

Abstract
Genetic instability has emerged as an important hallmark of human neoplasia. Although most types of cancers exhibit genetic instability to some extent, in colorectal cancers genetic instability is a distinctive characteristic. Recent studies have shown that deregulation of genes involved in sister chromatid cohesion can result in chromosomal instability in colorectal cancers. Here, we show that the replisome factor minichromosome maintenance complex–binding protein (MCMBP), which is directly involved in the dynamics of the minichromosome maintenance complex and contributes to maintaining sister chromatid cohesion, is transcriptionally misregulated in different types of carcinomas. Cellular studies revealed that both MCMBP knockdown and overexpression in different breast and colorectal cell lines is associated with the emergence of a subpopulation of cells with abnormal nuclear morphology that likely arise as a consequence of aberrant cohesion events. Association analysis integrating gene expression data with clinical information revealed that enhanced MCMBP transcript levels correlate with an increased probability of relapse risk in colorectal cancers and different types of carcinomas. Moreover, a detailed study of a cohort of colorectal tumors showed that the MCMBP protein accumulates to high levels in cancer cells, whereas in normal proliferating tissue its abundance is low, indicating that MCMBP could be exploited as a novel diagnostic marker for this type of carcinoma.
Abbreviations: CIN, chromosomal instability; MCM complex, minichromosome maintenance complex; MCMBP, MCM-binding protein
Introduction
For a cell to generate two daughter cells, faithful DNA replication and partitioning of the sister chromatids to each of the newly formed cells is of utmost importance, preventing the transmission of potentially harmful mutations to the daughter cells, which otherwise might result in developmental defects or cancer. The set of molecular events that lead to the generation of genetic abnormalities is known as genetic instability. Genetic instability can be broadly divided into two major groups. The first is chromosomal instability (CIN), which refers to specific chromosomal alterations that might lead to acquisition or loss of entire chromosomes, as well as duplication, translocation, inversion, or deletion of particular chromosomal segments. The second is instability at the nucleotide level due to defective DNA replication and faulty DNA repair pathways [1]. Defects in either replication or checkpoint responses increase the susceptibility to cancer by triggering genome instability [2].
Genetic instability has emerged as a new hallmark of human cancers [3]. The development of new technologies such as comparative genomic hybridization has extensively demonstrated gain and loss of gene copy numbers in malignantly transformed cells in many different types of carcinomas [4]. Different tumors can harbor drastically different genome alterations. These include a large number of defects in the cellular machinery that regulates DNA replication and DNA repair. Together, these observations suggest that genome instability is rather widespread in cancer cells. Although some degree of genetic instability is seen in different types of cancers, it is a distinctive characteristic of colorectal cancers [5], [6]. Strikingly, only a few genes that might cause genetic instability have been identified and no general mechanism that can explain the origin of this phenotype has emerged.
The most distinctive characteristic of some types of cancers, including colorectal carcinomas, is CIN due to loss of sister chromatid cohesion [7], [8]. This clearly illustrates that defects in sister chromatid cohesion can progress to tumorigenesis and cancer.
A new member of the minichromosome maintenance (MCM) complex was identified in humans and denoted as MCM-binding protein (MCMBP) [9]. The MCM complex is considered the main replicative helicase of the cell and it is highly conserved throughout eukaryotes. The MCM protein family is characterized by the AAA ATPase motif. The replicative helicase consists of six related core subunits (MCM 2-7) that form a hexameric ring. Metazoans and plants have additional MCM subunits, as well as developmentally specific versions of the complex. In the late M and early G1 cell cycle phases, the MCM complex is recruited onto chromatin as part of the prereplication complex, where it functions as the molecular engine that unwinds the duplex DNA and also powers fork progression during DNA replication [10].
It was proposed that MCMBP can replace MCM2, which would lead to the formation of a different complex with MCM 3 to 7 [9]. Additionally, recent studies have postulated that MCMBP can disassemble the MCM2–7 complex and might function as an unloader of the MCM complex from chromatin, redistributing the MCM proteins at the end of the S phase [11], [12]. Complementing previous observations, our experiments demonstrated that depletion of the plant MCMBP orthologue, the ETG1 protein, resulted in late G2 cell cycle arrest, correlating with a partial loss of sister chromatid cohesion. Similarly, cohesion defects were observed upon knockdown of the MCMBP gene in human cell cultures, suggesting that this evolutionarily conserved protein has an equally important role in mammals [13], [14].
Here, we show that deregulation of MCMBP in different breast and colorectal cancer cell lines is associated with the emergence of a subpopulation of cells with abnormal nuclear morphology that emerges as a consequence of aberrant cohesion events. Association analysis integrating gene expression data with clinical information revealed that alteration of MCMBP transcript levels correlates with an increment in the probability of relapse risk in different types of human carcinomas. Finally, a detailed study of different colorectal tumor cohorts showed that the MCMBP protein is highly abundant in colorectal adenocarcinomas. These data suggest that deregulation of MCMBP can drive the oncogenic transformation of different cancer types.
Materials and Methods
Gene Ontology Analysis
To identify significantly overrepresented Gene Ontology (GO) categories among the MCMBP coexpression neighborhood, the 300 genes most coexpressed with MCMBP were retrieved from COXPRESdb [15]. We then, used the BiNGO plugin from Cytoscape [16] to determine the enriched GO categories, using a P value < .01 according to a multiple t test with correction for false positives.
MCMBP Antibody Generation
To produce recombinant MCMBP protein, the cDNA sequence encoding the human MCMBP was polymerase chain reaction (PCR) amplified and cloned between the BamHI and NotI sites into the pGEX-6P-2 vector by using the In-Fusion PCR cloning system. pGEX-6P-2–hMCMBP plasmid was transformed in Escherichia coli strain MC1061 containing the transcription regulatory plasmid pICA2, which allows tight Isopropyl-beta-D-thiogalactoside (IPTG)-inducible expression regulation [17]. Exponentially growing cultures (28°C) were induced with 1.0 mM isopropyl-β-d-thiogalactopyranoside and incubated overnight at 20°C. Cell pellets were resuspended in buffer A [phosphate-buffered saline (PBS, pH 7.4), DNase I (1 mg/100 ml; Roche Diagnostics, Indianapolis, IN), and complete EDTA-free Protease Inhibitor Cocktail Tablets (Roche Diagnostics)] and lysed by sonication. Insoluble proteins were removed by centrifugation. The supernatant was applied to a glutathione sepharose 4FF column (GE Healthcare, Little Chalfont, Buckinghamshire, UK) pre-equilibrated with buffer B (PBS, pH 7.4). Glutathione S-transferase (GST)–tagged hMCMBP was eluted from the column with buffer C [50 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 10 mM reduced glutathione]. Elution fractions containing GST-hMCMBP were pooled and dialyzed against buffer D [50 mM Tris-HCl (pH 7.0), 150 mM NaCl, 1 mM EDTA, and 1 mM DTT]. The GST-hMCMBP fusion protein was digested with the PreScission Protease (GE Healthcare) to clip off GST. The digested sample was run on a glutathione sepharose 4B column (GE Healthcare) pre-equilibrated with buffer B for removal of the GST tag and the PreScission Protease. Flow-through fractions containing hMCMBP were pooled. The purified recombinant hMCMBP protein was dialyzed to buffer B. The purity of the fractions was checked by means of sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Endotoxin removal was obtained by applying the purified hMCMBP sample to an ActiClean Etox column (Sterogene, Carlsbad, CA) pre-equilibrated with buffer B.
Anti-human MCMBP antisera were obtained by immunizing rabbits with full-length MCMBP protein. Two different rabbits were immunized with 0.5 μg of antigen in a volume of 0.5 ml. The immunizations were carried out subcutaneously 14, 28, and 56 days after the first preimmune bleeding. Blood was collected on days 38, 66, and 80. Recombinant hMCMBP was immobilized to NHS-activated sepharose (GE Healthcare). Rabbit anti-hMCMBP serum was run on the affinity resin pre-equilibrated with buffer B (PBS, pH 7.4). Bound rabbit IgGs were eluted from the column with buffer E [100 mM glycine (pH 3.0) and 50 mM NaCl] and immediately neutralized with 1 M Tris. Elution fractions containing the rabbit IgG were pooled. The purity of the fractions was checked by means of sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Generation of Stable MCF7 and MDA-MB-231 Lines with Constitutive Knockdown of MCMBP or with MCMBP Overexpression
The lentiviral construct V2HS-158067 (Catalog No. RHS4430-99167940) to knock down MCMBP (C10ORF119) was purchased from Open Biosystems (Lafayette, CO). Similarly, for the overexpression of MCMBP, a lentiviral construct containing the complete ORF of the MCMBP gene (C10ORF110) was obtained from Open Biosystems (MHS1011-58896) and cloned in the pDWPITetoMCMBPv5His vector. Following the manufacturer's instructions, the Qiagen midi-preps extraction kit was used to obtain 100 μl of V2HS-158067 construct at 1 μg/μl, for transfecting human embryonic kidney–293 (HEK-293) cells to produce viral particles.
Three different constructs were used to produce the final DNA mix for transfecting HEK-293 cells. Three microliters of pCMVD8-9-MCO23, 1.5 μl of pMG-MCO22, and 3 μl of V2HS-158067 (each at a concentration of 1 μg/μl) were mixed with 1 μl of 1 M sodium acetate, 3 μl of absolute ethanol, and 8.5 μl of bi-distillated water to obtain a final volume of 20 μl. The material was gently mixed by pipetting and placed at − 70°C for 30 minutes, after which it was centrifuged at maximum speed for 30 minutes at 4°C. The supernatant was removed and the pellet was washed with 250 μl of 70% ethanol. The mixture was again centrifuged at maximum speed for 5 minutes at 4°C, and the supernatant was discarded. The pellet was dried at room temperature and resuspended in 10 μl of bi-distillated water. HEK cells were grown in 25-cm2 flasks in 5 ml of complete medium (Dulbecco's modified Eagle's medium with 10% fetal calf serum) at 37°C in 5% CO2. Two days before transfection, HEK cells were trypsinized, split into six-well plates containing 4 ml of fresh culture medium, and incubated at 37°C in 5% CO2 for 2 days. At the time of transfection, the cells were still subconfluent.
A calcium phosphate precipitate suspension was prepared in two 5-ml tubes. In one of these tubes, 250 μl of Hepes-buffered saline solution was dispensed (16.4 g of NaCl, 11.9 g of Hepes acid, and 0.21 g of Na2HPO4 per liter, pH 7.05). In the other tube, a solution containing 10 μl of the lentiviral mix, 190 μl of TE buffer, and 50 μl of 2.5 M CaCl2 was prepared; it was then added dropwise to the tube containing the Hepes buffer. Afterward, the solution was vortexed gently for 30 seconds and then left for 10 minutes at 37°C to allow the calcium phosphate to precipitate. Then, 4 μl of 25 mM chloroquine was added to the mix. Subconfluent HEK cells were transfected by adding 250 μl of the DNA mix to the cultured cells. HEK cells were grown for 8 hours at 37°C in 5% CO2, and the culture medium was refreshed. Finally, the HEK cells were grown for 2 days to produce viral particles. They were then trypsinized, collected in 15-ml tubes, and filtered through a syringe and a 0.45-μm filter. The filtered suspension of viral particles was divided into aliquots of 1 ml in 1.5-ml centrifuge tubes and stored at − 80°C.
MCF7 (or MDA-MB-231) cells were grown in 25-cm2 flasks in 5 ml of medium (10% bovine calf serum and 10 mM Hepes) at 37°C in 5% CO2. Before infecting the MCF7 cells with the viral particles, they were trypsinized and counted. Aliquots of 4 ml containing 70,000 to 80,000 cells per milliliter were placed in 15-ml tubes. MCF7 cells were precipitated by centrifugation at 2000 rpm for 5 minutes. The supernatant was removed and cells were gently resuspended in 600 μl of viral particle suspension. The mixture was divided in triplicates of 200 μl in 96-well plates and centrifuged at 2000 rpm for 90 minutes at 32°C. The plates were then placed at 37°C in 5% CO2 for 2 days to let the viruses infect the cells. The triplicates of cells were then trypsinized and combined in cells of a 12-well plate. After 2 days of incubation, the cells were transferred in the same way to six-well plates and kept at 37°C in 5% CO2 for a quarantine period of 4 weeks. The MCF7 medium was replaced twice a week.
Flow Cytometry Experiments
About 250,000 MCF7 cells were seeded in 5 ml of MCF7 medium in a six-well plate. Then, they were grown in 25-cm2 flasks in 5 ml of medium (10% bovine calf serum and 10 mM Hepes) at 37°C in 5% CO2. Each cell line (MCF7 control, empty vector, and MCMBP knockdown) was seeded in triplicate. After 48 hours, cells were trypsinized, pelleted (110g for 5 minutes), and resuspended in 800 μl of staining solution (http://www.partec.com). The cells were filtered through a 30-μm mesh and the nuclei were analyzed using the CyFlow cytometer and FloMax software (http://www.partec.com).
Immunofluorescence Analysis of Breast Cancer Lines
MCF7 parental, empty vector control, and MCMBP knocked down cells were grown in 25-cm2 flasks in 5 ml of MCF7 medium at 37°C in 5% CO2. Two days before the experiment, cells were trypsinized and resuspended in 4.5 ml of fresh medium. Small coverslips were placed inside 24-well plates and 1 ml of the cell suspension was added to every coverslip. Cells were grown at 37°C in 5% CO2 for 2 days. Afterward, cells were washed with 1 ml of PBS three times and fixed in paraformaldehyde. Subsequently, 200 μl of 0.02% gelatin in PBS were added to the coverslips and cells were incubated for 1 hour at room temperature. A solution of 200 μl of gelatin in PBS buffer containing 1 μl of primary antibody anti-MCMBP, anti-MCM4 (ab4459), anti-MCM6 (ab4458), anti-MCF7 (ab52489) (Abcam, Cambridge, UK), or anti–E-cadherin was made. Gelatin was removed from the coverslips and 200 μl of the primary antibody solution was added to the cells; they were incubated for 1 hour at room temperature. Excess primary antibody was removed by washing the cells three times with 1 ml of PBS buffer. A solution of 200 μl of gelatin in PBS buffer containing 0.5 μl of secondary antibody, 2 mg/ml Alexa Fluor 488 goat anti-rabbit antibody, or 2 mg/ml Alexa Fluor 594 goat anti-mouse antibody (Invitrogen, Carlsbad, CA) was made, and 200 μl of the secondary antibody solution was added to the cells; incubation was continued for 1 hour at room temperature in the dark. Excess secondary antibody was removed by washing the cells three times with 1 ml of PBS buffer. Coverslips were mounted on microscopic glass slides and counterstained with VECTASHIELD (Vector Laboratories, Burlingame, CA) containing 4′,6-diamidino-2-phenylindole (DAPI). Cells were imaged using the green channel of a BX61 Olympus epifluorescence microscope equipped with a 1006/1.30 UPlan FLN objective coupled to a U-C MAD 3 imaging system with the Cell-M imaging software (Olympus, Tokyo, Japan). A minimum of 200 fields per construct was scored, and multinucleated and/or micronucleated cells were counted.
Immunofluorescence Analysis of Colorectal Cancer Lines
DLD1 parental cells were grown in 25-cm2 flasks in 5 ml of RPMI 1640 medium supplemented with 10% fetal calf serum at 37°C in 5% CO2. The following small interfering RNA (siRNA) sequences (DharmaFECT, Thermo Fisher Scientific, Waltham, MA) were used for the specific transfections: human MCMBP (C10ORF119) (SMARTpool; J-014474-09, J-014474-10, J-014474-11, and J-014474-12) and control (SMARTpool non-targeting pool). The final concentration of each siRNA was 30 nM. Immunofluorescence was carried out as described above for the breast cell lines.
Quantitative PCR Analysis of MCMBP Expression and Cell Cycle Analysis
Cells were collected with a rubber policeman 48 hours after transfection. RNA was extracted with an RNeasy Animal Mini Kit (Qiagen, Venlo, NL) and cDNA was prepared with the cDNA Synthesis System according to the manufacturer’s instructions (Roche Diagnostics). For quantitative PCR (qPCR), a LightCycler 480 SYBR Green I Master (Roche Diagnostics) was used with 100 nM primers and 0.1 mg of reverse transcription reaction product. Reactions were run and analyzed on the LightCycler 480 Real-Time PCR System according to manufacturer’s instructions (Roche Diagnostics). All quantifications were normalized to the expression levels of TATA box binding protein (TBP) and ubiquitin C (UBC). Quantitative reactions were done in triplicate and averaged. The primers were 5′ACTCTCCACGAAATACCACTTTG3′ and 5′GTAGGATGTTGAGGGACTGACTCG3′ for MCMBP; 5′TGAGCCAGTGCCAGAGCCAGA3′ and 5′GCTCCATCTTCTGCATCCACATC3′ for cyclin B1; 5′CCAAAGTTCCAGTTCAACCCACC3′ and 5′CAATCCACTAGGATGGCACGCA3′ for cyclin B2; 5′CCTTGCCAGAGCTTTTGGAATACC3′ and 5′CACACTTCATTATTGGGAGTGCCC3′ for Cyclin-dependent kinase 1 (CDK1); 5′GTGGAGTGTTGGCTGTATCTTTGC3′ and 5′GCTCCCGACTCCTCCATCTCAG3′ for Cyclin-dependent kinase 4 (CDK4); 5′TGCCGCTCTCCACCATCCG3′ and 5′GGTTTCAGTGGGCACTCCAGG3′ for Cycline-dependent kinase 6 (CDK6); 5′CGGCTGTTTAACTTCGCTTC3′ and 5′CACACGCCAAGAAACAGTGA3′ for TBP; and 5′ATTTGGGTCGCGGTTCTTG3′ and 5′TGCCTTGACATTCTCGATGGT3′ for UBC.
HEK-293T Cell Culture and Transfection
For the overexpression of MCMBP, a lentiviral construct containing the complete ORF of the MCMBP gene (C10ORF110) was purchased from Open Biosystems (MHS1011-58896) and cloned in the pDWPITetoMCMBPv5His vector. For the transfection experiments, the previously prepared lentiviral vector was used at a final concentration of 1 μg/μl. In an Eppendorf tube, 50 μl of CaCl2/Hepes, 197 μl of Tris-EDTA (TE), and 3 μl of the lentiviral construct containing MCMBP were mixed. This mix was added dropwise to 250 μl of Buffered Saline (BS)/Hepes in another Eppendorf tube. This final mixture was gently vortexed for 1 minute and incubated at 37°C for 10 minutes. After incubation, the mix was added dropwise to 1.0 × 106 HEK cells in a T25 flask. After 6 hours, the medium was refreshed.
Chromosome Spreads and DAPI Staining
Forty-eight hours after transfection, subconfluent MDA-MB-231, MCF10A, and HEK cells were treated with KaryoMAX Colcemid (Invitrogen) to enrich for mitotic chromosomes. The complete medium was replaced by 2 ml of medium at a final concentration of KaryoMAX of 0.6 mg/ml. Cells were incubated at 37°C in 5% CO2 for 5 hours, harvested, trypsinized, pelleted (110g for 5 minutes), and resuspended in 1 ml of a hypotonic solution of 60 mM KCl and left at room temperature for 30 minutes. After incubation, the different cell lines were pelleted twice (110g for 5 minutes) and resuspended in freshly made methanol/glacial acetic acid (3:1) added dropwise. Two or three drops of suspended cells were applied to pre-cleaned smear glass slides (Menzel-Glazer, Braunschweig, DE) and chromosomes were counterstained with VECTASHIELD (Vector Laboratories) containing DAPI. Mitotic spreads of each cell population were imaged with the DAPI channel of a BX61 Olympus epifluorescence microscope equipped with a 1006/1.30 UPlan FLN objective coupled to a U-C MAD 3 imaging system with the Cell-M imaging software (Olympus). A minimum of 200 metaphases was evaluated per construct and metaphases in which > 70% of the chromosomes had separated sister chromatids were scored as positives.
Immunohistochemistry for Tumor Samples
Tumor sections were deparaffinized and rehydrated as follows. Sections were washed twice for 3 minutes in xylene and twice for 1 minute in, successively, isopropanol, 100% ethanol, and 70% ethanol. They were finally rinsed with tap water and the antigens were retrieved for 2 hours in 1 × citrate buffer (Dako, Santa Clara, CA) using a Dako retriever. The samples were then cooled down and rinsed with tap water. Afterward, the tumor sections were immersed in a peroxidase-blocking solution (1:9 solution of H2O2/methanol) for 10 minutes. After incubation, the sections were washed three times for 5 minutes with PBS. They were dried gently and the tumor sample was circled using a Dako pen (Dako). Tumor samples were blocked for 45 minutes at room temperature with a blocking buffer (Dako) enriched with 5% goat serum. Afterward, the blocking buffer was removed by aspiration, the primary antibody (1/100 dilution of MCMBP antibody in blocking buffer) was added to the samples, and incubation was continued overnight at 4°C. Afterward, tumor sections were washed three times for 5 minutes with PBS. As a secondary antibody, EnVision anti-rabbit drop–HRP conjugated antibody was used; the section was covered with a few drops and incubated for 45 minutes at room temperature. Slides were developed by incubation with a liquid DAB solution for 30 seconds to 1 minute until a positive reaction was observed. The reaction was stopped by immersing the slides in bi-distillated water. The slides were counterstained with hematoxylin (Mayer-Fluka) for 40 seconds. Finally, the tumor samples were dehydrated by washing twice for 1 minute in, successively, 70% ethanol, 100% ethanol, and isopropanol and twice for 3 minutes in xylene. Finally, slides were mounted with Depex. For each tumor section, the normal tissue fraction and the tumoral fraction were identified. At least 200 nuclei were selected in each fraction, and nuclei that were stained were scored as positive for MCMBP expression.
Cox Survival Analyses
The microarray data analyzed in this study were downloaded from the NCBI-Gene-Expression-Omnibus (GEO) website (www.ncbi.nlm.nih.gov/geo). The Cel files of studies performed on the HG133A or HG133plus2 Affymetrix array platforms with samples from colorectal cancer (GSE17537), head and neck carcinoma (GSE10300), leukemia (GSE23501), or lung carcinoma (GSE3141) were extracted, background-subtracted, normalized, summarized (median polish option) using frozen robust multiarray analysis (fRMA) [18], and converted back to signal intensity values. Cox survival analysis was performed in R with the Survival package using the raw expression intensity converted in binary categories whether or not the expression value is above a threshold value. This threshold was defined as the expression value leading to the highest Chi-square value in a training Cox survival analysis. To this end, the range of expression values for the MCMBP probe set (217905 at) was divided in 50 interval values before a Cox analysis was run for each interval value with the data converted to 0 or 1, according to whether the MCMBP expression value is below or above the considered interval value.
Immunofluorescence Analysis of Histone H2A.X
MCF7 parental, empty vector control, and MCMBP knocked down cells were grown in 25-cm2 flasks in 5 ml of MCF7 medium at 37°C in 5% CO2. Two days before the experiment, cells were trypsinized and resuspended in 4.5 ml of fresh medium. Small coverslips were placed inside 24-well plates and 1 ml of the cell suspension was added to each coverslip. Cells were grown at 37°C in 5% CO2 for 2 days. Afterward, the cells were washed three times with 1 ml of PBS and fixed with 500 μl of methanol. The cells were then frozen at − 20°C for 10 minutes. Methanol was removed from the coverslips and the cells were washed three times with 1 ml of PBS buffer. Subsequently, 200 μl of 0.02% gelatin in PBS solution were added to the coverslips and the cells were incubated for 1 hour at room temperature. A solution of 200 μl of gelatin in PBS buffer and 1 μl of primary antibody (0.05 μg/ml anti–phospho-histone H2A.X–ser-139 antibody, Catalog No. 05-636; Millipore, Billerica, MA) was made. Gelatin was removed from the coverslips and 200 μl of primary antibody solution was added to the cells and incubation was continued for 1 hour at room temperature. Excess primary antibody was removed by washing the cells three times with 1 ml of PBS buffer. A solution of 200 μl of gelatin in PBS buffer and 0.5 μl of secondary antibody (2 mg/ml Alexa Fluor 488 goat anti-mouse antibody, Invitrogen) was made and 200 μl of this secondary antibody solution was added to the cells. Incubation was continued for 1 hour at room temperature in the dark. Excess secondary antibody was removed by washing the cells three times with 1 ml of PBS buffer. Coverslips were mounted on microscope glass slides and counterstained with VECTASHIELD (Vector Laboratories) containing DAPI. Cells were imaged using the green channel of a BX61 Olympus epifluorescence microscope equipped with a 1006/1.30 UPlan FLN objective coupled to a U-C MAD 3 imaging system with the Cell-M imaging software (Olympus). A minimum of 100 nuclei per construct were selected and the histone H2A.X foci were counted in each nucleus.
qPCR Analysis of Tumor Samples
Tumor RNA was extracted with an RNeasy Animal Mini Kit (Qiagen) and cDNA was prepared with the cDNA Synthesis System according to the manufacturer’s instructions (Roche Diagnostics). For qPCR, a LightCycler 480 SYBR Green I Master (Roche Diagnostics) was used with 100 nM primers and 0.1 mg of reverse transcription reaction product. Reactions were run and analyzed on the LightCycler 480 Real-Time PCR System according to the manufacturer’s instructions (Roche Diagnostics). All quantifications were normalized to the expression levels of TBP and UBC. Quantitative reactions were done in triplicate and averaged. The primers were 5′ACTCTCCACGAAATACCACTTTG3′ and 5′GTAGGATGTTGAGGGACTGACTCG3′ for MCMBP; 5′CGGCTGTTTAACTTCGCTTC3′ and 5′CACACGCCAAGAAACAGTGA3′ for TBP; and 5′ATTTGGGTCGCGGTTCTTG3′ and 5′TGCCTTGACATTCTCGATGGT3′ for UBC.
Results
Coexpression and GO Analyses Confirm that MCMBP Is Involved in Cohesion Processes
Our previous studies demonstrated that ETG1-deficient plants (plant orthologue of MCMBP) undergo endogenous DNA stress and display a transient cell cycle arrest. A microarray transcriptomic analysis of wild-type and ETG1-deficient Arabidopsis plants revealed in the knockout plants a significant enrichment of genes involved in the cell cycle, mitotic cell cycle, microtubule-based movement, and sister chromatid cohesion [14]. To explore whether these functional processes were also associated with MCMBP, the human orthologue of ETG1, we tested whether the coexpression neighborhood of MCMBP was functionally enriched in GO categories related to M-phase progression (see Materials and Methods section). Indeed, the MCMBP coexpression neighborhood was found to be significantly enriched in processes related to M-phase progression, chromosome segregation, and sister chromatid cohesion (P value < .01 according to a multiple t test, with false discovery rate corrected; Figure 1). Similarly, among the top 50 MCMBP coexpressed genes, it was possible to identify coexpression neighbors that were directly involved in cohesion processes (Table 1).
Figure 1.
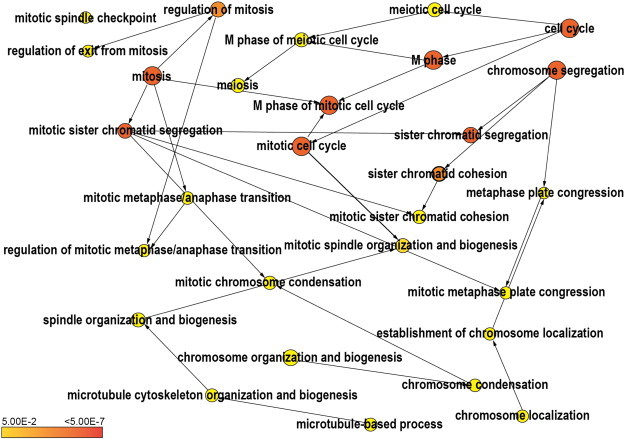
BiNGO (GO enrichment) analysis of the top 300 genes coexpressed with MCMBP. The yellow-to-orange color of the circles corresponds to the level of significance of the overrepresented GO category (P value < .01 according to a multiple t test with correction of false discovery rate). The size of the circle is proportional to the number of genes in the category. A clear overrepresentation of GO terms associated with M-phase processes and sister chromatid cohesion can be observed.
Table 1.
List of Genes Significantly Associated with the GO Term “Sister Chromatid Cohesion” (P value < .01) that Are Present in the MCMBP Coexpression Neighborhood (Top 50).
| Official Symbol | Functional Annotation |
|---|---|
| CTCF | CCCTC-binding factor (zinc finger protein) |
| MAD2 | MAD2 mitotic arrest deficient-like 1 (yeast) |
| SEH1L | SEH1-like (Saccharomyces cerevisiae) |
| SRPK1 | SFRS protein kinase 1 |
| BUB3 | Budding uninhibited by benzimidazoles 3 homolog (yeast) |
| CDC23 | Cell division cycle 23 homolog (S. cerevisiae) |
| NUP37 | Nucleoporin 37 kDa |
| STAG1 | Stromal antigen 1 |
| SMC1A | Structural maintenance of chromosomes 1A |
| SMC2 | Structural maintenance of chromosomes 2 |
| SMC3 | Structural maintenance of chromosomes 3 |
| SMC4 | Structural maintenance of chromosomes 4 |
MCMBP Depletion Causes Aberrant Cohesion Events in Different Cell Types
A role for MCMBP in sister chromatid cohesion was identified previously in HEK-293T cells. Depletion of MCMBP strongly affected sister chromatid cohesion, resulting in a higher proportion of nuclei with completely separated sister chromatids in comparison with controls [14]. To determine whether the effect on sister chromatid cohesion seen upon MCMBP knockdown in HEK cells is also present in other cell lines, MCMBP transcripts were knocked down in different breast cell lines. For the non-transformed epithelial cell line MCF10A, transient knockdown was achieved by using pooled short interference RNAs, whereas for the MDA-MB-231 breast cancer cell line, cellular cultures with a stable MCMBP knockdown were produced using lentiviral transduction (see Material and Methods section). MCMBP knockdown affected sister chromatid cohesion in both cell lines, suggesting that the effect is a specific phenotype caused by MCMBP depletion (Figure 2).
Figure 2.
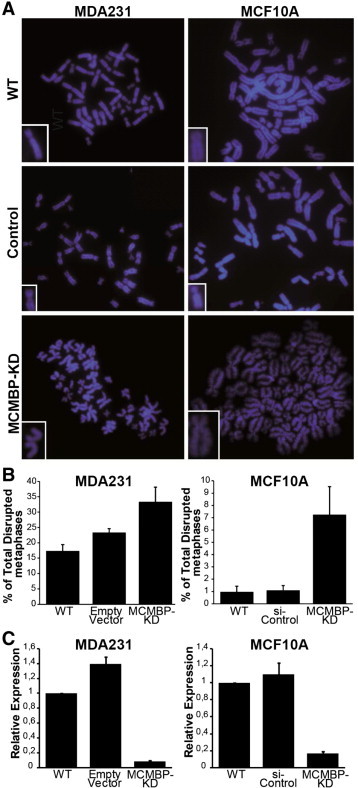
Knockdown of the MCMBP transcript has a direct effect on sister chromatid cohesion in different breast cancer cell lines. (A) Examples of metaphases wherein the sister chromatids are separated when the MCMBP transcript is deregulated, either transiently (MCF10A cell line) or in a stable manner (MDA-MB-231 cell line). Insets show higher magnification images of single sister chromatid pairs. For the MDA-MB-231 cells, control refers to the empty vector used for the lentiviral transfection, while for MCF10A cells, control refers to a pool of siRNAs designed not to target any gene in the human genome. (B) Quantification of disrupted metaphases for the analyzed cell lines. Disrupted metaphases refer to metaphases in which at least 70% of the chromosomes have totally separated sister chromatids. (C) qPCR analysis of the cultures showing depletion of the MCMBP transcript in the breast cancer cell lines.
MCMBP Misexpression Triggers Formation of Polylobed Nuclei and Micronuclei in Breast and Colorectal Cell Lines
We showed that high MCMBP gene expression levels are associated with a diminished probability of survival of breast carcinoma patients [19]. To identify the role of MCMBP in carcinogenesis, the effects of knocking down or overexpressing MCMBP were studied in the MCF7 breast cancer cell line. To that end, cell lines either constitutively overexpressing or silencing the MCMBP gene were generated. For studies at the protein level, a high-quality antibody against the MCMBP protein was generated (Supporting Information Figure S1).
MCMBP knocked down cultures were studied at the cellular level by flow cytometry, revealing a shoulder on the ploidy profile formed by a subpopulation of cells with a DNA content higher than 4C (Figure 3A). This cell population was correlated with the appearance of multinucleated (polylobed) and/or giant cells compared to the untransduced or control transduced cells. The number of multinucleated cells was almost five-fold higher in the knocked down cultures than in controls (Figure 3C). The aberrant nuclei stained negative for the MCMBP protein (Figure 3B). In MCF7 control cells, MCMBP was weakly present in a small proportion of multinucleated cells (Figure 3B). These cells might have arisen specifically because of MCMBP deregulation, given the presence of proteins belonging to the MCM complex in control multinucleated MCF7 cells (Supporting Information Figure S2). This phenotype was accompanied by the transcriptional up-regulation of specific cell cycle markers, such as cyclin B1, cyclin B2, and CDK1 (Figure 3D). Like the knockdown lines, MCMBP-overexpressing MCF7 cultures displayed multinucleated cells but accompanied by micronuclei (Figure 3E). The number of multinucleated cells was four-fold higher relative to controls (Figure 3F). Here, cell cycle marker genes were transcriptionally upregulated (Figure 3G).
Figure 3.
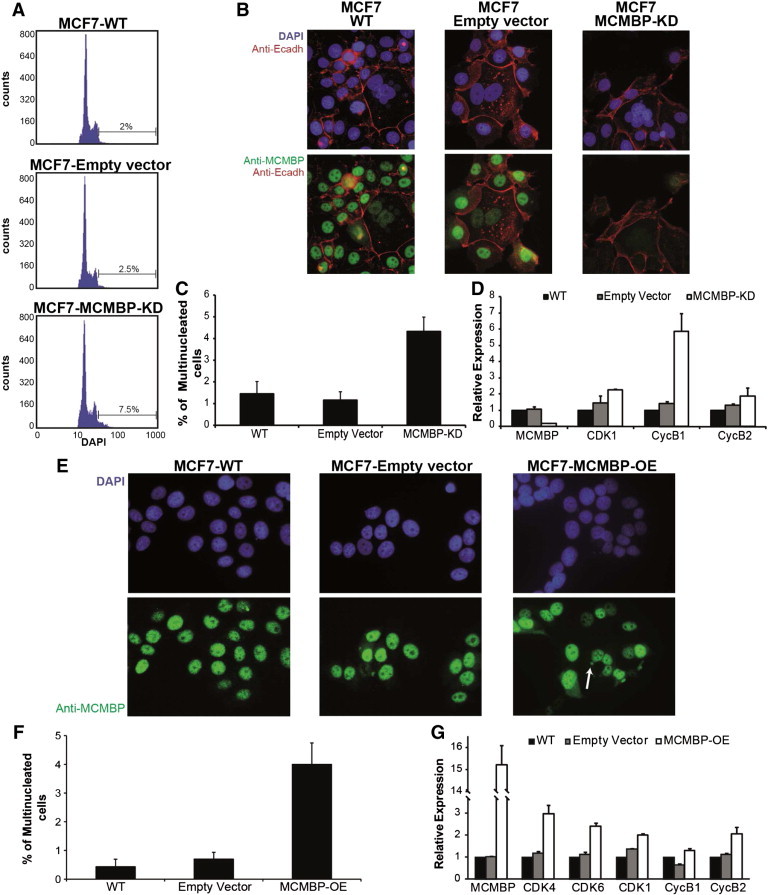
Cellular analysis of MCF7 cells in which MCMBP is knocked down or overexpressed. (A) Flow cytometry profiles of cells with MCMBP knockdown in comparison with controls. Upon knockdown, a subpopulation of cells with DNA content higher than 4C appeared in the culture. (B) A correlation exists between multinucleation and reduction or absence of MCMBP protein expression in those types of cells, both in control and knocked down cultures. (C) Quantification of the multinucleated cells present in the different cultures. (D) Up-regulation of specific cell cycle markers on MCMBP knockdown. (E) On MCMBP overexpression, multinucleated cells and micronuclei (arrows) can be observed. (F) Quantification of the multinucleated cells present in the different cultures. (G) Up-regulation of specific cell cycle markers on MCMBP overexpression.
It has been proven that the appearance of multinucleated cells and/or cells with micronuclei is directly linked with different forms of CIN, including cohesion defects [20]. Given the observations that MCMBP links DNA replication with sister chromatid cohesion [13], [14], the aberrant nuclei observed in the MCMBP misexpression cultures might have originated due to defects in sister chromatid cohesion. As the overexpression of MCMBP also has a direct effect on the generation of multinucleated cells and/or cells harboring micronuclear structures, we tested the effects of transient MCMBP overexpression in HEK-293T cells on chromosomal structure. Again, effects on sister chromatid cohesion were observed. When the cells overexpressing MCMBP were examined in detail at metaphase, close to 70% of the metaphases were found to be disrupted, illustrating that in human cells not only depletion but also overexpression of MCMBP has a direct effect on sister chromatid cohesion (Figure 4).
Figure 4.
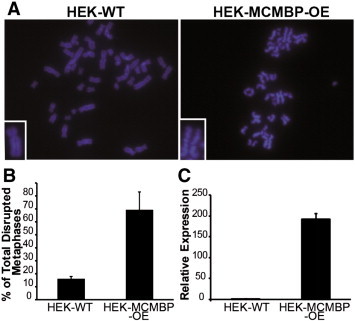
Overexpression of MCMBP has a direct effect on sister chromatid cohesion in HEK-293T cells. (A) Examples of metaphases in which the sister chromatids become separated when the MCMBP transcript is upregulated. Insets show higher magnification images of single sister chromatid pairs. (B) Quantitation of the phenotype: almost 70% of the metaphases are disrupted when MCMBP is overexpressed. (C) qPCR showing that the MCMBP transcript is upregulated almost 200-fold in the MCMBP-overexpressing cell line.
As mentioned before, colorectal carcinomas have high rates of genetic instability. Multinucleation and micronucleation represent common cellular aneuploidization processes derived from genetic instability events [21], [22]. Due to this, we decided to evaluate whether the phenotypes related to genetic instability seen in breast cancer lines are reproducible in the DLD1 colorectal cancer line.
The MCMBP transcript was transiently depleted by siRNA in the DLD1 cell line. As observed in the MCF7 cell lines, multinucleated and/or giant cells staining negative for the MCMBP protein were also observed in the colorectal cell line (Figure 5A). The number of multinucleated cells was almost six-fold higher in the DLD1 knocked down cultures than in controls (Figure 5B).
Figure 5.
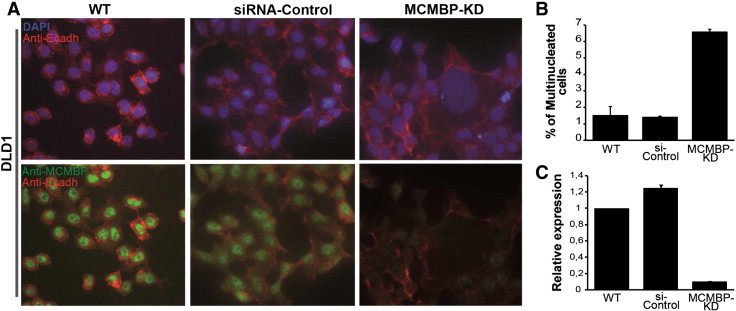
Cellular analysis of MCMBP-depleted DLD1 cell line. (A) There is a correlation between multinucleation and reduction or absence of MCMBP protein expression in MCMBP knocked down cultures. (B) Quantification of the multinucleated cells in the different analyzed cultures of the DLD1 cell line. (C) qPCR analysis showing depletion of the MCMBP transcript in the studied DLD1 cell lines.
Knockdown of MCMBP Triggers the Response of DNA Stress Genes
Aneuploidization events are accompanied by the induction of the DNA stress response machinery. To test whether MCMBP depletion could trigger specific DNA stress response events, immunolocalization of the phosphorylated H2A.X histone using a specific antibody was carried out in MCF7 cultures. Phosphorylation of histone H2A.X was rarely observed in parental MCF7 cells or in cells transformed with the empty vector. In contrast, histone H2A.X phosphorylation was enhanced in the MCMBP knocked down cells (Figure 6). These results suggest that the absence of a functional MCMBP protein triggers the DNA stress response in MCF7 cells. These data are in agreement with recent data illustrating that depletion of MCMBP triggers the replication stress response in HeLa cells [23].
Figure 6.
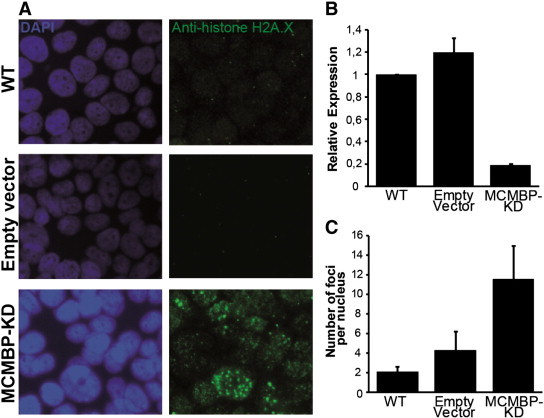
Immunostainings of phosphorylated histone H2A.X in MCF7 cells. (A) On MCMBP knockdown, the DNA stress response checkpoint is activated, as illustrated by the enhanced phosphorylation of the histone H2A.X. (B) qPCR analysis of the different MCF7 lines used in the experiment; 80% of the MCMBP transcript is reduced in the stable knocked down MCF7 line. (C) Quantitation of the histone H2A.X foci observed in the control nuclei in comparison with the MCMBP knocked down cells.
MCMBP Misexpression Correlates with Changes in Relapse Risk Probability of Different Carcinomas
Previously, we had shown the correlation that exists in breast carcinomas between MCMBP misexpression and relapse-free survival probability [19]. Concomitantly, experiments performed in breast tumor tissue revealed that MCMBP overexpression is directly linked with the estrogen receptor (ER) negative status, a hallmark of poor prognosis and difficult treatment of breast cancers (Supporting Information Figure S3), confirming the value of MCMBP as a prognostic marker.
To address the use of MCMBP transcript levels as a prognostic marker for cancer progression in different carcinomas, we extended our previously reported Cox analyses [19] to different types of cancers to detect whether in these neoplasias a change in MCMBP gene expression is associated with changes in relapse risk probability. To perform these analyses, our database of transcriptional profiles linked to well-annotated clinical information, including relapse events and relapse time, was extended to include different microarray expression experiments representing different human carcinoma types (see Materials and Methods section). The significance of a particular association between MCMBP gene expression and a relapse event was assessed by Cox regression analysis. Figure 7 shows that for different carcinomas both increased and reduced MCMBP gene expression are directly associated with an increment in relapse risk probability.
Figure 7.
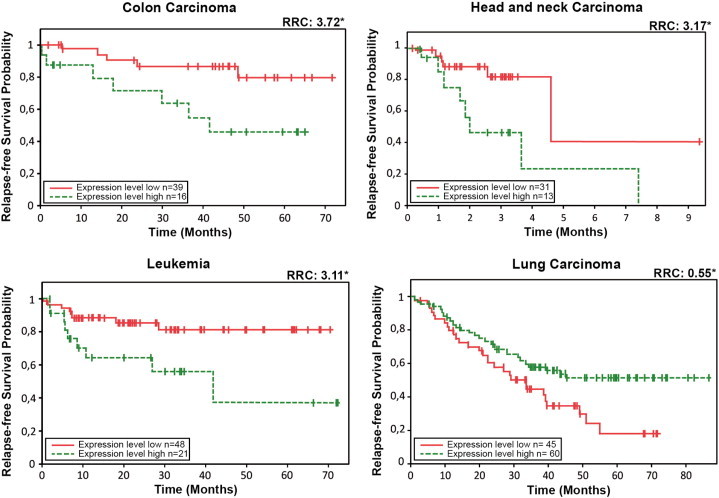
Examples of Cox survival plots for specific carcinomas. For colon and head and neck carcinomas and for leukemia, a clear association between increased MCMBP gene expression level and diminished probability of survival is illustrated. Contrary to lung carcinoma, in which when MCMBP is weakly expressed, the relapse-free survival probability is reduced (RRC, relative risk coefficient; asterisk represents statistically significant differences in the survival probability with P < .05, and n is the number of patients taken into account for the construction of the survival plots).
MCMBP Is Expressed More Strongly in Malignantly Transformed Cells than in Normal Proliferating Tissue
To study further the role of MCMBP in carcinogenesis, different tumor samples were studied at the protein level by immunohistochemistry. To determine whether MCMBP protein accumulation is associated with proliferation, normal tissues and tumor sections were analyzed and compared with samples stained with the Ki-67 antibody. The immunostainings revealed a strong correlation between MCMBP and Ki-67 accumulation: Ki-67–positive samples stained strongly positive for MCMBP, and samples negative for MCMBP were negative for Ki-67 (Figure 8). Interestingly, in breast cancer xenografts, Ki-67 identified proliferating cells in the tumor and also in the surrounding skin, whereas the MCMBP antibody identified proliferating cells in tumors, but its staining was weak in the proliferating cells of normal skin, suggesting that MCMBP staining predominantly marks cells that are malignantly proliferating (Figure 9).
Figure 8.
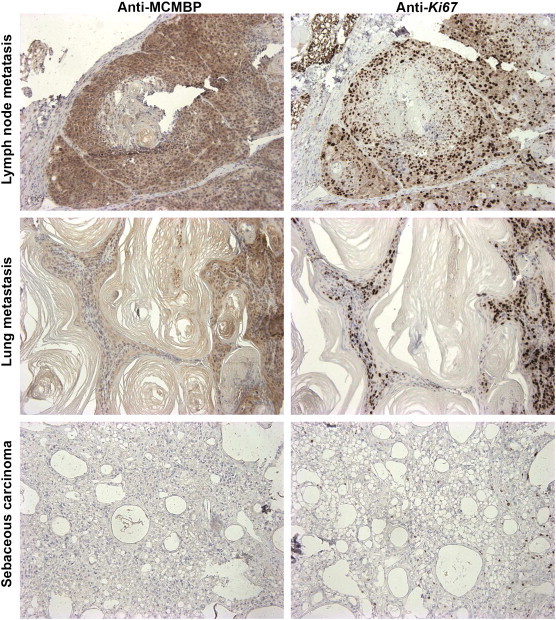
Immunohistochemical analysis of different tumor sections showing a high correlation between Ki-67 and MCMBP accumulation. Mouse tumors highly positive for Ki-67 are also positive for MCMBP and vice versa.
Figure 9.
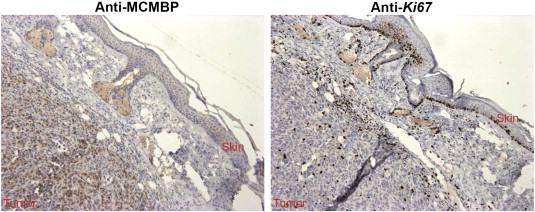
Breast cancer xenografts stained for Ki-67 and MCMBP. Ki-67 identifies proliferating cells from the tumor and also from the skin. Contrastingly, MCMBP identifies proliferating cells from the tumor, but its staining is weak in proliferating cells of normal skin, suggesting that MCMBP preferentially detects malignant cells.
To confirm a direct link between MCMBP levels and cancer progression and to validate the potential use of the MCMBP antibody in cancer diagnosis and/or prognosis, the immunohistochemical analysis was extended to a series of human tumors. We studied colon carcinomas because according to our data, MCMBP has a fundamental role in genetic instability originated mainly from cohesion defects. Compelling evidence suggests that cohesion is a key deregulated process, particularly in colorectal cancers [7], [8]. Twenty human colon adenocarcinoma samples were analyzed in detail. We selected tumor samples containing normal colon epithelium tissue in order to make direct comparisons between tumoral and normal tissues. In addition, this cohort contained tumors with different degrees of progression, ranging from well-differentiated colon adenocarcinomas to poorly differentiated carcinomas. Pathologic analysis of the samples revealed that normal epithelium had weak to moderate staining of MCMBP at the base of the crypts, the region where proliferative regeneration occurs. In contrast, MCMBP positivity disappeared toward the surface of the crypt at the site of the lumen, where cell division is not active and cells are differentiated (Figure 10B). Interestingly, in some other structures, MCMBP protein abundance was associated with proliferating cells in normal tissue. This was the case of the lymph nodules in peripheral lymph tissue in which the germinal centers, which contain B-cells that proliferate and mutate by somatic hypermutation, were moderately stained with the MCMBP antibody (Figure 10C). The same was observed in mucinous adenocarcinoma, where staining for MCMBP in the mucinous cancer cells, which are more differentiated, was weaker than in less differentiated cancer cells (Figure 10D). Strikingly, in most of the tumors, there was more abundant accumulation of MCMBP in comparison with the normal mucosa. In a few tumor samples, the staining for MCMBP was weak but still stronger than in normal tissue. In the tumoral fractions of the different types of carcinomas, MCMBP was remarkably abundant in the nuclei; the positivity was also detected to a lesser extent in the cytoplasm (Figure 10, E–H and Table 2). Finally, at the invasion front of the tumors, there was no clear difference in MCMBP abundance in comparison with the rest of the tumor mass.
Figure 10.
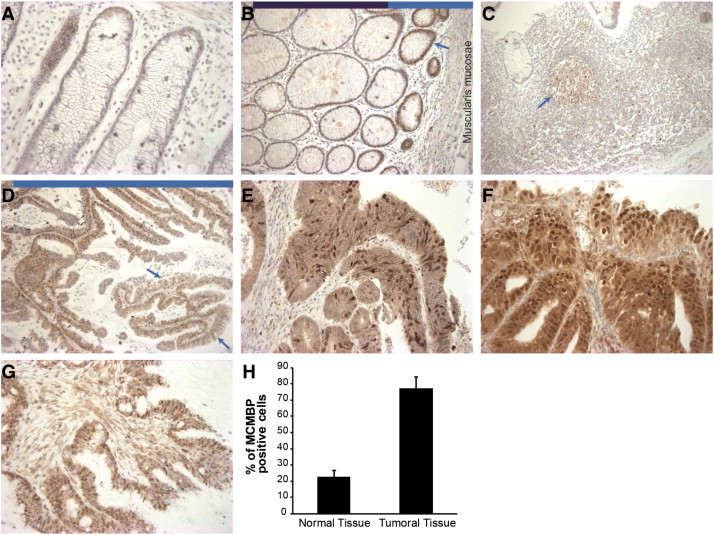
MCMBP protein levels in normal replicating cells and tumoral tissue. (A) In general, normal epithelium showed weak to moderate staining of MCMBP. (B) MCMBP protein is well expressed (arrow) at the base of the normal crypts (near the muscularis mucosae, light blue bar on top of the picture), corresponding to the proliferative stem and transient amplifying cell compartments from where regeneration happens. Contrastingly, MCMBP positivity disappears toward the surface of the crypt at the site of the lumen, where cell division is not active and cells are strongly differentiated (dark blue bar on top of the picture). (C) In lymphoid follicles, the germinal centers (arrow), which are sites that contain actively replicating B-cells, MCMBP is detected. (D) In the mucinous adenocarcinoma type, the mucinous cancer cells, which are more differentiated and responsible for the abnormal mucus production (red arrows), stained weaker for MCMBP in comparison with the highly MCMBP-positive cancer cells (light blue bar on top of the picture). There was a stronger expression of MCMBP in most tumors than in normal mucosa. MCMBP is abundant in the nuclei of tumor cells. (E) Well-differentiated adenocarcinoma. (F) Moderately differentiated adenocarcinoma. (G) Poorly differentiated adenocarcinoma. (H) Quantification of MCMBP-positive cells in the tumor mass of the carcinomas in comparison with the normal tissue.
Table 2.
Detailed Pathologic Analysis of the Colorectal Adenocarcinoma Samples.
| Tumor Information |
MCMBP Staining |
|||
|---|---|---|---|---|
| Tumor ID | Tumor Type | Nuclear Intensity | Cytoplasmic Intensity | Percentage of MCMBP-Positive Cells in the Tumor |
| 1586472 | MD after RCT | Strong | Strong | > 90 |
| 1588283 | WD to MD | Strong | Strong | > 90 |
| 1586788 | MD after RCT | Moderate | Light | 70 |
| 1587630 | MD | Strong | Strong | > 90 |
| 1587605 | WD to MD | Strong | Strong | 70 |
| 1588382 | MD | Strong | Strong | > 90 |
| 1588619 | MD to PD | Strong | Strong | 70 |
| 1588677 | MD | Moderate | Light | > 90 |
| 1586813 | MD | Moderate | Strong | 80 |
| 1588204 | WD | Strong | Strong | > 90 |
| 1589162 | PD | Light | Moderate | 80 |
| 1588915 | MD | Strong | Strong | > 90 |
| 1321831 | MD | Moderate | Light | 60 |
| 1324028 | MD | Strong | Light | 70 |
| 1324038 | MD | Moderate | Moderate | > 90 |
| 1324941 | MD | Strong | Strong | > 90 |
| 1328264 | MD + PD | Strong | Strong | > 90 |
| 1325706 | PD (mucinous type) | Strong | Strong | > 90 |
| 1335290 | MD | Strong | Moderate | > 90 |
| 1321369 | MD | Moderate | Light | 50 |
WD, MD, and PD, well-, moderately, and poorly differentiated adenocarcinomas, respectively.
RCT, radio-chemotherapy.
Given the positive direct association between MCMBP accumulation and the presence of malignantly proliferating cells, we decided to include more tumors in our histologic analyses. So, we selected 54 tumors (with the same characteristics and conditions used for the previously studied cohort) for which detailed clinical information was available (Table 3). The pathologic analysis of this new cohort of tumors confirmed the observations obtained with the previous one: the majority of the tumors had a clearly greater accumulation of MCMBP in comparison with the normal mucosa and the normally proliferating tissue. Additionally, in this new set of tumors, we found a significant association between the differentiation grade of the tumors and MCMBP accumulation. Most of the poorly differentiated adenocarcinomas retained MCMBP accumulation in > 90% of the tumor mass, representing a statistically significant association (P < .05 according to the hypergeometric distribution).
Table 3.
Detailed Pathologic Analysis of the Second Cohort of Colorectal Adenocarcinoma Samples.
| Tumor Information |
MCMBP Staining |
Patient Information |
|||||
|---|---|---|---|---|---|---|---|
| Tumor ID | Tumor Type | Nuclear Intensity | Cytoplasmic Intensity | Percentage of MCMBP-Positive Cells in the Tumor | Relapse-Metastasis | Disease-Free Survival | Overall Survival |
| 1 | MD | Moderate | Light | 50% | Liver-Lung | 6 | 25 |
| 2 | WD | Moderate | Light | 50% | Lung | 14 | 29 |
| 3 | MD | Strong | Light | 70% | None | 60 | 60 |
| 4 | MD | Light | Light | > 90% | Paravesical | 56 | 60 |
| 5 | MD | Light | Light | > 90% | Lung | 20 | 27 |
| 6 | MD | Strong | Moderate | > 90% | None | – | – |
| 7 | MD | Strong | Light | 60% | None | 60 | 60 |
| 8 | WD | Strong | Moderate | 70% | None | 60 | 60 |
| 9 | WD | Moderate | Light | > 90% | None | 60 | 60 |
| 10 | MD | Light | Light | 60% | None | 60 | 60 |
| 11 | MD | Strong | Moderate | > 90% | None | 71 | 71 |
| 12 | PD | Strong | Light | > 90% | Liver-Peritoneal | 22 | 60 |
| 13 | WD | Strong | Light | > 90% | Lung–Adrenal | 42 | 60 |
| 14 | PD | Strong | Light | > 90% | None | 60 | 60 |
| 15 | MD | Light | Light | 100% | None | 60 | 60 |
| 16 | Moderate | Light | 50% | None | 60 | 60 | |
| 17 | WD | Strong | Strong | 80% | None | 60 | 60 |
| 18 | PD | Strong | Light | > 90% | Bronchus–Lymph node | 65 | 65 |
| 19 | WD | Strong | Moderate | 70% | None | 5 | 5 |
| 20 | PD | Light | Moderate | 90% | None | 60 | 60 |
| 21 | MD | Moderate | Moderate | > 90% | None | 60 | 60 |
| 22 | PD | Light | Light | > 90% | Small intestine | 16 | 16 |
| 23 | MD | Light | Light | > 90% | None | 60 | 60 |
| 24 | WD | Moderate | Light | 50% | None | 60 | 60 |
| 25 | MD | Moderate | Light | 50% | None | 60 | 60 |
| 26 | MD | Moderate | Light | 80% | None | 60 | 60 |
| 27 | MD | Moderate | Moderate | 50% | None | 60 | 60 |
| 28 | WD | Moderate | Light | 50% | None | 60 | 60 |
| 29 | MD | Strong | Strong | > 90% | Liver-Lung-Brain | 23 | 43 |
| 30 | PD | Moderate | Moderate | 70% | Liver | 5 | – |
| 31 | MD | Light | Moderate | > 90% | None | 60 | 60 |
| 32 | WD | Strong | Moderate | 80% | None | 60 | 60 |
| 33 | WD | Strong | Light | 70% | None | 60 | 60 |
| 34 | WD | Strong | Light | 70% | Liver | 30 | 30 |
| 35 | MD | Strong | Strong | > 90% | None | 60 | 60 |
| 36 | MD | Moderate | Strong | 60% | None | 60 | 60 |
| 37 | PD | Light | Strong | 80% | None | 0 | 0 |
| 38 | MD | Light | Moderate | 70% | Mesenteric-Omental | 39 | 39 |
| 39 | MD | Light | Moderate | 80% | None | 38 | 38 |
| 40 | PD | Moderate | Light | 80% | None | 60 | 60 |
| 41 | WD | Moderate | Light | 70% | Liver | 8 | 14 |
| 42 | PD | Strong | Light | 60% | Liver | 60 | 60 |
| 43 | MD | Light | Light | > 90% | None | 60 | 60 |
| 44 | MD | Moderate | Light | 60% | Peritoneal | 30 | 60 |
| 45 | MD | Moderate | Light | 60% | Liver-Lung | 14 | 27 |
| 46 | WD | Strong | Light | 60% | Local tumor recurrence | 24 | 38 |
| 47 | MD | Moderate | Light | 80% | None | 60 | 60 |
| 48 | PD | Strong | Light | > 90% | Brain | 12 | 16 |
| 49 | PD | Moderate | Moderate | 80% | None | – | – |
| 50 | MD | Light | Light | 50% | None | 60 | 60 |
| 51 | PD | Light | Moderate | > 90% | Liver | 5 | 5 |
| 52 | MD | Moderate | Light | 70% | None | 60 | 60 |
| 53 | MD | Strong | Strong | > 90% | None | 10 | 10 |
| 54 | MD | Moderate | Light | 50% | Liver-Bone | 18 | 20 |
WD, MD, and PD, well-, moderately, and poorly differentiated adenocarcinomas, respectively.
Disease-free survival and overall survival are given in months.
Discussion
Colorectal cancer accounts for about one million new cases every year, and it is the second most frequently diagnosed internal malignancy worldwide [24]. These statistics stress out the necessity of looking for new strategies for better diagnosis and treatment of this disease.
Colorectal carcinomas display heterogeneous distinctive characteristics, and their origin is due to a compendium of diverse natural histories, pathologic features, and aberrations in distinct molecular pathways that converge to cause the malignancy [25], [26]. It has been proposed that three major drivers are involved in the generation of colorectal carcinomas: CIN, the CpG island methylator phenotype, and microsatellite instability. The predominant abnormality is CIN, accounting for up to 85% of cases [27]. Nevertheless, only few genes that might contribute to CIN governing colorectal cancers have been identified. It was shown that genes involved in sister chromatid cohesion are frequently mutated in colorectal carcinomas and their disruption leads to CIN in this type of neoplasias [7].
Here, we demonstrated that MCMBP knockdown in MCF7 cells induces the accumulation of a subpopulation of cells that are multinucleated with polylobed nuclei. When we studied the depletion of the MCMBP protein by using a specific antibody against MCMBP, we observed that the multinucleated cells in the MCMBP knocked down cultures were MCMBP negative. These observations were corroborated in different colorectal cell lines. Strikingly, also by overexpressing the MCMBP transcript in MCF7 cell cultures, we observed an increase in the number of multinucleated cells, together with the appearance of micronuclei. These observations confirm recent studies that identified a direct link between MCMBP expression and nuclear morphology [23].
It has been shown that a correlation exists between sister chromatid cohesion phenotypes, chromosome-segregation defects, and the appearance of multinucleated polylobed cells. Nguyen and collaborators showed that when the Aurora-B protein is misregulated, there is accumulation of multinucleated cells, which can promote tumorigenesis [28]. Similarly, in a genome-wide screening for genes related to cell division, Neumann and colleagues showed that genes that are necessary for the proper separation of the chromosomes could be clustered according to specific phenotypes that have in common the production of multinucleated cells [29]. Chromosomal missegregation and its direct consequence, the production of aneuploid cells, is a new hallmark of neoplastic transformation [3]. Aneuploid cells are prompted to become cancer cells due to the generation of an imbalanced genome, which amplifies oncogenes in some cells and restrains tumor suppressors in others [25]. The MCMBP protein might participate in oncogenic transformation by the generation of aneuploid multinucleated cells due to its effects on chromatid cohesion. Therefore, its deregulation might be regarded as a driver mutation in the oncogenic process. This is emphasized by the observation that even in MCF7 control cells MCMBP is scarce in a small proportion of spontaneously occurring multinucleated cells. These cells might arise specifically because of MCMBP deregulation, given that control multinucleated MCF7 cells express other proteins belonging to the MCM complex (Supporting Information Figure S2).
Several observations suggest that establishment of cohesion is coupled to the replication process and occurs very near the replisome. Cohesion is initiated by the acetyltransferase ECO1/CTF7, which travels along the DNA together with replication forks [30]. Furthermore, in yeast the depletion or mutation of several nonessential components of the replisome, such as the replication factor CTF18, causes cohesion defects [31], [32]. By associating with MCM proteins, MCMBP is very likely a component of the replisome [33], and according to the results presented here, it also plays a role in establishing cohesion during the replication process. MCMBP has been described as a new member of the MCM complex [14], and at the same time, it is strongly coexpressed with genes related to sister chromatid cohesion, such as MAD2 mitotic arrest deficient-like 1 (MAD2), budding uninhibited by benzimidazoles 3 homolog (BUB3), and the members of the structural maintenance of chromosomes (SMC) complex (Table 1).
Spindle checkpoint regulators, such as the BUB kinases and MAD2, protect cells from aberrant chromosome segregation and might therefore function as suppressors of malignant transformation. Interestingly, MAD2 is either upregulated or downregulated depending on the tumor type, and in both cases, these alterations result in chromosomal imbalances and tumor development [34], [35]. Some other genes related to cohesion and chromosome segregation processes behave like MAD2, being overexpressed in certain types of carcinomas and downregulated in others and, in both cases, resulting in abnormal chromosome structure. For example, this is true for the Aurora kinases [36], [37] and the Polo-like kinase 1 [38], [39]. Interestingly, down-regulation or overexpression of MCMBP in HEK-293T cells produced similar effects on the cohesion of sister chromatids, suggesting that regulation of molecules involved in this process must be tightly controlled in order to avoid problems in DNA replication and DNA repair that can result in carcinogenic events. Supporting this, recent evidence from human cell cultures suggests that MCMBP overexpression can lead to nuclear abnormalities similar to those associated with MCMBP depletion, indicating that an excess of MCMBP can be as detrimental as lack of it [23]. Thus, precise levels of MCMBP might be required to avoid CIN events.
However, no Arabidopsis thaliana overexpressing ETG1 could be obtained (unpublished data), suggesting that high ETG1 levels interfere with the plant generation process and signifying that overexpression of ETG1 and MCMBP is probably more deleterious than its depletion. Correspondingly, a larger number of disrupted metaphases in HEK cells were observed on MCMBP overexpression than on transcript depletion. Alternatively, as demonstrated recently, MCMBP overproduction could exert its phenotypic effects by creating a domain-negative version of the molecule [12].
Misregulation of the role of MCMBP in cohesion might not be the only factor that accounts for cancer progression. As mentioned above, defects either in the replication process or in checkpoint responses increase the susceptibility to cancer by triggering genome instability [2], [40]. MCMBP is physically part of the replication fork and its function links the replication process with sister chromatid cohesion. Therefore, it might act synergistically with both the origin and the progression of specific carcinomas.
It is known that an excessive amount of MCM2–7 complex is loaded onto the origins of replication before the replication process starts [41]. Although a huge number of potential replication origins exist throughout the genome, activation of only a fraction of them is sufficient for DNA replication [42]. Nevertheless, in the presence of replicative stress resulting from external sources or coming from the DNA replication process itself, many replication forks are stalled. For the replication process to continue, dormant origins licensed by excess MCM2–7 complexes are used as backups for these emergency situations, increasing the number of replication forks and promoting the completion of DNA replication [43]. Reduced levels of MCM2–7 proteins in mice causes spontaneous tumors with complete penetrance [44], suggesting that dormant origins play an important role in such conditions. These mouse models exhibited an abundance of spontaneous micronuclei, which is a characteristic of chromosome instability. Similarly, the MCM4chaos3 allele, encoding a point mutation in the MCM4 gene that compromises the stability of the MCM complex, leads to a reduced number of dormant origins. This reduction of dormant origins results in accumulation of stalled replication forks. Despite the activation of multiple DNA repair pathways, a significant fraction of stalled replication forks persist into the M phase, interfering with chromosome segregation [43], [45]. Interestingly, in our study, the knockdown of the MCMBP transcript also triggered the response of DNA stress genes.
It was shown that MCMBP regulates the unloading of the MCM complex during the late S phase. In Xenopus egg extracts, MCMBP can destabilize and disassemble the MCM2–7 complex and might function as an unloader of the MCMs from chromatin [11]. This phenotype is also seen in Schizosaccharomyces pombe [12]. Given that MCMBP is important for destabilization and disassembly of the MCM complex, the overexpression of MCMBP could lead to premature destabilization of the complex. This has a direct effect on the rescue of the stalled replication forks and compromises DNA replication and chromosome stability (Figure 11). The additive effect of a defect in the mechanism that rescues stalled replication forks, combined with the emerging cohesion defects caused by MCMBP overexpression, might have a strong effect on chromosomal stability. Due to this, it was expected that MCMBP would be strongly overexpressed in the colorectal adenocarcinoma tumors, which are the archetypical examples of carcinomas derived from CIN processes.
Figure 11.
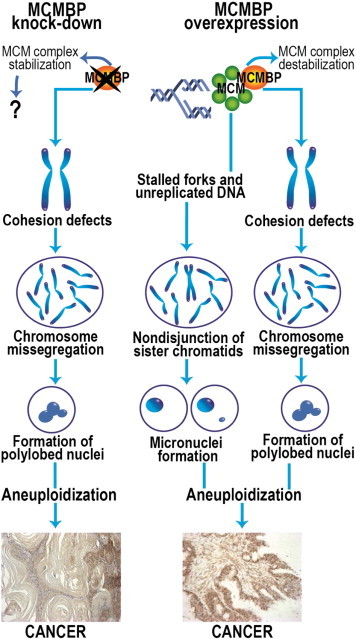
A model for chromosome instability caused by MCMBP misregulation. Both MCMBP down-regulation and overexpression result in loss of sister chromatid cohesion, causing chromosome missegregation and formation of polylobed aneuploid nuclei. When MCMBP is overexpressed, an additional mechanism for aneuploidization might exist, caused by destabilization of the MCM complex and generating problems when stalled replication forks occur. The presence of unreplicated DNA can cause nondisjunction of the sister chromatids in metaphase, producing cells with micronuclei, which can give rise to malignant cells. Still, it is not known whether the stabilization of the MCM complex by MCMBP down-regulation can directly affect the function of the replication machinery. The different mechanisms that emerge when MCMBP is upregulated or downregulated can explain why overexpression and knockdown of MCMBP can give rise to different carcinomas.
In the analyzed tumor samples, MCMBP accumulated in the nucleus, but in certain instances it also accumulated in the cytoplasm (Table 2). It has been documented that overexpression of the MCMBP orthologue in yeast results in delocalization and migration of the MCM subunits to the cytoplasm, causing abnormal replication [12]. Cytoplasmic presence of MCMBP might represent the premature destabilization of the whole MCM complex by its asynchronous action, another evidence of the role of MCMBP in carcinogenesis.
Evidence is accumulating for a tight relationship between members of the MCM complex and cancer. MCM2 was found to be a strong prognostic marker of breast cancer [46] and is also used to detect colorectal cancers [47]. Similarly, MCM proteins were considered as pre-cancer markers of esophageal cancers [48], and MCM2 and MCM4 were found to be independent predictors of survival in patients with non–small cell lung cancer [49].
Here, we have given consistent evidence illustrating that the novel replisome factor MCMBP can be considered a useful marker for cancer research. According to our Cox survival analyses, altered expression of MCMBP has a significant prognostic value in different types of cancers. Furthermore, its overexpression in breast carcinomas is directly linked with the estrogen receptor (ER) negative status, a hallmark of poor prognosis and difficult treatment of breast cancers (Supporting Information Figure S3). Altogether, our data suggest that deregulated MCMBP expression might be a cause of carcinogenesis in a wide spectrum of malignancies. MCMBP, its demonstrated prognostic value, and knowledge of its mechanism of action pave the way for its use in cancer diagnosis and maybe treatment.
Acknowledgements
We acknowledge Amin Bredan for critical reading of the manuscript and thank all members of the Molecular Oncology and Cell Cycle groups for fruitful discussions and suggestions and Lorena López for the help in preparing the manuscript. This research was funded by grants from the V.I.B.-International PhD Program in Life Sciences, the Research Foundation - Flanders (FWO), the geconcerteerde onderzoeksacties of Ghent University, the Stichting tegen Kanker, and the EU-FP7 Framework Program TuMIC 2008-201662.
Footnotes
This article refers to supplementary materials, which are designated by Figures S1 to S3 and are available online at www.neoplasia.com.
The authors declare that they have no conflicts of interests.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.07.011.
Appendix A. Supplementary data
Figure S1. Generation of a high-quality antibody against the MCMBP protein. (A) One microgram of proteins per well (plant extracts and MCMBP purified protein) was used to test MCMBP serum specificity by ELISA. (1) Arabidopsis protein extracts (negative control), (2) pre-immune control, (3) first bleeding, (4) second bleeding, and (5) last bleeding. The sera from the bleedings were diluted in different proportions (indicated on the right side of the ELISA reactions) and used to detect the MCMBP protein. (B) Western blot analysis of the purified MCMBP antibody in different cell lines: (1) MCF7-WT; (2) MCF7–empty vector; (3) MFC7–MCMBP-KD; (4) MCF7–MCMBP-OE (V5-tagged); (5) MDA-MB-231–WT; (6) MDA-MB-231–empty vector; (7) MDA-MB-231–MCMBP-KD; (8) MCMBP purified protein. Arrow at 75 kDa indicates the presence of the MCMBP protein. (C) Nuclear localization of the MCMBP protein in MCF7 cells. In MCF7 culture in which the MCMBP transcript has been constitutively knocked down, the MCMBP protein is not detected. (D) Localization of the MCMBP protein in overexpressing MCF7 cultures. MCMBP protein was detected using two different antibodies, the V5-tag antibody that detects the tagged version of MCMBP and the novel generated MCMBP antibody.
Figure S2. In multinucleated cells, the cells express MCM4 and MCM7, two of the core elements in the MCM complex. Contrastingly, MCMBP is poorly expressed in the same type of cells, suggesting that multinucleation is a direct consequence of MCMBP down-regulation, pinpointing a particular function for MCMBP in the MCM complex.
Figure S3. (A) MCMBP transcript levels in 56 primary human breast cancers, showing a broad variation in expression. (B) The 56 breast tumors were divided in quartiles according to MCMBP expression level. In tumors in which MCMBP is strongly expressed (Q3 and Q4 quartiles), the ER status is negative, a hallmark of poor prognosis and difficult treatment of breast cancers.
References
- 1.Jefford CE, Irminger-Finger I. Mechanisms of chromosome instability in cancers. Crit Rev Oncol Hematol. 2006;59:1–14. doi: 10.1016/j.critrevonc.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011;25:2041–2056. doi: 10.1101/gad.17010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Mathiesen RR, Fjelldal R, Liestol K, Due EU, Geigl JB, Riethdorf S, Borgen E, Rye IH, Schneider IJ, Obenauf AC. High-resolution analyses of copy number changes in disseminated tumor cells of patients with breast cancer. Int J Cancer. 2012;131:E405–E415. doi: 10.1002/ijc.26444. [DOI] [PubMed] [Google Scholar]
- 5.Anderson GR, Brenner BM, Swede H, Chen N, Henry WM, Conroy JM, Karpenko MJ, Issa JP, Bartos JD, Brunelle JK. Intrachromosomal genomic instability in human sporadic colorectal cancer measured by genome-wide allelotyping and inter-(simple sequence repeat) PCR. Cancer Res. 2001;61:8274–8283. [PubMed] [Google Scholar]
- 6.Dyrso T, Li J, Wang K, Lindebjerg J, Kolvraa S, Bolund L, Jakobsen A, Bruun-Petersen G, Li S, Crüger DG. Identification of chromosome aberrations in sporadic microsatellite stable and unstable colorectal cancers using array comparative genomic hybridization. Cancer Genet. 2011;204:84–95. doi: 10.1016/j.cancergencyto.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 7.Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbero JL. Sister chromatid cohesion control and aneuploidy. Cytogenet Genome Res. 2011;133:223–233. doi: 10.1159/000323507. [DOI] [PubMed] [Google Scholar]
- 9.Sakwe AM, Nguyen T, Athanasopoulos V, Shire K, Frappier L. Identification and characterization of a novel component of the human minichromosome maintenance complex. Mol Cell Biol. 2007;27:3044–3055. doi: 10.1128/MCB.02384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bochman ML, Schwacha A. The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiol Mol Biol Rev. 2009;73:652–683. doi: 10.1128/MMBR.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishiyama A, Frappier L, Mechali M. MCM-BP regulates unloading of the MCM2–7 helicase in late S phase. Genes Dev. 2011;25:165–175. doi: 10.1101/gad.614411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding L, Forsburg SL. Schizosaccharomyces pombe minichromosome maintenance-binding protein (MCM-BP) antagonizes MCM helicase. J Biol Chem. 2011;286:32918–32930. doi: 10.1074/jbc.M111.282541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi N, Lammens T, Boudolf V, Maes S, Yoshizumi T, De Jaeger G, Witters E, Inzé D, De Veylder L. The DNA replication checkpoint aids survival of plants deficient in the novel replisome factor ETG1. EMBO J. 2008;27:1840–1851. doi: 10.1038/emboj.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi N, Quimbaya M, Schubert V, Lammens T, Vandepoele K, Schubert I, Matsui M, Inze D, Berx G, De Veylder L. The MCM-binding protein ETG1 aids sister chromatid cohesion required for postreplicative homologous recombination repair. PLoS Genet. 2010;6:e1000817. doi: 10.1371/journal.pgen.1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obayashi T, Kinoshita K. COXPRESdb: a database to compare gene coexpression in seven model animals. Nucleic Acids Res. 2011;39:D1016–D1022. doi: 10.1093/nar/gkq1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 17.Mertens N, Fiers W. An advanced vector system for high-level recombinant gene expression in E. coli. Biotechnology International II: Latest developments in the Biotechnology industry and research. Universal Medical Press; 1999. pp. 165–172. [Google Scholar]
- 18.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quimbaya M, Vandepoele K, Raspe E, Matthijs M, Dhondt S, Beemster GT, Berx G, De Veylder L. Identification of putative cancer genes through data integration and comparative genomics between plants and humans. Cell Mol Life Sci. 2012;69:2041–2055. doi: 10.1007/s00018-011-0909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shekhar MP, Lyakhovich A, Visscher DW, Heng H, Kondrat N. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 2002;62:2115–2124. [PubMed] [Google Scholar]
- 21.Rosario CO, Ko MA, Haffani YZ, Gladdy RA, Paderova J, Pollett A, Squire JA, Dennis JW, Swallow CJ. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc Natl Acad Sci U S A. 2010;107:6888–6893. doi: 10.1073/pnas.0910941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JE, Woo SR, Kang CM, Juhn KM, Ju YJ, Shin HJ, Joo HY, Park ER, Park IC, Hong SH. Paclitaxel stimulates chromosomal fusion and instability in cells with dysfunctional telomeres: implication in multinucleation and chemosensitization. Biochem Biophys Res Commun. 2011;404:615–621. doi: 10.1016/j.bbrc.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Jagannathan M, Sakwe AM, Nguyen T, Frappier L. The MCM-associated protein MCM-BP is important for human nuclear morphology. J Cell Sci. 2012;125:133–143. doi: 10.1242/jcs.089938. [DOI] [PubMed] [Google Scholar]
- 24.Potter MB. Strategies and resources to address colorectal cancer screening rates and disparities in the United States and globally. Annu Rev Public Health. 2013;34:413–429. doi: 10.1146/annurev-publhealth-031912-114436. [DOI] [PubMed] [Google Scholar]
- 25.Chandhok NS, Pellman D. A little CIN may cost a lot: revisiting aneuploidy and cancer. Curr Opin Genet Dev. 2009;19:74–81. doi: 10.1016/j.gde.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worthley DL, Leggett BA. Colorectal cancer: molecular features and clinical opportunities. Clin Biochem Rev. 2010;31:31–38. [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen HG, Makitalo M, Yang D, Chinnappan D, St Hilaire C, Ravid K. Deregulated Aurora-B induced tetraploidy promotes tumorigenesis. FASEB J. 2009;23:2741–2748. doi: 10.1096/fj.09-130963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumann B, Walter T, Hériché JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner KP, Shirahige K, Uhlmann F. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell. 2006;23:787–799. doi: 10.1016/j.molcel.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 31.Hanna JS, Kroll ES, Lundblad V, Spencer FA. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 2001;21:3144–3158. doi: 10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, Newitt R, Aebersold R, Boone C, Brown GW. Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell. 2004;15:1736–1745. doi: 10.1091/mbc.E03-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santosa V, Martha S, Hirose N, Tanaka K. The fission yeast minichromosome maintenance (MCM)-binding protein (MCM-BP), Mcb1, regulates MCM function during prereplicative complex formation in DNA replication. J Biol Chem. 2013;288:6864–6880. doi: 10.1074/jbc.M112.432393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pérez de Castro I, de Cárcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- 35.Pinto M, Soares MJ, Cerveira N, Henrique R, Ribeiro FR, Oliveira J, Jeronimo C, Teixeira MR. Expression changes of the MAD mitotic checkpoint gene family in renal cell carcinomas characterized by numerical chromosome changes. Virchows Arch. 2007;450:379–385. doi: 10.1007/s00428-007-0386-7. [DOI] [PubMed] [Google Scholar]
- 36.Baldini E, Arlot-Bonnemains Y, Mottolese M, Sentinelli S, Antoniani B, Sorrenti S, Salducci M, Comini E, Ulisse S, D'Armiento M. Deregulation of Aurora kinase gene expression in human testicular germ cell tumours. Andrologia. 2010;42:260–267. doi: 10.1111/j.1439-0272.2009.00987.x. [DOI] [PubMed] [Google Scholar]
- 37.Wang LH, Xiang J, Yan M, Zhang Y, Zhao Y, Yue CF, Xu J, Zheng FM, Chen JN, Kang Z. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 2010;70:9118–9128. doi: 10.1158/0008-5472.CAN-10-1246. [DOI] [PubMed] [Google Scholar]
- 38.Grinshtein N, Datti A, Fujitani M, Uehling D, Prakesch M, Isaac M, Irwin MS, Wrana JL, Al-Awar R, Kaplan DR. Small molecule kinase inhibitor screen identifies polo-like kinase 1 as a target for neuroblastoma tumor-initiating cells. Cancer Res. 2011;71:1385–1395. doi: 10.1158/0008-5472.CAN-10-2484. [DOI] [PubMed] [Google Scholar]
- 39.Nihal M, Stutz N, Schmit T, Ahmad N, Wood GS. Polo-like kinase 1 (Plk1) is expressed by cutaneous T-cell lymphomas (CTCLs), and its downregulation promotes cell cycle arrest and apoptosis. Cell Cycle. 2011;10:1303–1311. doi: 10.4161/cc.10.8.15353. [DOI] [PubMed] [Google Scholar]
- 40.Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Immunol. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowers JL, Randell JC, Chen S, Bell SP. ATP hydrolysis by ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication. Mol Cell. 2004;16:967–978. doi: 10.1016/j.molcel.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 42.Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell. 2011;41:543–553. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kunnev D, Rusiniak ME, Kudla A, Freeland A, Cady GK, Pruitt SC. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene. 2010;29:3630–3638. doi: 10.1038/onc.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, Schimenti JC. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet. 2007;39:93–98. doi: 10.1038/ng1936. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez MA, Pinder SE, Callagy G, Vowler SL, Morris LS, Bird K, Bell JA, Laskey RA, Coleman N. Minichromosome maintenance protein 2 is a strong independent prognostic marker in breast cancer. J Clin Oncol. 2003;21:4306–4313. doi: 10.1200/JCO.2003.04.121. [DOI] [PubMed] [Google Scholar]
- 47.Davies RJ, Freeman A, Morris LS, Bingham S, Dilworth S, Scott I, Laskey RA, Miller R, Coleman N. Analysis of minichromosome maintenance proteins as a novel method for detection of colorectal cancer in stool. Lancet. 2002;359:1917–1919. doi: 10.1016/S0140-6736(02)08739-1. [DOI] [PubMed] [Google Scholar]
- 48.Alison MR, Hunt T, Forbes SJ. Minichromosome maintenance (MCM) proteins may be pre-cancer markers. Gut. 2002;50:290–291. doi: 10.1136/gut.50.3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramnath N, Hernandez FJ, Tan DF, Huberman JA, Natarajan N, Beck AF, Hyland A, Todorov IT, Brooks JS, Bepler G. MCM2 is an independent predictor of survival in patients with non-small-cell lung cancer. J Clin Oncol. 2001;19:4259–4266. doi: 10.1200/JCO.2001.19.22.4259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Generation of a high-quality antibody against the MCMBP protein. (A) One microgram of proteins per well (plant extracts and MCMBP purified protein) was used to test MCMBP serum specificity by ELISA. (1) Arabidopsis protein extracts (negative control), (2) pre-immune control, (3) first bleeding, (4) second bleeding, and (5) last bleeding. The sera from the bleedings were diluted in different proportions (indicated on the right side of the ELISA reactions) and used to detect the MCMBP protein. (B) Western blot analysis of the purified MCMBP antibody in different cell lines: (1) MCF7-WT; (2) MCF7–empty vector; (3) MFC7–MCMBP-KD; (4) MCF7–MCMBP-OE (V5-tagged); (5) MDA-MB-231–WT; (6) MDA-MB-231–empty vector; (7) MDA-MB-231–MCMBP-KD; (8) MCMBP purified protein. Arrow at 75 kDa indicates the presence of the MCMBP protein. (C) Nuclear localization of the MCMBP protein in MCF7 cells. In MCF7 culture in which the MCMBP transcript has been constitutively knocked down, the MCMBP protein is not detected. (D) Localization of the MCMBP protein in overexpressing MCF7 cultures. MCMBP protein was detected using two different antibodies, the V5-tag antibody that detects the tagged version of MCMBP and the novel generated MCMBP antibody.
Figure S2. In multinucleated cells, the cells express MCM4 and MCM7, two of the core elements in the MCM complex. Contrastingly, MCMBP is poorly expressed in the same type of cells, suggesting that multinucleation is a direct consequence of MCMBP down-regulation, pinpointing a particular function for MCMBP in the MCM complex.
Figure S3. (A) MCMBP transcript levels in 56 primary human breast cancers, showing a broad variation in expression. (B) The 56 breast tumors were divided in quartiles according to MCMBP expression level. In tumors in which MCMBP is strongly expressed (Q3 and Q4 quartiles), the ER status is negative, a hallmark of poor prognosis and difficult treatment of breast cancers.