional categorization
All-unigenes were first searched using the blastx tool against public protein databases such as Non-redundant protein sequences (nr, http://www.ncbi.nlm.nih.gov), Swiss-Prot (http://www.expasy.org/sprot/), Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/pathway.html) and Cluster of orthologous groups for eukaryotic complete genomes (KOG, http://genome.jgi.d oe.gov/Tutorial/tutorial/kog.html). An e-value<10−5 was used as the threshold. To further understand the distribution of gene function, the protein functional classification and pathway were annotated by Gene Ontology (http://www.geneontology.org/), KOGs and KEGG. With NR annotation, GO functional annotation and classification were obtained using the Blast2GO program [20] and WEGO software [21], respectively.
6. Differential expressed genes (DEGs) identification and enrichment analysis
Referring to the method described by Audic and Claverie [22], the Beijing Genomics Institute (BGI) developed a rigorous algorithm to identify DEGs from two samples of RNA-seq data. Because the expression of each gene occupies only a small part of the library, we denote the number of unambiguous clean tags from gene A as x, and the p(x) is in the Poisson distribution. The formula is as follows:
The total clean tag number of sample 1 is N1, and total clean tag number of sample 2 is N2; gene A holds x tags in sample 1 and y tags in sample 2. The probability of gene A expressed equally between two samples can be calculated with the following formula:
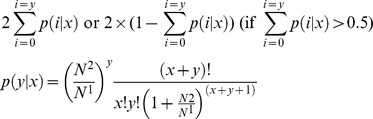 |
The P value corresponds to the differential gene expression test.
The false discovery rate (FDR) is a method used to determine the threshold of the P value in multiple tests. In our study, the reads per kilobase of exon model per million mapped reads (RPKM) value, was calculated referring to the formula described by Mortazavi et al. [23] and was used to quantify the transcript level of Z128 versus Z263. FDR≤10−3 and the absolute value of the log2Ratio (Z128_RPKM/Z263_RPKM) ≥1 as the threshold, were used to judge significant differences in gene expression. DEGs were then subjected to GO functional enrichment analysis and KEGG pathway enrichment analysis.
GO functional enrichment analysis provides GO terms, which significantly enrich in DEGs compared to the genome background, indicating that the DEGs are connected to interesting biological functions. All DEGs are firstly mapped to GO terms in the database, calculating gene numbers for every term, then the ultra-geometric test is used to find significantly enriched GO terms in DEGs compared to the genome background. The formula is:
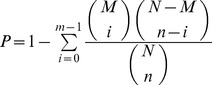 |
Where N is the number of genes with GO annotation; n is the number of DEGs in N; M is the number of genes that are annotated according to the GO terms; m is the number of DEGs in M. The calculated P value≤0.05 was taken as a threshold. GO terms fulfilling this condition are defined as significantly enriched GO terms in DEGs. This analysis recognizes the main biological functions that DEGs participate in.
Pathway enrichment analysis identifies significantly enriched metabolic pathways or signal transduction pathways in DEGs compared with the whole genome background. The formula is the same as that in GO analysis. Here, N is the number of genes with KEGG annotation, n is the number of DEGs in N, M is the number of genes related to specific pathways, and m is number of DEGs in M (Qvalue≤0.05). All DEGs are further mapped on to each pathway; up-regulated genes are marked with red borders while down-regulated genes are marked with green borders.
7. qRT-PCR analysis for genes related to flavonoid synthesis
The genes and primers used for the gene expression analysis related to flavonoid synthesis were listed in Table 1. qRT-PCR was performed in a total volume of 20 µL with 10 µL SYBR Premix Ex Taq(2×) (Takara, Japan), PCR forward primer (10 µM) 0.4 µL,PCR reversed primer 0.4 µL (10 µM),ROX Reference Dye II (50×) 0.4 µL,cDNA template 2.0 µL and ddH2O 6.8 µl on a 7500 real-time PCR machine (Applied Biosystems) according to the manufacturer’s instructions. PCR amplification employed a 10 s denaturing step at 95°C, followed by 5 s at 95°C and 40 s at 60°C with 40 cycles. Relative mRNA levels were calculated by the 2−ΔΔCT method with Gh18S (accession number: L24145) as an internal control.
Table 1. The primers used in this study.
| Genename | NCBIaccession no. | Direction | PrimerSequence (5′→3′) | Productlength |
| CHS | EF643507 | F | GGTGTGGACATGCCTGGGGC | 265 |
| R | CAGCTGCGGCACCATCACCA | |||
| CHI | EF187439 | F | ATCCGTTGAGTTTTTCAGAG | 127 |
| R | CCAAATAGCAACGCAAT | |||
| F3′H | F | CGAGGAGATGGATAAGGTGATTG | 128 | |
| R | GCAAGTTCAAGGGAGTAGATGGA | |||
| F3H | EF187440 | F | GCTTCTTGAGGTGTTGTCAGAGG | 116 |
| R | CAGGTTGAGGGCATTTAGGATAG | |||
| DFR | FJ713480 | F | CGCGACCCTGGCAACTCGAA | 417 |
| R | CCAGGCTGCTTGCTCTGCCA | |||
| ANS | EU921264 | F | AAGAGAAGTATGCCAACGAC | 102 |
| R | AGAAGTAGTCCTCCCACTCA | |||
| ANR | FJ713479 | F | TCCTCAACAAAAGATACCCTGACTT | 147 |
| R | CGGTTTGGTCGTAGATTTCCTC | |||
| LAR | F | AAAGTAGCCAAAGCCCTTCA | 260 | |
| R | TAACAGTGCCGACAGAGTGAA | |||
| Gh18S | L24145 | F | TGACGGAGAATTAGGGTTCGA | 100 |
| R | CCGTGTCAGGATTGGGTAATTT |
8. Statistical analysis
All of the experiments concerning data comparisons were performed three times. Statistical analyses were performed using the S-N-K method of independent-samples t-test at 95% confidence with IBM SPSS Statistics 11.0 (SPSS Inc., Chicago, USA). Values with different lowercases represent a significant difference at P<0.05.
Results
1. The differences in fiber color and quality between Z263 and Z128
Line Z128 was a selected mutant from Z263 and both lines have similar genetic backgrounds. However, their fibers were different. As shown in Fig. 1, the fiber of Z263 was dark brown while that of Z128 was light. Though the primary walls of the fiber cell in both Z263 and Z128 contain pigments (Fig. 1), more pigments were laid in the lumen of the fiber cell in Z263 compared with that Z128. This resulted in a darker color in the Z263 fiber. However, the fiber yield and quality of Z128 was better than that of Z263. The lint percentages (%), upper half mean length (mm), micronaire and fiber strength in Z128 were also better than those in Z263 (Fig. 1). Furthermore, Z128 contained more cellulose (98.5%) than Z263 (94.5%).
Figure 1. The fiber color, yield, quality and microstructure of Z128 and Z263.

Statistical analyses were performed at 95% condence with IBM SPSS Statistics 11.0 (SPSS Inc., Chicago, USA). Values with an asterisk represented a significant differe- nce at P<0.05. The fiber microstructure of Z263 (C I) and Z128 (C II) was observed under a microscope (×3000 and ×2500, respectively).
2. The de novo assembled transcriptome of the fibers in Z263 and Z128
By removing useless sequences, a total of 38,114,054 (2,858,554,050 nucleotides) and 39,355,642 (2,951,673,150 nucleotides) 75 bp-length clean reads were obtained from the Z128 and Z263 mRNA libraries, respectively. A total of 170,201 and 182,404 contigs, 143,588 and 151,478 scaffolds and 59,926 and 71,895 unigenes were assembled in the Z128 and Z263 library, respectively. Because both of the samples for library construction were collected from the same tissue (ovule and fiber), two unigene libraries were taken forward for sequence clustering and redundancy removal to generate a new unigene library (All-unigene library) to make the non-redundant unigenes as long as possible. The All-unigene library contained 71,895 unigenes with an average length of 533 bp, which was obviously greater and longer than the other two libraries. The length of most of the unigenes in the three libraries was in the range of 100–1,000 bp, accounting for 93.08%, 92.44% and 88.21% in Z128-, Z263- and all-unigene library, respectively.
Since the all-unigene library contained the most complete and longest sequences, it was used to run batch alignment with a cut-off E-value of 10−5 on online public databases. A total of 49,941 (69.46%) and 31,714 (44.11%) unigenes received annotations from the NCBI non-redundant (nr) and Swiss-Prot databases, respectively. KEGG, KOGs and GO similarity analyses indicated that 20,241 (28.15%), 14,333 (19.94%) and 7,757 (10.79%) unigenes matched these databases, respectively. To investigate the genomic similarity between Gossypium and other species, we estimated all annotations of unigenes from nr. The result showed that the most abundant unigenes were annotated as “Vitis vinifera”, “Ricinus communis” and “Populus trichocarpa”, which accounted for 29.71%, 29.59% and 24.63%, respectively. Furthermore, we annotated 68.2% and 80.2% unigenes on the recently r- eleased A and D genome of diploid cotton (which were considered to be two donor g- enomes of the tetroploid cotton subgenome), respectively.
GO classification analysis showed that 7,757 all-unigenes were categorized into three main ontologies: biological process, cellular component and molecular function, which were further categorized as 54 terms, and all-unigenes were classified into different terms. One unigene might be repeatedly classified in different terms, therefore, a total of 32,935 all-unigenes (including 75 unigenes repeated in different categories) were distributed over 42 terms, 14,867 (45.14%) of them were categorized in cellular component ontology, 10,229 (31.06%) in biological process and 7,839 (23.80%) in molecular function (Fig. 2). The detailed classification demonstrated that the term “cell” (4,876) and “cell part” (4,876) in “cellular component” ontology, “metabolic process” (3,026) and “cellular process” (3,002) in “biological process” ontology, “binding” (3,538) and “catalytic” (3,030) in “molecular function” contained the most unigenes, respectively. Furthermore, 764 unigenes under the term “pigmentation” indicated that an abundance of pigment-related biological processes were involved in the development of colored cotton fiber.
Figure 2. All-unigenes classified by GO analysis.

The all-unigenes library was aligned to the KOG database to predict and classify possible functions. A total of 14,333 unigenes were matched and categorized into 24 classes (Fig. 3). The function class defined as “general function prediction” (code: R) had the most unigenes (24.61%), followed by “transcription” (K: 13.33%), “replication, recombination and repair” (L: 12.86%), “signal transduction mechanisms” (T: 10.95%) and “posttranslational modification, protein turnover, chaperones” (O: 10.70%).
Figure 3. COG classification of all-unigenes.

A–Z represented different functions classified by GO analysis, respectively.
The all-unigene library was aligned with the KEGG pathway database, and the result showed a total of 38,645 unigenes which were classified into six categories, mostly concentrated in three of them: metabolism (11,371; 29.42%), protein families (10,638; 27.53%) and cellular processes (6,959; 18.01%). Furthermore, 1,315 unigenes were clustered in the “biosynthesis of secondary metabolites” category (Table S1).
3. Differentially expressed genes (DEGs) analysis
The unigene expression level in the Z128 and Z263 libraries were compared. A total of 13,278 (20.26%) unigenes were significantly differentially expressed when these two non-redundant libraries were compared (FDR≤0.001, |log2Ratio|≥1). A total of 5,345 (8.16%) of them were up-regulated, while 7,933 (12.10%) of them were down-regulated; the others were not DEGs (Fig. 4).
Figure 4. Mapping of all expressed unigenes.

This figure was created by comparing gene expression levels of Z128 to Z263. FDR≤0.001, |log2Ratio| ≥1 was used as threshold, red dots represented up-regulated genes, green dots represented the down-regulated genes, and blue dots represented gene expression with no significant difference.
The GO analysis results showed that when corrected (P-value≤1), seven enriched terms belonged to three ontologies, one of them was categorized in “biological process”, three were in “cellular component” and three were in “molecular function” (Table 2).
Table 2. The DEGs enriched terms in GO analysis (P-value<1).
| Ontology | Gene Ontology term | Clusterfrequency | GenomeFrequencyof use | CorrectedP-value |
| Biologicalprocess | RNA-dependent DNA replication | 15/597, 2.5% | 47/4110, 1.1% | 0.82163 |
| Cellularcomponent | cytoplasmic vesicle | 137/696, 19.7% | 750/4911, 15.3% | 0.06344 |
| cytoplasmicmembrane-bounded vesicle | 135/696, 19.4% | 745/4911, 15.2% | 0.10093 | |
| spliceosome | 7/696, 1.0% | 14/4911, 0.3% | 0.23912 | |
| Molecularfunction | oxidoreductase activity, actingon paired donors, withincorporation or reductionof molecular oxygen,2-oxoglutarate as one donor,and incorporation of oneatom each of oxygen intoboth donors | 7/769, 0.9% | 13/5273, 0.2% | 0.32203 |
| RNA-directed DNApolymerase activity | 15/769, 2.0% | 48/5273, 0.9% | 0.76482 | |
| DNA polymerase activity | 15/769, 2.0% | 49/5273, 0.9% | 0.95719 |
Table 3 showed the top 10 DEGs enriched pathways, seven of which were categorized as a “metabolism” pathway, two were categorized as “genetic information processing” pathways and the other one was categorized as “environmental information processing”. Further identification indicated that five of the “metabolism” terms belonged to the “biosynthesis of secondary metabolites” sub-category.
Table 3. The top 10 DEGs enriched pathways in KEGG analysis.
| No. | Pathway | DEGs withpathwayannotation(3,213) | All genes withpathwayannotation(20,242) | P-value | Pathway ID |
| 1 | Zeatinbiosynthesis | 16 (0.5%) | 41 (0.2%) | 0.000299 | ko00908 |
| 2 | Anthocyaninbiosynthesis | 4 (0.12%) | 5 (0.02%) | 0.002767 | ko00942 |
| 3 | ABC transporters | 40 (1.24%) | 164 (0.81%) | 0.003 | ko02010 |
| 4 | Diterpenoidbiosynthesis | 15 (0.47%) | 47 (0.23%) | 0.004717 | ko00904 |
| 5 | Phenylpropanoidbiosynthesis | 56 (1.74%) | 255 (1.26%) | 0.006239 | ko00940 |
| 6 | 3-Chloroacrylicacid degradation | 18 (0.56%) | 63 (0.31%) | 0.007641 | ko00641 |
| 7 | Flavone andflavonol biosynthesis | 14 (0.44%) | 45 (0.22%) | 0.007987 | ko00944 |
| 8 | Ubiquitinmediatedproteolysis | 108 (3.36%) | 550 (2.72%) | 0.00975 | ko04120 |
| 9 | Taurine andhypotaurinemetabolism | 5 (0.16%) | 10 (0.05%) | 0.012555 | ko00430 |
| 10 | Base excisionrepair | 36 (1.12%) | 158 (0.78%) | 0.014168 | ko03410 |
4. Expression of related genes for color and fiber development in two cotton lines of different fiber color and quality
The pigmentation deposits in the brown colored cotton fiber were closely related to the flavonoid and proanthocyanidins biosynthesis. In this study, the “anthocyanin biosynthesis” and “flavone and flavonol biosynthesis” appeared on the top 10 list of the KEGG enrichment analysis (Table 3). Further analysis using the KEGG database indicated that a total of 14 unigenes were involved in the “flavone and flavonol biosynthesis” pathway (Table 4), which could be further classified as three orthology categories. “K05280” contained one up-regulated unigene which encoded flavonoid 3′-hydroxylase (F3′H). In addition, 10 down-regulated unigenes that encoded flavonol 3-O-methyltransferase (OMT) belonged to “K05279”; in “K10757”, all unigenes encoded flavonol 3-O-glucosyltransferase (FOGT), two of them were up-regulated, and one was down-regulated. A total of four unigenes were mapped in the “anthocyanin biosynthesis” pathway, all of them were up-regulated. All four unigenes were involved in anthocyanin 5-O-glucosyltransferase (5GT) encoding.
Table 4. The involved unigenes in Flavone and flavonol biosynthesis and Anthocyanin biosynthesis pathways.
| Orthology | Entry ofenzyme | Unigenes | Ratio1 | Status2 | Encoded protein | |
| Flavonoidbiosynthesis | K00660 | 2.3.1.74 | Unigene56280_All | −1.9 | D | chalcone synthase (CHS) |
| K00588 | 2.1.1.104 | Unigene34142_All | −1.4 | D | caffeoyl-CoA O-methyltransferase | |
| Unigene42690_All | −3.6 | D | ||||
| Unigene10308_All | −1.1 | D | ||||
| K01859 | 5.5.1.6 | Unigene69406_All | −1.6 | D | chalcone isomerase (CHI) | |
| K05277 | 1.14.11.19 | Unigene56780_All | −2.3 | D | anthocyanidin synthase (ANS) | |
| Unigene57093_All | −3.0 | D | ||||
| Unigene5167_All | −1.6 | D | ||||
| K08695 | 1.3.1.77 | Unigene43073_All | −1.1 | D | anthocyanidin reductase (ANR) | |
| K05280 | 1.14.13.21 | Unigene71334_All | 2.8 | U | flavonoid 3'-hydroxylase (F3'H) | |
| K00475 | 1.14.11.9 | Unigene70267_All | 3.9 | U | flavanone 3-hydroxylase (F3H) | |
| Unigene71550_All | 4.2 | U | ||||
| K05278 | 1.14.11.23 | Unigene70581_All | 4.1 | U | flavonol synthase (FLS) | |
| Unigene6472_All | 1.0 | U | ||||
| Flavone andflavonolbiosynthesis | K05280 | 1.14.13.21 | Unigene71334_All | 2.8 | U | flavonoid 3'-hydroxylase (F3'H) |
| K05279 | 2.1.1.76 | Unigene13368_All | −2.2 | D | flavonol 3-O-methyltransferase (FOMT) | |
| Unigene21288_All | −1.4 | D | ||||
| Unigene38018_All | −1.2 | D | ||||
| Unigene47446_All | −2.4 | D | ||||
| Unigene47563_All | −3.7 | D | ||||
| Unigene50636_All | −2.7 | D | ||||
| Unigene57809_All | −1.7 | D | ||||
| Unigene58594_All | −1.9 | D | ||||
| Unigene70908_All | −1.7 | D | ||||
| Unigene9272_All | −1.1 | D | ||||
| K10757 | 2.4.1.91 | Unigene50839_All | 1.0 | U | flavonol 3-O-glucosyltransferase (FOGT) | |
| Unigene8998_All | 2.1 | U | ||||
| Unigene54635_All | −4.3 | D | ||||
| Anthocyaninbiosynthesis | K12338 | 2.4.1.298 | Unigene18122_All | 2.2 | U | anthocyanin 5-O-glucosyltransferase (5GT) |
| Unigene59143_All | 12.0 | U | ||||
| Unigene67316_All | 12.5 | U | ||||
| Unigene71660_All | 12.9 | U |
1: ratio indicated log2(z128_RPKM/Z263_RPKM).
2: “U” indicated that this unigene was up-regulated; “D” indicated down-regulated.
Another important pigmentation pathway in plants is the “flavonoid biosynthesis pathway”, which is also considered to be a key pathway for pigment formation in brown fiber cotton. Although it was not shown in Table 3, eight important genes were involved in this pathway. Chalcone synthase (CHS), chalcone isomerase (CHI), leucoanthocyanidin reductase (LAR), anthocyanidin reductase (ANR) and anthocyanidin synthase (ANS) were down-regulated in this pathway, while flavonoid 3′-hydroxylase (F3′H), flavanone 3-hydroxylase (F3H) and flavonol synthase (FLS) were all up-regulated. According to the distribution of DEGs in the entire pathway, most of the down-regulated DEGs (CHS, CHI, LAR, ANR, ANS) were enriched in upstream and downstream pathways, and the up-regulated DEGs were in the middle of the flavonoid biosynthetic pathway (F3H, F3′H, FLS). However, another gene in the middle of the pathway, the dihydroflavonol 4-reductase (DFR), was unchanged (Fig. 5). Furthermore, a gene that encodes 5-O-glucosyltransferase in the anthocyanin biosynthesis pathway was up-regulated. The down-regulated genes in the flavonoid biosynthesis pathway suggested that there were less pigments in Z128 compared with that in Z263. This was confirmed by the lighter brown fiber color in Z128 compared to the dark brown fiber in Z263.
Figure 5. The schematic of pigment formation in cotton fiber.

The red and green arrows indicated the up- and down-regulated status detected by comparing gene expression in Z128 with that in Z263. chalcone synthase (CHS); chalcone isomerase (CHI); flavanone 3-hydroxylase (F3H); flavonol synthase (FLS); dihydroflavonol 4-reductase (DFR); leucoanthocyanidin reductase (LAR); anthocyanidin synthase (ANS); UDP-favonoid glucosyl transferase (UFGT); anthocyanidin reductase (ANR); anthocyanin 5-O-glucosyltransferase (5GT).
To test the reliability of comparative transcriptional data, qRT- PCR analysis was performed for CHS, CHI, LAR, DFR, F3H, F3′H, ANR and ANS. Samples were selected from the flavonoid biosynthesis pathway across five developmental time points from 0 DPA to 20 DPA. Overall, the results of the qRT-PCR analysis were consistent with the results from the transcriptome for the mixed mRNAs of five developmental time points of the eight selected genes. However, when Z128 was compared to Z263 at 10 DPA, the selected eight genes had significantly higher transcript levels in Z128 (Fig. 5). This suggests that the genes involved in the flavonoid biosynthesis pathway at 10 DPA are related to better fiber quality formation. Moreover, the genes involved in flavonoid biosynthesis affected the fiber color and fiber quality of the brown fiber cotton. Some other genes such as the ethylene related factors also regulated fiber development [24]. According to Table 5, all of the ethylene related factors had higher expression levels in Z128 than in Z263. The DEGs analysis also revealed that transcript levels of different fiber development related factors varied dramatically in cotton fibers, such as reactive oxygen [25], ethylene [24], Ca2+/CaM [26], and sucrose phosphate synthase [27] (Table 5).
Table 5. Fiber development related DEGs of brown and white fibers.
| genes | Z263 (RPKM) | Z128 (RPKM) | p-value | FDR | Ratio1 | Status2 | Homologous proteins |
| Unigene31293_All | 40.4792 | 19.2683 | 5.06E-15 | 2.40E-16 | −1.1 | D | extracellular Cu/Zn superoxide dismutase |
| Unigene15009_All | 36.5823 | 3.0274 | 1.54E-52 | 2.92E-54 | −3.6 | D | class III peroxidase |
| Unigene22859_All | 1.8516 | 5.7002 | 4.97E-04 | 1.53E-04 | 1.6 | U | glutathione peroxidase |
| Unigene25776_All | 2.1838 | 9.8675 | 1.78E-09 | 2.04E-10 | 2.2 | U | peroxisomal membrane ABC transporter family |
| Unigene40537_All | 80.13 | 230.84 | 2.93E-14 | 1.51E-15 | 1.5 | U | fiber quinone-oxidoreductase |
| Unigene50168_All | 2.78 | 1.20 | 3.83E-02 | 2.08E-02 | −1.2 | D | calcium-transportingATPase(ACA9) |
| Unigene1176_All | 16.85 | 45.17 | 2.03E-13 | 1.24E-14 | 1.4 | U | calcium ion transmembrane transporter(ACA2) |
| Unigene55005_All | 3.59 | 0.50 | 1.55E-03 | 5.46E-04 | −2.8 | D | calcium ion binding/transporter(ATNRT1∶2) |
| Unigene15884_All | 3.32 | 14.7 | 2.71E-14 | 1.38E-15 | 2.1 | U | autoinhibited calcium ATPase |
| Unigene61435_All | 1.1851 | 5.9315 | 3.37E-04 | 1.00E-04 | 2.3 | U | ethylene receptor |
| Unigene40770_All | 5.06 | 14.98 | 2.02E-07 | 3.10E-08 | 1.5 | U | ethylene responsive element binding protein |
| Unigene26099_All | 4.86 | 61.24 | 0.00 | 0.00 | 3.7 | U | ethylene responsive transcription factor 2b |
| Unigene37372_All | 22.708 | 90.2862 | 0.00 | 0.00 | 1.9 | U | ethylene-responsive element binding protein ERF2 |
| Unigene62869_All | 0.86 | 9.38 | 2.65E-05 | 6.01E-06 | 3.4 | U | Ethylene-responsive transcription factor 1B |
| Unigene30365_All | 6.59 | 21.61 | 6.11E-14 | 3.33E-15 | 1.7 | U | sucrose phosphate synthase |
1: ratio indicated log2(z128_RPKM/Z263_RPKM);
2: “U” indicated that this unigene was up-regulated; “D” indicated down-regulated.
Discussion
The RNA-seq approach based on next generation sequencing technology provided us with a new method to study the transcriptome of developing cotton fibers. It was not dependent on existing genome information and was an efficient way to quantify the expression level of a single gene without high background noise [28]. In recent years, this technology has been successfully applied in transcriptome studies for many non-model organisms [13], [17], [29]–[30].
In this study, we mixed the RNA from four important fibers at various developmental stages (5, 10, 15 and 20 DPA) to de novo assemble the transcriptome of developmental colored cotton fiber. A total of 125,014 unigenes were generated in two sequencing libraries, which were further assembled into 71,895 all-unigenes with an average length of 533 bp, 69.46% of which could be matched in the NCBI database (E-value≤10−5). This volume of data was greater than that reported in previous studies on other species [17], [30]. Approximately 20,000 unigenes identified from G. hirsutum were recorded in the NCBI database (Nov 2011). In this study, approximately 50,000 unigenes (EST) were matched with nr database records. Therefore, we believe that our unigene library contained almost all of the known unigenes from G. hirsutum. In conclusion, we acquired a high quality and well-assembled transcriptome library for developing colored cotton fibers.
In all nr-annotated unigenes, only 1,537 (3.08%) were directly annotated with the field of “Gossypium hirsutum”; most other unigenes were assigned to other species, which mainly included “Vitis vinifera”, “Ricinus communis” and “Populus trichocarpa”. This implies that the genome of cotton may be very similar to these species and this could be a reference for a prospective cotton sequencing project.
Z263 was the offspring which derived from a cross between white fiber cotton and dark brown cotton, and Z128 was an inbred line selected from Z263 with a lighter color and better fiber quality. Therefore, the similar genetic backgrounds provided a fine model with which to study the mechanism underlying the formation of brown color in cotton fiber. We compared the whole transcriptome for each and, unexpectedly, an abundance of unigenes (20.26%) revealed significant differential expression between Z263 and Z128 (FDR≤0.001, |log2Ratio|≥1). This evidence demonstrates that all DEGs are relatively evenly distributed in most of the relevant metabolism pathways. Namely, the divergence of cotton fiber color and quality was the result of complex processes generated by multiple metabolic processes.
There is limited information in the literature on the molecular mechanism that underlies the formation of fiber color in cotton. Xiao et al [6] cloned five flavonoid structure genes from brown cotton fiber and found that all the cloned genes could be involved in pigmentation metabolism for brown fiber. Several studies focused on chemicals also suggested that proanthocyanidins (condensed tannins) might be the precursor of pigmentation in natural colored cotton fiber [7]–[8]. Here, almost of all the unigenes which encoded the key enzymes (CHS, CHI, LAR, ANS and ANR) of the flavonoid biosynthesis pathway were down-regulated in Z128 compared with that in Z263, implying that accumulation of proanthocyanidins in Z263 might be more than that in Z128. Zhan et al. [7]–[8] suggested that the depth of brown color might be closely related to the accumulated quantity of condensed tannins. Another unexpected finding in this study was related to the “anthocyanin biosynthesis” pathway. As a downstream metabolic pathway, it is one of the most important elements of pigment biosynthesis in plants [31]–[33]. We found that all unigenes involved in this pathway were significantly up-regulated (read in Z263 = 0) in Z128 and homologous with anthocyanin 5-O-glucosyltransferase (5GT), which could make anthocyanin more stable by modification [34]. Another recent study demonstrated that the lack of glucose at the 5 position of anthocyanin could lead to color variation in carnations [35]. This result implied that the depth of brown cotton fiber color variation might be the consequence of an interaction between oxidized tannins and glycosylated anthocyanin.
F3H has been thoroughly studied for decades and it is predominantly expressed during the fiber elongation stage in G. barbadense, a process that has no relation to pigment formation [36]. Furthermore, when the F3H–RNAi segment was transferred into brown fiber plants, they yielded more stunted fibers. Transgenic analysis showed that the suppression of F3H not only inhibited fiber elongation but also retarded fiber maturation [37]. F3H was suppressed in Z263 but up-regulated in Z128. This evidence suggests that F3H is important in fiber development. Like F3H, F3′H and FLS were very important in the pigments synthesis. Therefore, it is possible that the genes in the middle were up-regulated. According to Xiao et al [6] and Zhan et al [7], tannins could be the key chemical responsible for the brown color in cotton fiber. As shown in Fig. 5, LAR, ANR, and ANS were the key enzymes to accumulate the tannins. The down-regulated CHS, CHI, LAR, ANR, and ANS genes in the main pathway of pigment formation should be related to lighter fiber color of Z128 than that of Z263.
The pigmentation related genes not only regulated the fiber color but also influenced the fiber quality. The flavonoids are abundant and widely distributed plant secondary metabolites, and known to be an active participant in fiber development [38]. Previous studies showed that in fiber cells, the flavonoid genes were dominantly expressed in the fiber elongation stage [39]–[41]. In our study, CHS, CHI, LAR, DFR, F3H, F3′H, ANR and ANS showed higher expression levels at 10 DPA in Z128, thus highlighting that flavonoid metabolism represents a novel pathway with the potential for cotton fiber improvement. Our GWAS analysis of SNP in cotton germplasm indicated that the genes involved in flavonoid biosynthesis were also associated with fiber quality traits (unpublished data). Therefore, the highly expressed flavonoid gene in the fiber elongation stage in Z128 should be related to better fiber quality.
Cotton fibers are single-celled trichomes that differentiate from the ovule epidermis, including fiber initiation, elongation, secondary cell wall biosynthesis and maturation, leading to mature fibers. Ca2+ and ROS are two important factors involved in fiber cell growth [42]. Ca2+/Calmodulin (CaM) is involved in plant growth and development through interaction with ROS signaling [43]. Based on gene expression profile analysis, Ca2+/CaM is implicated in cotton fiber elongation. However, currently, there remains little direct evidence of the mechanism of Ca2+/CaM on cotton fiber development. In our study, Ca2+ related genes were either down-regulated or up-regulated in Z128 compared with that in Z263.
Recent literature indicates that ethylene acts as a positive regulator of root hair, apical hook, and hypocotyl development [44]–[46]. Furthermore, Shi et al. [24] found that ethylene biosynthesis was one of the most significantly up-regulated biochemical pathways during fiber elongation. Exogenously applied ethylene promoted robust fiber cell expansion, whereas its biosynthetic inhibitor L-(2-aminoeth oxyvinyl)-glycine (AVG) specifically suppressed fiber growth. The ethylene biosynthesis pathways in our data were not shown in Table 3 as the “top 10 DEGs enriched pathways in KEGG analysis”, however, a number of ethylene related genes were up-regulated in Z128 compared with that in Z263, such as Unigene61435_All, Unigene40770_All, and Unigene62869_All. This suggests that ethylene related genes may contribute to better fiber quality in Z128.
Supporting Information
Classification of all-unigenes by KEGG analysis.
(XLS)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by the “Twelfth Five-Year Plan” of the National Science and Technology Support Project (2011BAD35B05 and 2013BAD01B03) and Basic Scientific Research funds in National Nonprofit Institutes (SJA0608). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vreeland JM (1999) The revival of colored cotton. Sci Am 280: 90–96. [Google Scholar]
- 2.Dickerson DK, Lane EF, Rodriguez DF (1999) Naturally colored cotton: resistance to changes in color and durability when refurbished with selected laundry aids. California Agric Tech Inst, California State University, Fresno, California. 42p.
- 3.Murthy MSS (2001) Never say dye: the story of coloured cotton. Resonance 6, 29–35.
- 4. Feng HJ, Sun JL, Wang J, Jia YH, Zhang XY, et al. (2011) Genetic effects and heterosis of the fibre colour and quality of brown cotton (Gossypium hirsutum). Plant Breeding 130: 450–456. [Google Scholar]
- 5. Kohel R (1985) Genetic analysis of fiber color variants in cotton. Crop Sci 25: 793–797. [Google Scholar]
- 6. Xiao YH, Zhang ZS, Yin MH, Luo M, Li XB, et al. (2007) Cotton flavonoid structural genes related to the pigmentation in brown fibers. Biochem Biophys Res Commun 35: 873–78. [DOI] [PubMed] [Google Scholar]
- 7. Zhan SH, Lin Y, Cai YP, Li ZP (2007) Preliminary deductions of the chemical structure of the pigment brown in cotton fiber. Chin Bull Bot 24: 99–104 (Chinese with english abstract).. [Google Scholar]
- 8. Zhan SH, Lin Y, Cai YP, Li ZP (2007) Relationship between the pigment in natural brown cotton fiber and the condensed tannin. Cotton Sci 19: 183–188 (Chinese with english abstract).. [Google Scholar]
- 9. Haas BJ, Zody MC (2010) Advancing RNA-seq analysis. Nat Biotechnol 28: 421–423. [DOI] [PubMed] [Google Scholar]
- 10. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-SNat Methods. 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 11. Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, et al. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hittinger CT, Johnston M, Tossberg JT, Rokas A (2010) Leveraging skewed transcript abundance by RNA-Seq to increase the genomic depth of the tree of life. Proc Natl Acad Sci USA 107: 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo S, Zheng Y, Joung JG, Liu S, Zhang Z, et al. (2010) Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genomics 11: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tisserant E, Da Silva C, Kohler A, Morin E, Wincker P, et al. (2011) Deep RNA sequencing improved the structural annotation of the Tuber melanosporum transcriptome. New Phytol. 189: 883–891. [DOI] [PubMed] [Google Scholar]
- 15. Ru Z, Wang G, He S, Du X (2013) The difference of fiber quality and fiber ultrastructure in different natural colored cotton. Cotton Sci 23: 184–188. [Google Scholar]
- 16. Wan CY, Wilkins TA (1994) A Modified Hot Borate Method Significantly Enhances the Yield of High-Quality RNA from Cotton (Gossypium hirsutum L.). Anal Biochem 223: 7–12. [DOI] [PubMed] [Google Scholar]
- 17. Z Xia, H Xu, J Zhai, D Li, H Luo, C He, X Huang (2011) RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis . Plant Mol Biol 77: 1–10. [DOI] [PubMed] [Google Scholar]
- 18. Li R, Zhu H, Ruan J, Qian W, Fang X, et al. (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pertea G, Huang X, Liang F, Antonescu V, Sultana R, et al. (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- 20. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 21. Ye J, Fang L, Zheng H, Zhang Y, Chen J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34: 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7: 986–995. [DOI] [PubMed] [Google Scholar]
- 23. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-SNat Methods. 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 24. YH Shi, Zhu SW, Mao XZ, Feng JX, Qin YM, et al. (2006) Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 18(3): 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaudhary B, Hovav R, Flagel L, Mittler R, Wendel J (2009) Parallel expression evolution of oxidative stress-related genes in fiber from wild and domesticated diploid and polyploid cotton (Gossypium). BMC Genomics 10: 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang W, Tu L, Yang X, Tan J, Deng F, et al. (2014) The calcium sensor GhCaM7 promotes cotton fiber elongation by modulating reactive oxygen species (ROS) production. New Phytol 202: 509–520. [DOI] [PubMed] [Google Scholar]
- 27. Haigler CH, Singh B, Zhang D, Hwang S, Wu C, et al. (2007) Transgenic cotton over-producing spinach sucrose phosphate synthase showed enhanced leaf sucrose synthesis and improved fiber quality under controlled environmental conditions. Plant Mol Biol 63: 815–832. [DOI] [PubMed] [Google Scholar]
- 28. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1): 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, et al. (2010) Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-SPlant Physiol. 152: 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei W, Qi X, Wang L, Zhang Y, Hua W, et al. (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12: 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dooner HK, Robbins TP, Jorgensen RA (1991) Genetic and developmental control of anthocyanin biosynthesis. Ann Rev Genet 25: 173–199. [DOI] [PubMed] [Google Scholar]
- 32. Halloin JM (1982) Localization and changes in catechin and tannins during development and ripening of cottonseed. New Phytol 90: 651–657. [Google Scholar]
- 33. Grotewold E (2006) The genetics and biochemistry of floral pigments. Annu Rev Plant Biol 57: 761–780. [DOI] [PubMed] [Google Scholar]
- 34. Yamazaki M, Gong Z, Fukuchi-Mizutani M, Fukui Y, Tanaka Y, et al. (1999) Molecular cloning and biochemical characterization of a novel anthocyanin 5-O-glucosyltransferase by mRNA differential display for plant forms regarding anthocyanin. J Biol Chem 274: 7405–7411. [DOI] [PubMed] [Google Scholar]
- 35. Nishizaki Y, Matsuba Y, Okamoto E, Okamura M, Ozeki Y, et al. (2011) Structure of the acyl-glucose-dependent anthocyanin 5-O-glucosyltransferase gene in carnations and its disruption by transposable elements in some varieties. Mol Genet Genomics 286: 383–394. [DOI] [PubMed] [Google Scholar]
- 36. Tu LL, Zhang XL, Liang SG, Liu DQ, LF Zhu, et al. (2007) Gene expression analyses of sea-island cotton (Gossypium barbadense L.) during fiber development. Plant Cell Rep 26: 1309–1320. [DOI] [PubMed] [Google Scholar]
- 37. Tan J, Tu L, Deng F, Hu H, Nie Y, et al. (2013) A genetic and metabolic analysis revealed that cotton fiber cell development was retarded by flavonoid naringenin. Plant Physiol 162: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Owens DK, Crosby KC, Runac J, Howard BA, Winkel B (2008) Biochemical and genetic characterization of Arabidopsis flavanone 3beta-hydroxylase. Plant Physiol Biochem 46: 833–843. [DOI] [PubMed] [Google Scholar]
- 39. Gou JY, Wang LJ, Chen SP, Hu WL, Chen XY, et al. (2007) Gene expression and metabolite profiles of cotton fiber during cell elongation and secondary cell wall synthesis. Cell Res 17: 422–434. [DOI] [PubMed] [Google Scholar]
- 40. Hovav R, Udall JA, Hovav E, Rapp R, Flagel L, et al. (2008) A majority of cotton genes are expressed in single-celled fiber. Planta 227: 319–329. [DOI] [PubMed] [Google Scholar]
- 41. Rapp R, Haigler C, Flagel L, Hovav R, Udall J, et al. (2010) Gene expression in developing fibres of Upland cotton (Gossypium hirsutum L.) was massively altered by domestication. BMC Biol. 8: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qin YM, Zhu YX (2011) How cotton fibers elongate: a tale of linear cell-growth mode. Curr. Opin. Plant Biol. 14: 106–111. [DOI] [PubMed] [Google Scholar]
- 43. Lee SH, Seo HY, Kim JC, WD Heo, Chung WS, et al. (1997) Differential activation of NAD kinase by plant calmodulin isoforms. The critical role of domain I. J Biol Chem 272: 9252–9259. [DOI] [PubMed] [Google Scholar]
- 44. Achard P, Vriezen WH, Van Der Straeten D, Harberd NP (2003) Ethylene regulates Arabidopsis development via the modulation of DELLA protein growth repressor function. Plant Cell 15: 2816–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seifert GJ, Barber C, Wells B, Roberts K (2004) Growth regulators and the control of nucleotide sugar flux. Plant Cell 16: 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grauwe LD, Vandenbussche F, Tietz O, Palme K, Straeten DVD (2005) Auxin, ethylene and brassinosteroids: Tripartite control of growth in the Arabidopsis hypocotyl. Plant Cell Physiol 46: 827–836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Classification of all-unigenes by KEGG analysis.
(XLS)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.