

Abstract
The Nup98-HoxD13 (NHD13) fusion gene was identified in a patient with therapy-related myelodysplastic syndrome (MDS). When transgenically expressed in hematopoietic cells, mice faithfully recapitulate human disease with serial progression from peripheral blood (PB) cytopenias and increased bone marrow (BM) blasts to acute leukemia. It is well accepted that genomic instability in dysplastic hematopoietic stem/progenitor cells (HSPC) drives the evolution of MDS to acute leukemia. Findings here demonstrate that reticulocytes, myeloid and lymphoid PB cells of NHD13 mice, display an increase in the age-associated loss of glycosylphosphatidylinositol-linked surface proteins versus wild type controls. These data correlate with a progressive increase in the DNA damage response as measured by γ-H2AX activity, accumulating BM blasts as the disease progresses and finally development of acute leukemia. These findings clearly demonstrate a state of progressive genomic instability that increases the likelihood of a “second hit” or complimentary mutation later in the disease to trigger development of acute leukemia and underscores the mechanistic nature of how the NUP98-HoxD13 transgene induces progression of MDS to acute leukemia. Additionally, these data support the use of the PIG-A assay as an efficient, real-time surrogate marker of the genomic instability that occurs in the MDS HSPCs.
Key Point
The PIG-A assay is a sensitive, nonlethal method for the serial assessment of genomic instability in mouse models of MDS.
Introduction
Myelodysplastic syndrome (MDS) is a disease of the hematopoietic stem/progenitor cell (HSPC) whose pathogenesis is closely tied to genomic instability. A complex milieu of deficits in DNA repair, oxidative stress, and epigenomic instability underpin the pathogenesis of this heterogeneous disease as it evolves to acute leukemia. Additionally, cytogenetic abnormalities are measured in the majority of MDS patients [1], higher rates of MDS are reported in diseases with established genomic instability, and exposures to genotoxic agents may result in secondary MDS [2], [3], [4].
The Nup98-HoxD13 (NHD13) transgenic mouse was developed from a patient with therapy-related MDS and displays key features of the disease with high penetrance [5]. Investigators have hypothesized that the development of acute leukemia results from a complimentary, pro-leukemic mutation that occurs later in the disease course of this mouse [6]. In support of this, recent work shows increased bone marrow (BM) γ-H2AX activity suggesting genomic instability is an important driver of these secondary mutations [7]. In vivo analysis of the BM HSPCs, however, is invasive, technically challenging, and impractical for serial sampling [8], [9]. As a consequence, investigators must utilize large animal numbers that allow only a solitary, static assessment of the BM and add expense to the study. A minimally invasive assay that facilitates the serial measurement of genomic instability, as it parallels the disease course, would be valuable in the study of MDS and prediction of progression to leukemia.
In this report, we have applied testing of the phosphatidylinositol glycan anchor (PIG-A) gene product to the NHD13 MDS/AML mouse model. The loss of the X-linked PIG-A gene activity will result in the absence of glycosylphosphatidylinositol (GPI)–linked proteins expressed on peripheral blood (PB) cells and may temporally correlate with increased BM blasts and the progression of MDS to acute leukemia. As a potential mechanism by which the NHD13 transgene drives this disease, we find that the loss of GPI-linked surface proteins on PB cells was found to be closely associated with increased γ-H2AX, a measure of the DNA damage response. These data strongly indicate that the NHD13 transgene functions, at least in part, by inducing a progressive form of genomic instability and these results offer important new insight about the timing of acquisition of leukemogenic mutations. While the results do not identify a specific mutation that causes evolution of MDS and acute leukemia, these data indicate that the PIG-A assay represents a novel, non-lethal approach for studying MDS progression in this model.
Materials and Methods
Mice
C57BL/6 Nup98-HOXD13 (NHD13) transgenic mice were obtained from Jackson Laboratory, (Stock # 010505, Bar Harbor, ME) and aged alongside non-transgenic wild type (WT) littermates. Complete blood counts were performed using a Hemavet hematology analyzer (Drew Scientific, Waterbury, CT). Mice were housed in the University of Florida, Cancer and Genetics Research Complex vivarium under SPF conditions. The UF IACUC approved this study (protocol #s 201102224 and 201203669).
DNA damage assays
Assessment γ-H2AX was carried out using an established technique [10] using P-Histone H2AX-Alexa Flour (Cell Signaling, Danvers, MA). Samples were analyzed on an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA).
A neutral comet assay was used to measure DNA repair in HSPCs. NHD13 and WT mice, aged 6 months, were euthanized, BM collected from femurs and lineage (lin−) negative cells isolated by immunomagnetic negative selection (Miltenyi, San Diego, CA). Following irradiation with 1 Gy, Lin− BM cells were mixed with LM Agarose, applied to slides and lysed at 4°C overnight (Trevigen, Gaithersburg, MD). Slides were then electrophoresed at 21 V for 45 minutes and DNA was precipitated followed by immersion in 70% ethanol for 30 minutes. Slides were stained by SYBR green and observed under fluorescence microscope. Comet analysis and Olive tail moment calculations were performed by Wimasis Image analysis (Munich, Germany). Results represent an average of 50 randomly chosen comets for each sample.
PIG-A Assay
Testing for GPI-linked proteins on reticulocytes was performed by adding 2 μl of anti-CD24-FITC, 0.5 μl of anti-CD71-PE, and 1 μl of anti-TER-119-PE-Cy5 to 37.5 μl cell staining buffer and 9 μl of peripheral blood collected from mice (eBioscience, San Diego, CA) [11]. For GPI-linked protein testing on monocytes and T-cells, erythrocytes were lysed with buffer (BioLegend, San Diego, CA), resuspended in 100 μl staining buffer, and either 1 μl F/480-FITC and 1 μl Ly6C-PE (for monocytes) or 1 μl CD3-FITC and 1 μl CD90-APC (for T-cells) were added. In all cases, samples were incubated on ice for 30 minutes, washed, and resuspended. Samples were analyzed on an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Up to 1 × 106 events were captured for reticulocyte analysis. For monocytes and T-cells up to 2 × 104 events were analyzed.
Sequencing of PIG-A cDNA
CD24-negative reticulocytes were collected by sorting on a FACsAria II at the UF ICBR Flow cytometry core. RNA was prepared from approximately 2000 CD24-negative reticulocytes from each mouse using a RiboPure Blood RNA isolation kit (Life Technologies, Grand Island, NY) and cDNA was created with a High Capacity RNA-to-cDNA kit (Life Technologies, Grand Island, NY) according to manufacturer protocols. As a control, cDNA was also prepared from CD24-positive reticulocytes. A two-round, nested PCR strategy was used to amplify and isolate the PIG-A open reading frame using the primers and conditions described [12]. Gel isolated PCR products were cloned into the pCR4-Topo vector by Topo-TA cloning according to the manufacturer’s protocol (Life Technologies, Grand Island, NY). Sanger sequencing was done at the UF ICBR using primers in flanking the insert and internal primers previously described [12]. Mutations were identified by comparing the sequence of individual clones to WT using CLC sequence viewer 7.0.2 software (CLCbio, Boston, MA).
Results
In the disease paroxysmal nocturnal hemoglobinuria, hemolytic manifestations arise from an acquired somatic mutation of the X-linked PIG-A gene [13]. The loss of this gene product results from a spontaneous mutation of the HSPC which is subsequently observed in its progeny circulating in the PB [14]. As the result of the mutational inactivation of PIG-A, affected erythrocytes that arise from the affected BM HSPC do not express necessary GPI-linked complement regulatory proteins (CD55 and CD59) which renders them susceptible to clinically significant complement mediated hemolysis. We hypothesize that this association will also be present and can be detected in the terminally differentiated PB cell progeny of such affected MDS BM HSPCs.
To study any progressive nature of genomic instability over the natural history of MDS, we selected the Nup98-HOXD13 (NHD13) transgenic mouse model of human MDS. This mouse model demonstrates the key clinical characteristics of MDS with evolution to acute leukemia as observed in the PB and BM of humans with this disease [5]. The mice develop leukopenia early in life as evidenced by significantly reduced white blood cell counts (WBC) compared to WT mice (Figure 1A, P < .01). In addition, NHD13 mice display a progressive macrocytic anemia characterized by a trend of decreasing hemoglobin concentration as mice age (Figure 1B). Following this, NHD13 mice display a significant, serial increase of BM blasts upon aging that precede the development of acute leukemia (Figure 1C, P < .01). In addition, NHD13 mice develop a characteristic anemic phenotype with noticeable pallor of their fore- and hind limbs and of the nasal region (Figure 1D). Late in the disease course, the PB of NHD13 mice becomes hypochromic and noticeable weight loss occurs (Figure 1D and data not shown).
Figure 1.
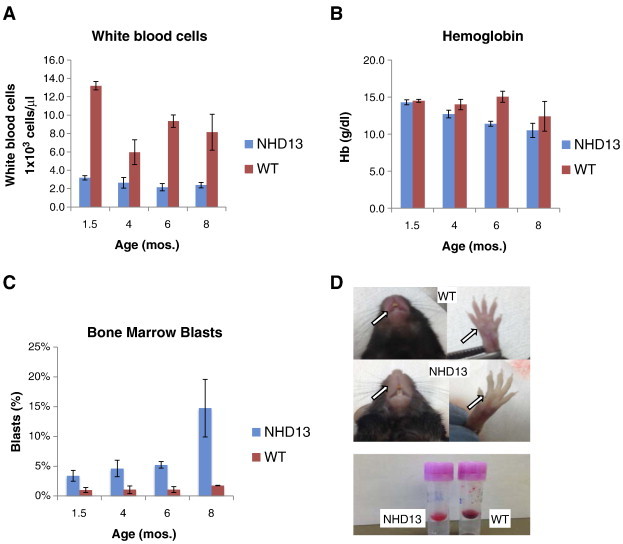
The NHD13 transgenic mouse develops a highly penetrant form of progressive myelodysplastic syndrome (MDS). (A) White blood cell count (WBC) is lower in the NHD13 transgenic compared to WT controls. (B) Hemoglobin values trend downward over time indicating progressive, age associated anemia. (C) Immature cells (blasts) within the bone marrow, measured by side scatter vs. CD45, are increased over WT controls at all time points and further increase with aging of the NHD13 mice. (D) NHD13 mice develop physical exam findings consistent with MDS including pallor of the nasal region and paws. Peripheral blood is hypochromic late in the disease course.
To measure progressive genomic instability that may occur in NHD13 mice as they age, we first evaluated γ-H2AX activity in PB mononuclear cells (PBMCs) to detect activation of the DNA damage response that may occur due to increased genomic instability. Significantly, at 6 months of age γ-H2AX activity is increased by almost 5 fold in PBMCs of NHD13 mice compared to WT controls (Figure 2A, P < .01). Furthermore, both NHD13 and WT mice, display a progressive age-associated increase of γ-H2AX in PBMCs (Figure 2A). Second, we determined whether any such increased genomic instability observed in cells from NHD13 mice may be due to failure of DNA repair in HSPCs. For this, the lineage-negative HSPC BM population was isolated from WT and NHD13 mice and a Neutral Comet assay was performed to measure the extent and rate of DNA double-strand break repair following irradiation of these cells with 1 Gy (Figure 2B). The repair of double-strand breaks was quantified by measuring the Comet Olive Tail Moment (OTM) for 50 randomly chosen cells (Comets) in each sample. Significantly, at 1 hour following irradiation, HSPCs from NHD13 mice display a significant increase in the average OTM compared to cells from WT mice, indicating that NHD13 HSPCs are more susceptible to DNA damage (Figure 2C, 131 vs. 83, P < .01). Furthermore, 24 hours following irradiation, the lineage-negative HSPCs from NHD13 mice display an increased number of Comet cells with an increased OTM compared to cells from WT mice (Figure 2, B and C). Taken together these results suggest that the progressive genomic instability observed in the NHD13 model may be due to failure of DNA repair in HSPCs from transgenic mice and may explain why leukemogenic mutations are serial and accumulate with time to herald disease progression [6], [7].
Figure 2.
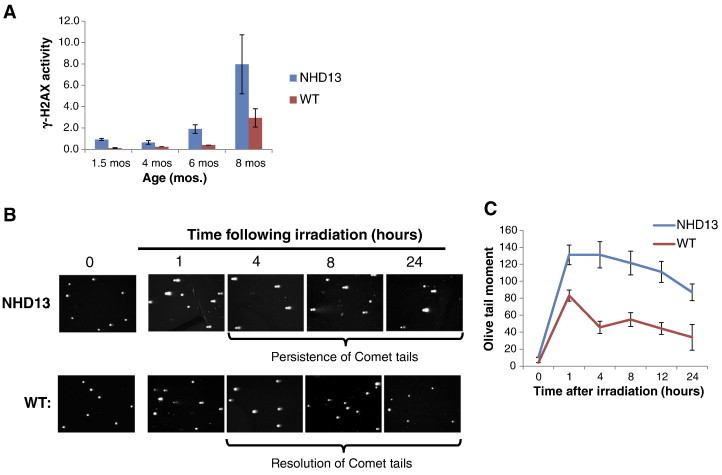
The NHD13 mouse displays progressive genomic instability. (A) γ-H2AX is increased in NHD13 mice compared to controls. (B) Representative Comet assays of Lineage-negative bone marrow cells following irradiation with 1 Gy. (C) Following treatment with 1 Gy ionizing radiation, the average Comet olive tail moment was calculated from 50 randomly chosen cells by Wimasis.
As an efficient, real-time means to measure genomic instability of the MDS mouse in vivo, a flow cytometry assay was used to serially measure spontaneous mutations of the X-linked phosphatidylinositol glycan anchor (PIG-A) gene that results in the absence of glycosylphosphatidylinositol (GPI)-linked anchor proteins on surface of peripheral blood cells (Figure 3) [11], [15]. We first measured loss of the GPI-linked protein CD24 on the surface of peripheral reticulocytes since these cells are abundant and easily collected from mice without the need for euthanasia. In addition, due to their short lifespan (48–72 hours), reticulocytes are an attractive surrogate for the real-time measurement of HSPC genomic instability, since mutations observed over serial measurements may represent mutations occurring in the HSPC population(s) that constantly replenishes these cells [12], [16]. When compared to WT, NHD13 mice have a significantly higher number of PB reticulocytes on which the GPI-linked protein CD24 is absent (Figure 4A; 1072 vs 20 per 2 × 105 reticulocytes at 8 months, P < .05). To account for any variability of reticulocytosis, a mutation frequency was calculated by dividing the number of CD24-negative reticulocytes by the total number of reticulocytes. Importantly, the frequency of CD24 negative cells significantly increases as NHD13 mice age (Figure 4B). Second, we also evaluated other PB cells that arise from the myeloid or lymphoid lineage to confirm that the loss of GPI-linked proteins was universal among PB circulating cells and therefore likely due to mutation of PIG-A in HSPCs. We tested for loss of Ly6C on monocytes (granulocyte-monocyte progenitor) and CD90 on T-cells (common lymphoid progenitor). Similar to reticulocytes, NHD13 mice have approximately twice the frequency of somatic mutations in T-cells and monocytes compared to WT control cells (Figure 4, C and D). While the differences in T-cells was significant, due to the low numbers of monocytes available for analysis in the PB sample collected from a cheek bleed and the low frequency of Ly6C-negative cells, the differences between the NHD13 and WT, although showing a trend, did not reach statistical significance. Collectively, these results strongly suggest that the loss of GPI-linked proteins on PB cells represents a mutation at the level of the HSPC.
Figure 3.
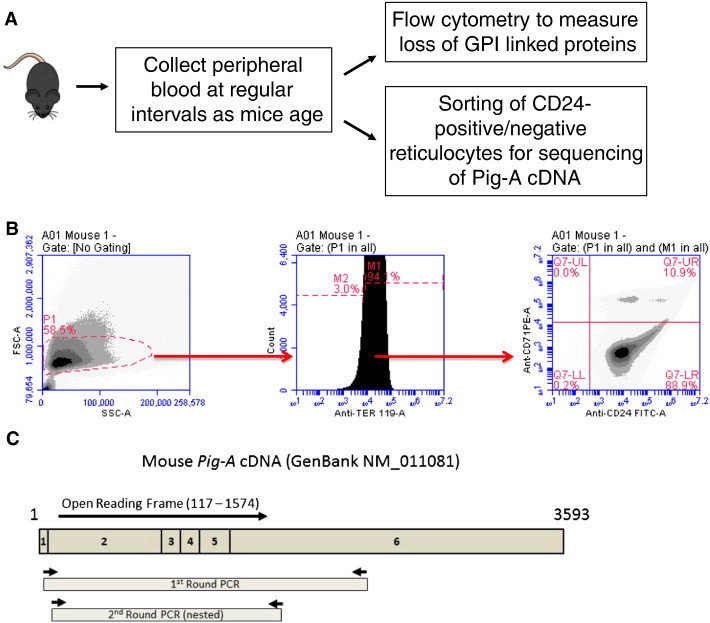
Evaluation of PIG-A loss in peripheral blood cells to measure genomic instability. (A) Experimental design for measuring PIG-A cDNA mutations and loss of GPI-linked proteins. (B) Typical flow cytometry gates and graphs to enumerate CD24-deficient reticulocytes detected in the CD24-CD71+Ter119+ quadrant of peripheral blood. (C) Schematic of the mouse PIG-A cDNA amplification and sequencing strategy.
Figure 4.
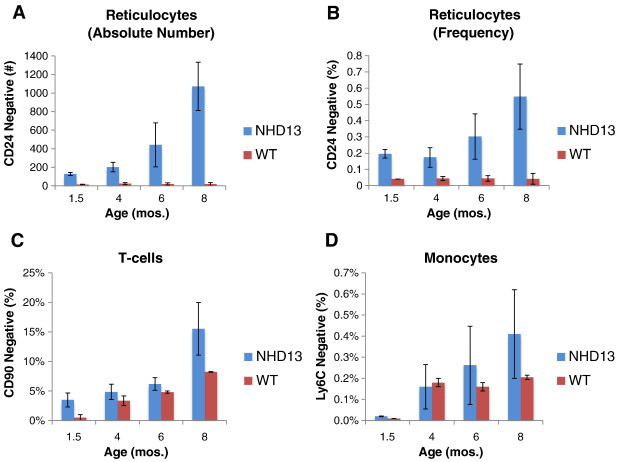
Frequency of GPI-linked protein loss progresses with age and correlates with disease phenotype. (A) Absolute number of CD24-negative reticulocytes is increased in PB of NHD13 mice compared to WT mice and increases with age. 1 × 106 cells were analyzed for each sample. (B) Frequency of CD24 loss (CD24 mutants/total reticulocytes x 100) increases with age. (C) In PB of NHD13 mice CD90-negative T-cells increase with age and are increased compared to WT controls. 2 × 104 events were analyzed. (D) Loss of the monocyte GPI-linked protein Ly-6C increases over disease course in NHD13 mice. 2 × 104 events were analyzed.
In order to confirm that any loss of CD24 was due to a mutation in the PIG-A gene, the PIG-A cDNA from CD24-negative reticulocytes sorted from the PB of three 8-month-old NHD13 mice was amplified and sequenced (Figure 3). Four PIG-A cDNA clones were analyzed from the CD24-negative reticulocytes of each mouse. As a control, PIG-A cDNA was also amplified from CD24-positive reticulocytes of each mouse. All four CD24-negative clones from each of the three mice contained mutations in the PIG-A cDNA sequence (Table 1). Most of the mutations were single base pair substitutions resulting in amino acid replacement. However, two mice had clones that had a nucleotide deletion resulting in a frameshift mutation (Table 1). Significantly, 3 of 4 separate clones from mouse 1 carried the same mutation at nucleotide 1191 and all 4 clones of mouse 3 had identical PIG-A mutations. These identical mutations may be due to expansion of PIG-A mutations in HSPCs while non-identical mutations likely arise in lineage committed cells. Importantly, PIG-A cDNA from all of the CD24-positive reticulocytes sequenced as WT PIG-A (data not shown). These data demonstrate that the PIG-A mutant phenotype detected in PB reticulocytes by loss of CD24 is caused by mutation of the PIG-A gene.
Table 1.
Mutations in the PIG-A open reading frame of CD24-negative reticulocytes.
| Mouse | Clone | cDNA positiona (exon) | Mutation b | Consequence |
|---|---|---|---|---|
| 1 | A | 307 (exon 2) | G to A | Gly-64 to Arg |
| 802 (exon 2) | T to C | Val-229 to Ala | ||
| 1191 (exon 5) | G to A | Gly-359 to Ser | ||
| 1385 (exon 6) | T to C | Silent | ||
| 1490 (exon 6) | T to C | Silent | ||
| 1509 (exon 6) | T to C | Trp-465 to Arg | ||
| 1555 (exon 6) | A to G | Lys-480 to Arg | ||
| B | 710 (exon 2) | G to T | Silent | |
| 914 (exon 3) | deletion of A | Frameshift | ||
| 1191 (exon 5) | G to A | Gly-359 to Ser | ||
| 1531 (exon 6) | A to G | Asp-427 to Gly | ||
| C | 155 (exon 2) | T to C | Silent | |
| 767 (exon 2) | A to G | Silent | ||
| 1021 (exon 4) | A to G | Gln-302 to Arg | ||
| 1104 (exon 5) | G to C | Val-330 to Leu | ||
| 1269 (exon 6) | T to C | Phe-385 to Leu | ||
| D | 756 (exon 2) | G to A | Asp-214 to Asn | |
| 1191 (exon 5) | G to A | Gly-359 to Ser | ||
| 2 | A | 227 (exon 2) | G to A | Met-37 to Ile |
| 235 (exon 2) | A to G | Asp-40 to Gly | ||
| 722 (exon 2) | A to G | Ile-202 to Met | ||
| 742 (exon 2) | C to T | Ala-209 to Val | ||
| 1270 (exon 5) | T to C | Phe-385 to Ser | ||
| B | 328 (exon 2) | C to T | Thr-71 to Ile | |
| 527 (exon2) | deletion of T | Frameshift | ||
| 992 (exon 4) | A to G | Silent | ||
| 1253 (exon 5) | C to t | Silent | ||
| C | 537 (exon 2) | C to T | Silent | |
| 542 (exon 2) | C to T | His-141 to Tyr | ||
| D | 1389 (exon 6) | A to G | Thr-425 to Ala | |
| 3 | A | 301 (exon 2) | A to G | Glu-62 to Gly |
| 571 (exon 2) | C to T | Thr-152 to Ile | ||
| 660 (exon 2) | A to G | Ile-182 to Val | ||
| 1335 (exon 6) | A to G | Silent | ||
| B | 301 (exon 2) | A to G | Glu-62 to Gly | |
| 571 (exon 2) | C to T | Thr-152 to Ile | ||
| 660 (exon 2) | A to G | Ile-182 to Val | ||
| 1335 (exon 6) | A to G | Silent | ||
| C | 301 (exon 2) | A to G | Glu-62 to Gly | |
| 571 (exon 2) | C to T | Thr-152 to Ile | ||
| 660 (exon 2) | A to G | Ile-182 to Val | ||
| 1335 (exon 6) | A to G | Silent | ||
| D | 301 (exon 2) | A to G | Glu-62 to Gly | |
| 571 (exon 2) | C to T | Thr-152 to Ile | ||
| 660 (exon 2) | A to G | Ile-182 to Val | ||
| 1335 (exon 6) | A to G | Silent |
The nucleotide position is based on the numbering of the mouse PIG-A cDNA sequence (Genbank NM_011081).
Sequence alterations in the nontranscribed cDNA strand compared to WT PIG-A.
Discussion
In MDS, genomic instability is a complex and multifactorial process with microsatellite instability and aberrant DNA repair [4], [17], [18]. Specifically, reduced expression of non-homologous end joining DNA repair genes in the NHD13 model may promote genomic instability and increase translocation frequency [18], [19]. Additionally, it is clear that point mutations, alterations in telomere regulation, and epigenetic changes can all contribute to genomic instability in both the NHD13 model and MDS patient samples [20], [21], [22]. These genotoxic influences appear to intensify with aging and can result in the loss of important tumor suppressor genes or activation of an oncogene to drive leukemia. Here we report that the loss of the PIG-A phenotype with age mirrors the age associated increases of both γ-H2AX and BM blasts in the NHD13 mice that precedes evolution of MDS to acute leukemia. We recognize that the loss of GPI-linked proteins in MDS is unlikely to be the catalyst for the progression to frank leukemia. Instead, we propose that the increased rate of GPI-linked protein loss in the PB cells, which correlates with disease-specific findings of the PB and BM, is indicative of the progressive genomic instability inherent to this disease. Thus, the strength of the PIG-A assay is its ability to allow the non-lethal, serial measurements of genomic instability that may be representative of any gene at the level of the MDS HSPC.
Future directions stemming from these findings may involve use of the PIG-A assay to: 1) discriminate mice that will develop acute leukemia in the NHD13 background; 2) evaluate genomic instability in other animal BM failure/hematologic malignancy models; and 3) perform serial measurements of genomic instability in patients with MDS that are at risk of developing acute leukemia.
Acknowledgments
This work was supported by a UF Health Cancer Center Team Science Award and NIH/NHLBI R01 HL054083.
Footnotes
Conflict of interests
none
Contribution
M.B. designed the experiments, performed research, analyzed results, and wrote the manuscript. R.L.B analyzed results and wrote the manuscript. X.C. designed the experiments, analyzed results, and wrote the manuscript. W.S.M. designed the experiments, analyzed results and wrote the manuscript.
References
- 1.Raza A, Galili N. The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat Rev Cancer. 2012;12(12):849–859. doi: 10.1038/nrc3321. [DOI] [PubMed] [Google Scholar]
- 2.Cioc AM, Wagner JE, MacMillan ML, DeFor T, Hirsch B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: morphologic and cytogenetic characteristics. Am J Clin Pathol. 2010;133(1):92–100. doi: 10.1309/AJCP7W9VMJENZOVG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gadner H, Haas OA. Experience in pediatric myelodysplastic syndromes. Hematol Oncol Clin North Am. 1992;6(3):655–672. [PubMed] [Google Scholar]
- 4.Zhou T, Hasty P, Walter CA, Bishop AJ, Scott LM, Rebel VI. Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp Hematol. 2013;41(8):665–674. doi: 10.1016/j.exphem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slape C, Lin YW, Hartung H, Zhang Z, Wolff L, Aplan PD. NUP98-HOX translocations lead to myelodysplastic syndrome in mice and men. J Natl Cancer Inst Monogr. 2008;39:64–68. doi: 10.1093/jncimonographs/lgn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slape C, Liu LY, Beachy S, Aplan PD. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras, and Cbl. Blood. 2008;112(5):2017–2019. doi: 10.1182/blood-2008-01-135186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung YJ, Robert C, Gough SM, Rassool FV, Aplan PD. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk Res. 2014;38(1):95–102. doi: 10.1016/j.leukres.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamous M, Al-Zoubi A, Khabaz MN, Khaledi R, Al Khateeb M, Al-Zoubi Z. Purification of mouse bone marrow-derived stem cells promotes ex vivo neuronal differentiation. Cell Transplant. 2010;19(2):193–202. doi: 10.3727/096368910X492599. [DOI] [PubMed] [Google Scholar]
- 9.SUNDBERG RD, HODGSON RE. Aspiration of bone marrow in laboratory animals. Blood. 1949;4(5):557–561. [PubMed] [Google Scholar]
- 10.Huang X, Darzynkiewicz Z. Cytometric assessment of histone H2AX phosphorylation: a reporter of DNA damage. Methods Mol Biol. 2006;314:73–80. doi: 10.1385/1-59259-973-7:073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtani S, Unno A, Ushiyama A, Kimoto T, Miura D, Kunugita N. The in vivo Pig-a gene mutation assay is useful for evaluating the genotoxicity of ionizing radiation in mice. Environ Mol Mutagen. 2012;53(8):579–588. doi: 10.1002/em.21724. [DOI] [PubMed] [Google Scholar]
- 12.Kimoto T, Suzuki K, Kobayashi XM, Dobrovolsky VN, Heflich RH, Miura D, Kasahara Y. Manifestation of Pig-a mutant bone marrow erythroids and peripheral blood erythrocytes in mice treated with N-ethyl-N-nitrosourea: direct sequencing of Pig-a cDNA from bone marrow cells negative for GPI-anchored protein expression. Mutat Res. 2011;723(1):36–42. doi: 10.1016/j.mrgentox.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 13.Risitano AM, Rotoli B. Paroxysmal nocturnal hemoglobinuria: pathophysiology, natural history and treatment options in the era of biological agents. Biologics. 2008;2(2):205–222. doi: 10.2147/btt.s1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–3709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peruzzi B, Araten DJ, Notaro R, Luzzatto L. The use of PIG-A as a sentinel gene for the study of the somatic mutation rate and of mutagenic agents in vivo. Mutat Res. 2010;705(1):3–10. doi: 10.1016/j.mrrev.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Noble NA, Xu QP, Ward JH. Reticulocytes. I. Isolation and in vitro maturation of synchronized populations. Blood. 1989;74(1):475–481. [PubMed] [Google Scholar]
- 17.Kuramoto K, Ban S, Oda K, Tanaka H, Kimura A, Suzuki G. Chromosomal instability and radiosensitivity in myelodysplastic syndrome cells. Leukemia. 2002;16(11):2253–2258. doi: 10.1038/sj.leu.2402703. [DOI] [PubMed] [Google Scholar]
- 18.Puthiyaveetil AG, Reilly CM, Pardee TS, Caudell DL. Non-homologous end joining mediated DNA repair is impaired in the NUP98-HOXD13 mouse model for myelodysplastic syndrome. Leuk Res. 2013;37(1):112–116. doi: 10.1016/j.leukres.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrne M, Wray J, Reinert B, Wu Y, Nickoloff J, Lee SH, Hromas R, Williamson E. Mechanisms of oncogenic chromosomal translocations. Ann N Y Acad Sci. 2014;1310(1):89–97. doi: 10.1111/nyas.12370. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty S, Sun CL, Francisco L, Sabado M, Li L, Chang KL, Forman S, Bhatia S, Bhatia R. Accelerated telomere shortening precedes development of therapy-related myelodysplasia or acute myelogenous leukemia after autologous transplantation for lymphoma. J Clin Oncol. 2009;27(5):791–798. doi: 10.1200/JCO.2008.17.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Issa JP. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am. 2010;24(2):317–330. doi: 10.1016/j.hoc.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto M, Shichishima T, Noji H, Ikeda K, Nakamura A, Akutsu K, Maruyama Y. High frequency of several PIG-A mutations in patients with aplastic anemia and myelodysplastic syndrome. Leukemia. 2006;20(4):627–634. doi: 10.1038/sj.leu.2404135. [DOI] [PubMed] [Google Scholar]