

Abstract
Purpose
Retinitis pigmentosa (RP) is a photoreceptor disease that affects approximately 100,000 people in the United States. Treatment options are limited, and the prognosis for most patients is progressive vision loss. Unfortunately, understanding of the molecular underpinnings of RP initiation and progression is still limited. However, the development of animal models of RP, coupled with high-throughput sequencing, has provided an opportunity to study the underlying cellular and molecular changes in this disease.
Methods
Using RNA-Seq, we present the first retinal transcriptome analysis of the rd10 murine model of retinal degeneration.
Results
Our data confirm the loss of rod-specific transcripts and the increased relative expression of Müller-specific transcripts, emphasizing the important role of reactive gliosis and innate immune activation in RP. Moreover, we report substantial changes in relative isoform usage among neuronal differentiation and morphogenesis genes, including a marked shift to shorter transcripts.
Conclusions
Our analyses implicate remodeling of the inner retina and possible Müller cell dedifferentiation.
Introduction
Retinitis pigmentosa (RP) is a degenerative eye disease characterized by progressive loss of photoreceptors in the outer retina. Degeneration is triggered by one of more than 200 gene mutations that disrupt RPE or photoreceptor function [1-3]. Genetically heterogeneous in nature, RP is attributed to autosomal-dominant, recessive, and X-linked mutations. However, in all RP variants, the disease converges, with varying rapidity, on the same outcome: progressive and largely irreversible loss of vision. This devastating disorder has been the focus of considerable research, and much has been learned about the genetic determinants of RP [1]. However, we hope our study comparing the retinal transcriptomes of diseased and wild-type mice will further the knowledge of the molecular mechanisms underlying disease progression, particularly for the responses of the inner retina.
Although the molecular disruption of RP begins in the outer retina, significant changes also occur throughout the inner retina as a result of the loss of photoreceptors. The inner nuclear layer, consisting of bipolar and amacrine cells, suffers significant cell loss of approximately 20–60% of their population [4-8]. The structure of bipolar cell dendritic harbors change as well, including 1) formation of recurrent bipolar-bipolar synapses [7], 2) sprouting of neurites [9-11], and 3) near-complete retraction of dendrites in later stages of the disease [6,12-15]. However, some cells maintain their normal structure. Glycinergic amacrine cells maintain connections to ON bipolar cells, suggesting that amacrine-bipolar cell circuitry is preserved during disease progression [7]. Additionally, despite the substantial loss of retinal ganglion cells (25–80%) [4,5,16], the morphology and stratification of ON and OFF ganglion cell dendrites are somewhat preserved [17].
In addition to anatomic changes that occur within the inner retina, physiologic changes have also been reported. The responsiveness of ON bipolar cells to glutamate decreases, while their GABAA receptor-mediated signaling increases [18,19]. This could be the result of the diminished expression of mGluR6 and Goα, two glutamate receptors found in ON bipolar cells [8,14,20]. Taken together, these data suggest a shift of ON bipolar cell behavior to one that physiologically resembles that of OFF bipolar cells. In fact, OFF responses in the diseased retina are preferentially preserved [21]. Additionally, the formation of bipolar-bipolar recurrent connections is thought to corrupt inner retinal circuitry and signaling [22]. Downstream ganglion cells have also demonstrated higher levels of spontaneous activity during disease, which suggests intrinsic modifications to ion-channel receptors and/or neural circuitry [17,21,23].
In contrast, data also indicate that inner retinal circuitry is well conserved. First, ON and OFF ganglion cells maintain their intrinsic firing properties [17]. When exposed to electrical current injections in the presence of synaptic blockers, the intrinsic firing behavior of ON, OFF-transient, and OFF-sustained ganglion cells was indistinguishable between wild-type and rd1 retinal ganglion cells. OFF transient cells showed rebound firing to negative current injections, whereas this behavior was absent in ON ganglion cells. Second, the balance of excitatory and inhibitory currents to the ganglion cell response is conserved in rd1 retinas [17,24]. Third, selectively stimulating ON bipolar cells using exogenously expressed channelrhodopsin-2 (ChR2) leads to natural ganglion cells responses, including transient spiking and center-surround organization [24,25].
Since several therapeutic approaches to vision restoration (e.g., microelectronic retinal prostheses [26,27] and optogenetics [25,28]) target the inner retina, there is a need for a more comprehensive understanding of molecular changes that occur as a function of photoreceptor degeneration. Additionally, most studies have focused on analyzing the changes of a small set of genes and proteins with known relevance to the disease. However, the recent development of RNA-Seq has opened the door to transcriptome-wide profiling of gene expression, with a higher degree of accuracy than microarrays, which promises a more thorough understanding of changes in gene regulation as a result of RP [29,30]. One recent study used RNA-Seq to profile the transcriptome of the murine retina and determined that genes associated with retinal diseases are among the most highly expressed within the tissue [31]. However, to date, it appears there has been no comparative profiling of the rd10 mouse model. Several other mouse models of RP exist, but the rd10 model closely mirrors human autosomal-recessive RP due to the relatively delayed onset of photoreceptor loss and slow disease progression.
Here, we present an analysis of the transcriptome for the isolated rd10 mouse retina and identify genes undergoing changes in expression and splicing compared to wild-type retinas. As expected, our results indicated a loss of rod cells and a corresponding downward change in expression levels for photoreceptor and vision-related genes. However, we also observed the increased relative expression of a significant number of genes falling within the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of complement and coagulation cascades, dysregulation of which has been implicated in age-related macular degeneration (AMD) [32,33]. In addition, we found that Müller-specific transcripts displayed large increases in expression, indicating reactive gliosis. We also observed a significant switch in splicing that favored isoforms of reduced 3′ untranslated region (UTR) length, primarily within genes involved in neurogenesis and differentiation. Finally, we have made our raw and processed data available as a valuable resource for other investigators.
Methods
Animals
This study adhered to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision research as well as the University of Southern California regulations for use of animals in medical research. All animal procedures were performed according to the Ophthalmic and Vision Research of the Association of Research in Vision and Ophthalmology, and the guidelines approved by the Institutional Animal Care and Use Committees at the University of Southern California. The study protocol was approved by the institutional animal care and use committee of USC. Wild-type (WT) mice (C57BL/6) and rd10 (B6.CXB1-Pde6brd10/J on a C57BL/6 background) mice were purchased from Jackson Laboratories (Bar Harbor, ME). The activity of the beta subunit of Pde6 is compromised in the rd10 mouse model of RP [34]. A spontaneous mutation in the gene encoding Pdeβ generates a faulty protein, causing an inability to catalyze the Pde6 complex. As a result, cGMP accumulates in photoreceptor cells to eventually reach toxic levels, reducing their ability to process light [35]. The Gγ13 mouse is a BAC transgenic mouse line created through the Gene Expression Nervous System Atlas (GENSAT) project, that expresses GFP exclusively in the ON bipolar cells within the retina. The Gγ13 mice were a generous gift from Dr. Alapakkam Sampath at the University of Southern California. Retinas were dissected from mice 48 to 120 days old. Typically, samples were collected in rd10 mice at at P61, wild-type at P48, and Gγ13 at P42 (Table 1). Animals were raised in a 12 h:12 h light-dark cycle with unlimited access to food and water, and all were euthanized at the same time of day to minimize circadian effects.
Table 1. Sample collection details of each genotypic group.
| Sample ID | Mouse ID | Genotype | DOB | Date isolated | Age (days) | Retinas |
|---|---|---|---|---|---|---|
| WT-S1 |
2356 |
WT |
1-Oct-2011 |
18-Nov-2011 |
48 |
2 |
| WT-S2 |
2357, 2358 |
WT |
1-Oct-2011 |
18-Nov-2011 |
48 |
4 |
| WT-S3 |
2410 |
WT |
7-Oct-2011 |
7-Feb-2012 |
120 |
2 |
| G-S1 |
2360 |
Gg13 |
7-Oct-2011 |
18-Nov-2011 |
42 |
2 |
| G-S2 |
2366 |
Gg13 |
7-Oct-2011 |
18-Nov-2011 |
42 |
2 |
| G-S3 |
2361, 2365 |
Gg13 |
7-Oct-2011 |
18-Nov-2011 |
42 |
4 |
| rd-S1 |
2316 |
rd10 |
3-Jun-2011 |
3-Aug-2011 |
61 |
2 |
| rd-S2 |
2318 |
rd10 |
3-Jun-2011 |
3-Aug-2011 |
61 |
2 |
| rd-S3 | 2317, 2319 | rd10 | 3-Jun-2011 | 3-Aug-2011 | 61 | 4 |
Cell dissociation
Unless otherwise stated, all chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO). Mice were anesthetized with intraperitoneal ketamine/xylazine (100 mg/kg ketamine and 16.5 mg/kg xylazine) followed by cervical dislocation. Retinas were quickly removed within 10 min post-mortem and bathed in cold HEPES-buffered Ames medium (adjusted to pH of 7.4 with 1 N NaOH) within a Petri dish. After the whole retina was isolated, sample tissue was lysed using an RNAse-free mortar and pestle rotor in 100 μl TRIzol reagent (Invitrogen, Carlsbad, CA) within a 1.5 ml Eppendorf (Qiagen, Valencia, CA). Each sample contained two or four whole retinas from the same mouse. We dissociated the retina with brief pulsations for 60 sec until the sample was homogenous, followed by gentle trituration.
Immunohistochemistry and confocal microscopy of retinal tissues
Expression of GFP and PKCα proteins in the retinas of the control and treated animals was evaluated using immunohistochemistry combined with confocal imaging as previously described [25]. Briefly, the eyecups were dissected, immersed in 4% paraformaldehyde, and sucrose-infiltrated overnight. The following day, they were frozen in optimal cutting temperature compound on dry ice, and 10 μm serial sections of tissues were prepared on a Leica CM 3050 S cryostat (Leica, Mannheim, Germany). The tissue sections were immunolabeled with rabbit antibodies (IgG fraction; dilution 1:500; Invitrogen) against green fluorescent protein (GFP) and mouse anti-protein kinase α (PKCα) IgG (dilution 1:100; Santa Cruz Biotechnology, Santa Cruz, CA) diluted in the blocking solution containing 3% bovine serum albumin (BSA) plus 5% normal goat serum. The sections were then incubated with secondary antibodies against rabbit IgG (Invitrogen; Alexa Fluor 488, dilution, 1:1,000) and mouse IgG (Invitrogen; Alexa Fluor 555, dilution, 1:200). Prolong gold antifade mounting media (Invitrogen) containing 4',6-diamidino-2-phenylindol (DAPI) was used to mount the sections and to stain cell nuclei. Antibody distribution was visualized using a TCS-SP5 Broadband Spectra laser confocal microscope equipped with a 20X and 63X (NA=1.2) objective (Leica Microsystems, Deerfield, IL).
mRNA-Seq Library construction
Total RNA was extracted using Illumina’s RNA Solexa Library protocol using 450 μl TRIzol (Invitrogen) per sample. RNA isolation was performed under RNase-free conditions, and samples with spectrophotometry A260/A280 ratios >1.75 with total RNA >2 μg were accepted for library generation. Samples were stored at −20 °C until the transcriptomes were generated. Two hundred base pair short-insert libraries were constructed, with 91 pair-end sequencing on Illumina Hiseq2000, generating 12 million clean reads (1.1 Gb clean data) per sample. This process was performed with DNase I treatment for degradation of ss- and ds-DNA in RNA samples. Poly (A)-containing mRNA molecules were purified from total RNA using poly (T) oligoattached magnetic beads, followed by mRNA fragmentation using the protocol from Covaris, Inc. (Woburn, MA). First-strand cDNA was generated using random hexamer-primed reverse transcription followed by second-strand cDNA synthesis and cDNA purification using a purification kit (Illumina, San Diego, CA). Synthesized cDNA was then subject to end-repair, 3′ adenylated, with adaptor ligation to the ends. Products were purified by TAE agarose gel electrophoresis and PCR-amplified under Illumina mRNA-Seq PCR amplification protocol conditions. Library validation and quality control were performed on the Agilent Technologies 2100 Bio-analyzer (Agilent Technologies, Inc., Santa Clara, CA) and ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). RINs over 7 were considered acceptable in RNA samples.
Computational methods
Reads were mapped to mm9 using RMAP [36]. We used a modified version of Ref-Seq for read-counting in which we collapsed isoforms to produce a single supertranscript for each gene. We then counted the number of reads in each gene, and differential expression was determined from these counts using EdgeR [37]. We used a stringent p value threshold of 1×10−4 for identifying differentially expressed genes. The same was done with exon-level counts to determine changed inclusion ratios, where Fisher’s exact test was used to determine the significance of the change in odds. The list of retinal cell-specific genes was compiled from Gamsiz at al. [31]. The set of mouse transcription factors was collected from the RIKEN Mouse transcription factor database [38], and the set of RNA-binding proteins was compiled from Galante et al. [39]. The KEGG pathway and biologic process gene-set enrichment was performed using DAVID [40]. Sequence data were deposited in GEO (accession number: GSE56473).
Results
We performed RNA-Seq on whole-retina samples from rd10 and wild-type mice of similar age. At the P61 time point, rd10 mice have undergone major photoreceptor death with few photoreceptor cell bodies remaining [41]. This was confirmed through immunostaining of the rd10 retinas with a rod bipolar cell-specific antibody (i.e., PKCα) and DAPI staining, which revealed virtually no marked cells (Figure 1A). We also profiled the transcriptome of Gγ13 transgenic mice that express enhanced green fluorescent protein (GFP) in the rod and ON cone bipolar cells. This was performed to evaluate differences against the wild-type samples, to deduce whether Gγ13 may substitute for C57BL/6J in the future, since selective GFP expression is advantageous for imaging purposes. An analysis of correlation between replicates (Figure 1D) confirmed that within-group correlations for wild-type and diseased samples were high, while between-group correlations were substantially lower. We also observed that the Gγ13 samples (GFP expressing) were equally correlated with wild-type samples, and that the mean expression levels of the wild-type retina samples were highly correlated (Pearson correlation coefficient of 0.9) with previous studies [31]. No significant changes in expression between the wild-type and GFP-expressing replicates were found (data not shown); as a result, we consider the Gγ13 samples additional wild-type replicates.
Figure 1.
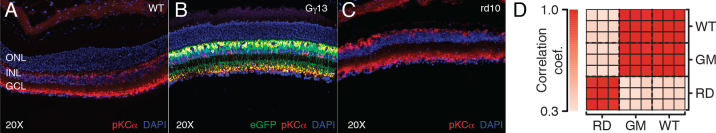
Confocal images of the retina showing GFP and PKCα expression in the ON bipolar cells of the mouse retina in 60 day old mice. A: PKCα stain of a normal retina in rod bipolar cells and the presence of the normal photoreceptor layer. B: GFP expression and PKCα in the bipolar cell layer of the Gγ13 retina and (C) the absence of photoreceptors in the rd10 retina with an intact bipolar cell layer. GFP is indicated in green in the confocal images. Red represents PKCα, a marker for rod bipolar cells, a type of ON bipolar cell in the inner nuclear layer (INL). Blue is 4',6-diamidino-2-phenylindole (DAPI), which stains for cell nuclei. Yellow indicates colocalization of GFP and PKCα expression. D: Correlation coefficients (Pearson’s) between replicates (each replicate is from a unique mouse) reflect high correlation within groups; WT = wild-type mice, RD = rd10 mice, GM = Gγ13 mice.
Retinas from rd10 mice show loss of high-expression vision- and RP-related genes
We identified a total of 1,079 genes with significant changes in mRNA levels with the conservative threshold of corrected p<1×10−5. Of these, 385 showed increased relative expression and 694 showed decreased relative expression in the rd10 retina. The complete list is provided in Appendix 1.
Gamsiz et al. reported that a significant proportion of the most highly expressed retinal genes have disease association [31]. Likewise, our data demonstrated that highly expressed vision- and disease-associated genes showed the most prominent changes in expression—predominantly decreased relative expression (Figure 2A). Of the top ten significant changes (Table 2), nine show decreases in relative expression levels, five have clear associations with phototransduction, and seven have been previously characterized as RP-causing mutations. In addition, genes with high-confidence decreases in relative expression were those with high expression levels in WT retinas (Figure 2B). In contrast, genes with high-confidence increases in relative expression showed no such bias toward having either high or low expression in the rd retina (Figure 2C).
Figure 2.

Changes in mRNA levels in rd10 retina. A: The proportion of genes with increased versus decreased expression in the rd10 retina when compared to wild-type, broken into False discovery rate (FDR) quantiles based on confidence of change (genes are considered to have increased or decreased expression if p<1×10−4). B: The expression level, as measured by RPKM (reads per kilo base of sequence per million reads mapped), in wild-type retinas for differentially expressed genes (p<1×10−4) that showed decreased relative expression in the rd10 retina, broken into quantiles by confidence of expression change. High confidence reductions in relative expression occur in genes with high expression in wild-type retina. C: As in B, but expression (RPKM) in the rd10 retina for genes that show significant increases in relative expression. High confidence increases in relative expression do not show any trend towards low- or high-expression genes in rd10 retina. D: The proportions of genes observed with increased and decreased relative expression that have been described as cell-type specific. E: Log-odds and corrected p-values for differentially expressed transcription factors (TFs) and RNA-binding proteins (RBPs). Transcription factors and RNA-binding proteins show both increases and decreases in relative abundance, though the strongest changes observed are decreased relative mRNA levels for a subset of transcription factors. Increased relative mRNA levels of transcription factors and RNA-binding proteins was observed in a number of genes associated with disease progression (discussed in text).
Table 2. Most significant expression changes in rd10 transcriptome.
| Gene | Direction | WT RPKM | rd10 RPKM | Function | Implicated in RP |
|---|---|---|---|---|---|
| Prph2 (Retinal degeneration slow) |
Down |
2310.8 |
75.9 |
Disc morphogenesis |
Evidence [108] |
| Fscn2 |
Down |
90.8 |
0.7 |
Disc morphogenesis |
Evidence [109] |
| Guca1b (RP48) |
Down |
651.8 |
6.6 |
Photoresponse [110] |
Evidence [111] |
| Serpina3n |
Up |
2.7 |
76.4 |
Inflammation |
Evidence [112] |
| Reep6 |
Down |
1143.8 |
31.2 |
Unknown |
No evidence |
| Pde6g (Phosphodiesterase) |
Down |
3971.3 |
117.6 |
Phototransduction |
Evidence [113] |
| Sag (Arrestin) |
Down |
4121.9 |
192.8 |
Phototransduction |
Evidence [114] |
| Rho (Rhodopsin) |
Down |
11,271.4 |
68.9 |
Phototransduction |
Evidence [115] |
| Pdc (Phosducin) |
Down |
1479.1 |
42.0 |
Phototransduction [116] |
No evidence [117] |
| Ccdc24 | Down | 120.5 | 5.2 | Unknown | No evidence |
Shown are the top ten most significant changes in gene expression between wild-type and rd10 retinas. RPKM values are means across all replicates. A direction of ‘up’ indicates increased relative expression in rd10 over wild-type, while ‘down’ indicates decreased relative expression in rd10 over wild-type.
Increased expression of Müller-specific genes accompanies decrease in rod-specific genes
Genes with decreased relative expression were mainly rod-specific (for example, Rho); their extreme decrease is expected and driven by the loss of rod photoreceptors. In contrast, we also observed a predominance of Müller-specific genes among those that showed increased relative expression (Figure 2D); the proportion was significantly higher than expected under the null hypothesis that increases in transcript expression do not favor any particular cell type (p<6.5×10−3, Fisher’s exact test). This suggests either increased numbers or modified activity of the glia. Despite the prominence of these two cell types, we also observed relative expression decreases in a few amacrine and bipolar cell-specific genes, and increases in amacrine, ganglion, and bipolar specific genes. This indicates transcriptional changes in cells other than rods and Müller glia.
Changes in transcriptional and post-transcriptional control
Despite changes in transcriptomic profiles resulting from different relative abundances of retinal cells, we also suspect that some changes were driven by shifts in individual cellular activity. In pursuit of this, we looked for expression changes in transcription factors (TFs) and RNA-binding proteins (RBPs) that may drive transcriptional or post-transcriptional regulatory changes. We identified significant changes in 76 TFs and 14 RBPs (Figure 2E and Appendix 1). Most dramatic changes were decreases in relative expression, predominantly among TFs. Although it is difficult to disambiguate responsive downregulation of genes from loss of photoreceptors, there were several TFs and RBPs with significantly increased relative expression, which provide more concrete evidence of transcriptomic changes in response to rod death. Among the RBPs, we observed increased relative expression of Rbpms, Cpeb2, Apobec1, and Rnasel. Rbpms encodes for a retinal ganglion cell marker [42], further suggesting transcriptional changes within ganglion cells, while Cpeb2 is involved in polyadenylation. Rnasel, in its active form, degrades RNA and is a component of immune response to viruses [43]. Finally, Apobec1 is an RNA-editing enzyme. Its canonical target is apolipoprotein B [44], where its editing results in a truncated protein. The association of apolipoprotein B deficiency with atypical RP has been previously established [45].
Among the transcription factors with increased relative expression, we observed several proteins with clear association to retinal function and development, and known roles in RP progression. Egr1, a nuclear transcriptional regulator, showed more than a 4× increase in relative levels [46]. Egr1 has recently been proposed as a protective response to photoreceptor death in the retinal degeneration slow (rds) mouse model [47] and may play a similar role in the rd10 model. Sox and Notch family proteins also showed increased relative expression, both of which are necessary for Müller glia development in the mouse retina [48], suggesting evidence of Müller glia activity. We also observed a relative increase in Cebpd, a regulator of macrophage activity, further implicating gliosis. Interestingly, we observed an increase in Vsx1 transcript levels, a gene primarily involved in differentiation and function of bipolar cells [49,50], but also associated with corneal wound healing [51]. The nuclear receptor transcripts Nr2e1 and Nr2e3 also showed a relative increase. Nr2e3 plays a role in determining the fate of retinal cells and can harbor RP-causing mutations in humans [52,53]. Murine Nr2e1 has been shown to control retinal progenitor proliferation and differentiation through regulation of cell-cycle proteins, in particular cyclin D1 [54].
Retina from rd10 mice show increased relative expression of immune response genes
We explored the sets of genes with increased and decreased relative expression to determine their function and relation to specific biologic processes and pathways. We observed that genes with increased relative expression were heavily associated with immune and inflammatory responses, particularly innate immune responses. Figure 3A shows the biologic processes and KEGG pathways that were significantly enriched for genes with increased relative expression levels (corrected p<0.001; several redundant terms have been omitted; see Appendix 2 for details), as well as the associated genes and the strength of change observed for each gene. Interestingly, the strongest changes were observed in genes that participate in many significant processes and pathways. Elements of the innate immune response, namely, the complement system, showed substantial evidence of activation. Figure 3B shows the KEGG pathway for the complement system, with the genes that show increased relative expression highlighted. No specific KEGG pathways were enriched within the genes that show decreased relative expression, but as expected, there was a predominance of vision-related biologic processes (Figure 3C). The complete set of enriched KEGG pathways and biologic processes for differentially expressed genes (increased and decreased relative expression) is provided in Appendix 2.
Figure 3.

Significantly enriched biologic processes and KEGG pathways among differentially expressed genes. A: Significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and biologic processes among genes that show increased relative expression and their corrected p value are shown. For each pathway or process, the genes present in it are listed, and the strength of the fold-change for each gene is shown. B: The alternative, lectin, and classical pathways of the complement cascade (top enriched KEGG pathway among genes with increased relative expression). Highlighted are all genes showing significant differential expression (in all cases, increased relative mRNA levels in rd10 retina versus wild-type retina). C: Significantly enriched biologic processes among those genes with a significant decrease in their relative expression, and the corrected p values for each term.
Degenerated retina exhibit changes in splicing
We observed significant changes in relative exon usage for 417 exons between the healthy and diseased samples (full list in Appendix 3), occurring in 284 unique genes. There was a significant preference toward increased inclusion (Figure 4A), with 67% of changes reflecting greater usage (p<2.2×10−12; binomial test). Decreased exon usage was primarily in 3′ UTRs, suggesting a global relaxation of post-transcriptional regulation, while increased exon use was primarily within the coding and 5′ UTR exons (Figure 4B–C). Most genes showed significant changes in only a single exon (220 of 284 genes), although we observed some genes with changes in multiple exons and in both directions (Figure 4D). One example is the cell adhesion protein Ncam1, which showed a decrease in the use of a promoter-proximal 3′ UTR exon in favor of a more distal one (Figure 4E–F). Notably, genes that demonstrated significant changes in exon use were enriched for biologic processes associated with neurogenesis, morphogenesis, and differentiation (Figure 4G).
Figure 4.

Changes in exon-inclusion rates for rd10 mice occur in neuron morphogenesis and differentiation related genes. A: Ratio of increased and decreased inclusion rates for exons stratified by confidence (FDR); the majority of cases promote increased exon usage, though this is somewhat relaxed in the highest confidence changes. B: The proportion of exons with significantly decreased usage that are categorized as coding exons, 3′ or 5′ untranslated region (UTR) exons. C: As in B, but for exons with significantly increase usage. D: The number of genes with a given number of exons showing increased relative inclusion (y-axis) and a given number of exons showing decreased relative inclusion (x-axis). E: The UCSC genome browser track shows changes in usage of exons within the Ncam1 transcript. F: Schematic representation of the data from panel E, showing the average proportion of reads falling within each exon and the corrected p value from a Fisher’s exact test. G: Gene Ontology (GO) terms associated with those genes that exhibit changes in splicing between the control and the rd10 samples.
Discussion
Retinitis pigmentosa remains a challenging problem from clinical and research perspectives, with many questions unanswered about disease progression. Our study sheds some light on several of these quandaries at the genome level within the rd10 mouse. Several will inform our evolving understanding of more general concepts, such as the role of glial cells in the central nervous system and the stimulation of stem cell properties and dedifferentiation. There are challenges in comparing results from such different tissues; the rd10 retina is essentially devoid of photoreceptors. For example, disambiguating changes in gene expression from differences in relative cell-type abundance is difficult. However, our analysis approach uses normalization to account for large changes in expression for some genes, and we feel several important observations can be made, which we now detail.
Müller gliosis and dedifferentiation
The role of the glia in retinitis pigmentosa is complex. The main resident glial cells in the retina are the Müller glia; within healthy retina, they are responsible for providing nutrients and filling various support roles for neurons. The response of these glial cells to insult, termed gliosis, varies dramatically between species [55,56]. Within the mammalian retina, gliosis is characterized by the proliferation of Müller glia, accompanied by changes in gene expression [57]. It has been proposed that this protective mechanism is intended to shield and repair neurons, but substantial evidence also links gliosis with negative outcomes [58]. Roesch et al. [59] recently noted gene expression changes occurring in Müller glia from the rd1 and Rho knockout mouse models using an microarray, but to date, no deep sequencing has characterized changes in rd10 mice. Our data indicate that Müller cells are increasingly active in rd10 murine models of RP. Gliosis, marked by upregulation of glial fibrillary acidic protein (Gfap), was significantly increased. Müller-exclusive transcripts Sox8 and Sox9 also showed increased relative expression. These factors are required for Müller glial development and linked to Notch signaling implicated in glial proliferation via a fibroblast growth factor 2/mitogen-activated protein kinase (FGF2/MAPK) path [48,60]. In our studies, Fgf2 expression was enriched as well. Additional relative-expression increases in Müller-specific genes Apoe, Aqp4, Ccnd3, Cspg5, Dbi, Dkk3, Gstt1, Kctd2, Mt1, Prdx2, and Vim also reflect changes in Müller gene expression during gliosis.
Remarkably, gliosis in zebrafish (among other vertebrates) is thought to be central to their capacity to regenerate retinal neurons through the dedifferentiation of Müller glia into neuronal progenitors [56]. Several research groups have also demonstrated that mammalian Müller glia can be stimulated to follow a similar process, providing substantial hopes for their use in retinal regeneration therapies [61]. In our data, we observed increased relative expression of genes encoding for embryonic retinal progenitor and cell cycle markers, including Bmp1, Bmpr2, Cdc14a, Ctsb, Gas1, Gli2, Gsx2, Jag2, Metrn, Pax6, Pdgfa, Rpa1, and Spred1. Despite decreased expression of NeuroD1 critical for neuronal differentiation, we found increased expression levels of D-type cyclins (cyclin D1, cyclin D3), suggesting attempts to reenter the cell cycle. Increases in the relative expression of neurogenic Sox2 and Notch signaling genes (Notch 1, 2, 3, and Cntn1) also mirrored this finding. SOX2 and Notch factors regulate reentry of Müller glia into the cell cycle, maintain progenitor cell quiescence, and prevent dedifferentiation [48,62,63]. However, downstream Notch effectors showed mixed results. Id1, Hes5, and Notch-inhibitor Nrarp did not show significant changes in expression, although Id3 and Hes1 expression increased. Id factors maintain cells in undifferentiated, progenitor states, but also directly induce Müller glial and bipolar fates. Conversely, transcription factors Hes1 and Hes5 downregulate the expression of proneural bHLH proteins that are crucial for the formation of varying retinal cell types [64,65]. In addition, we observed significant evidence to suggest a global shortening of transcripts: A high proportion of exons with decreased inclusion were 3′ UTR exons, suggesting a global shortening of 3′ UTRs. This shortening is a sign of decreased post-transcriptional regulation and indicates a shift toward a more stem-like state. Splicing changes were also observed in genes associated with neuron development, differentiation, and morphology (Figure 4). In light of these findings, we hypothesize that a population of Müller cells are undergoing dedifferentiation, as observed in other vertebrates.
Activation of the complement cascade and related cell-death pathways
We observed that genes with increased relative expression were strongly associated with the immune and inflammatory response. In particular, several components of the complement cascade, the most strongly enriched KEGG pathway identified in our analysis, showed increased mRNA levels in the rd10 mouse retina. Recently, upregulation of complement genes has also been reported in retinitis pigmentosa in microarray studies, specifically Bf (Cfb) and C2 [66]. Interestingly, we did not detect significantly increased expression of C2, although our RNA-Seq data confirmed increased relative expression of Bf. However, we detected previously unreported increases in expression for complement factors C1q (C1qa, C1qb, C1qc), C1r, C3, C4 (C4b), and Cfi as well as the C1 inhibitor Serping1, suggesting a more pervasive change in the activities of the complement system, at least in the rd10 murine model.
Apart from complement-mediated cell lysis, additional mechanisms of cell death contribute to photoreceptor death. Previous studies have established that apoptosis, autophagy, and complement are triggered in parallel within the rd/rd (rd1) mouse model [67]. Our studies mirrored these findings, with notable relative-expression increases in key caspase and cathepsin family proteases. In particular, caspase (Casp) -1, -8, -9, -12, and cathepsins (Cts) B, C, D, L, and S demonstrated significant increases. Despite differences in the method of activation, all caspases result in apoptosis [68]. Caspase-9 subsequently activates effector caspase-3, -6, and -7, but interestingly, our data did not show statistically significant increases in the three effector proteins [68]. Similar to caspases, lysosomal cathepsin proteases amplify apoptotic signals but uniquely contribute to autophagy, an event leading to caspase-independent cell death [69]. However, certain cathepsins, such as cathepsin S, may regulate proinflammatory cytokine expression and autophagic inflammation. A recent study discovered that inhibition of cathepsin S elicited the production of reactive oxygen species, activation of nuclear factor-kB (NF-kB), and induction of prodeath autophagy [70]. In any case, although it appears that cathepsins may play an important role in apoptosis and autophagy, further research is needed to better characterize cathepsins and their contributions to RP.
Response to oxidative stress
Historically, one of the more puzzling aspects of RP progression has been the eventual death of cone cells although causative mutations do not directly impact them. One explanation is that decreased rod oxygen consumption with rod death causes hyperoxia within the retina. In response, vascular pruning and vessel attenuation occur, resulting in cone death and subsequent hypoxia in the inner retina [71,72]. As a result, therapies aimed at reducing oxidative stress have been shown to slow the progression of the RP phenotype [71,73]. In support of this model, we observed slightly less than a twofold increase in expression for one of the hypoxia-induced VEGF proteins (Vegfc). VEGF family proteins are active in angiogenesis and induce expression of adhesion molecules [74]. Interestingly, although Vegf expression is driven by hypoxia-inducible factor alpha (Hif1a), the master regulator of oxygen tension, we did not detect increased expression of Hif1a despite the hypoxic environment associated with RP [75]. However, relative-expression increases in genes encoding for oxidative stress markers such as NADPH oxidase 2 (Nox2), nitric oxide synthase (Nos1), and neutrophil cytosolic factor 1 (Ncf1) were detected. These major sources of reactive oxygen species (ROS) are known to aggravate cellular inflammation and injury [76]. To combat oxidative stress, neuroprotective responses included increased expression levels of clusterin (Clu), peroxiredoxin family antioxidants (Prdx), glutamine synthetase (Glul), and superoxide dismutase 1 (Sod1). Heightened levels of ROS also led to upregulation of ceruloplasmin (Cp) in our study. Cp protein is often elevated in inflammation and known to scavenge for superoxide anions to prevent the oxidation of lipids. As a ferroxidase, Cp is also crucial for regulating intracellular iron levels [77-79]. This is clinically relevant to retinal health, since mutations in Cp result in the toxic interaction of excess iron and ROS that gives rise to neural and retinal degeneration [78].
Increased expression of cellular adhesion molecules
Within the rd10 retina, one of the strongest enrichments we detected in genes with increased expression was for cellular adhesion related processes. We observed increased VEGF-mediated expression of E-selectin (Sele), vascular cell adhesion molecule 1 (Vcam1), but most significantly a more than fourfold increase in intercellular adhesion molecule 1 (Icam1). ICAM1 promotes leukocyte adhesion to the retinal vasculature, inducing cytokine activation and initiating inflammation [80]. Increased relative expression of Vcam1, an adhesion molecule that promotes neovascularization under oxidative stress, was also observed [81]. Additionally, increased relative expression of cell surface adhesion receptor Cd44 was also found. This was consistent with findings in studies analyzing rds and light-induced murine models of retinal degeneration, in which Cd44 protein and protein distribution were increased throughout the retina [82]. CD44 is involved in angiogenesis and suggested to contribute to vascular endothelial stability. Changes in Cd44 expression markedly impact leukocyte-endothelial interactions and subsequent leukocyte migration into the bloodstream [83]. Relatively little is known about the role of adhesion molecules in the progression of RP [84], but mutations in two adhesion genes (protocadherin 15 and cadherin 23) cause Usher syndrome, a disorder leading to hearing and vision loss highly similar to RP [85], as well as other disorders affecting retinal integrity [84]. Our data also showed increased expression of cadherin 11 and protocadherins 7, B5, 9, and 17, indicating an important role for cell adhesion molecules and the potential for regulation of cellular adhesion to contain retinal inflammation.
Immune response and microglial activation
Within the eye, the innate immune response involves photoreceptor- and Müller glia-mediated microglial activation, in which resident macrophages are recruited to phagocytose cellular debris [86]. In RP, this process has been linked to toll-like receptor (Tlr) proteins, pattern recognition receptors that stimulate innate immune cells through MAPK and nuclear factor-kB (NF-kB) pathways [87]. Here, diseased mice demonstrated elevated Tlr2, 3, 4, 5, 9, and 13 levels. In a recent study, Tlr2 and Tlr4 receptor increases were attributed to the release of dying photoreceptor proteins [86]. Furthermore, microglial phagocytosis of retinal proteins resulted in the production of inflammatory chemokines and cytokines that aggravated retinal cell death. In our study, enhanced expression of chemokines (Ccl2, Ccl5, Tnfaip1, Cxcl1, Cxcl16) and cytokines from the transforming growth factor beta family (Tgfβ1, Tgfβ2, Tgfβ3) support this claim. We also report increased expression in suppressor of cytokine signaling (Socs) and tissue inhibitors of metalloproteinases (Timp) genes involved in countering cytopathic damage. SOCS proteins regulate the intensity and duration of cytokine signals, while TIMP proteins activate pathways that curb inflammation and cytokine biosynthesis [88,89]. In our findings, we observed enriched Socs2, 3, and 5 alongside increased relative expression of Timp1, 2, 3, and 4. Recently, induction of Socs was also linked to Vegf expression under hypoxic conditions, suggesting that SOCS proteins may also mitigate oxidative stress and confer neuroprotective abilities [88]. To much surprise, we did not find increased expression of the proinflammatory cytokines Ifnγ or Il4, whose protein products are normally produced and abundantly secreted during retinal insult [88].
Remodeling of the inner retina
RP primarily affects rods and cones in the outer retina, but substantial evidence indicates that the inner retinal network of bipolar, amacrine, and ganglion cells also undergo changes once photoreceptor afferents are lost [66,90,91]. These changes in morphology include inner retinal reorganization as well as neuronal sprouting, dendritic arborization, and synaptogenesis [92]. Our data support retinal remodeling, showing changes in splicing and increased relative expression for genes associated with neurogenesis and structural support. In particular, significant changes in Pten [93], Gpr98 [94,95], and Crb [96,97] were observed. These have established connections to RP and similar retinal dystrophies [98,99]. Phosphate and tensin homolog (Pten) encodes a lipid and protein phosphatase that functions in cell migration and cell positioning during embryonic development [93,100]. Similarly, Gpr98 (Vlgr1) encodes for a large G-protein coupled receptor, highly expressed during neural development, with levels diminishing following neurogenesis [99,101]. However, it is unclear whether upregulated Pten and Gpr98 reflect additional functions of maintenance and neural repair during late RP.
In the mouse retina, Crumbs (Crb) polarity proteins interact with adherens junction proteins at the external limiting membrane, situated between photoreceptors and Müller glia [98]. Crb1 and Crb2 are essential to photoreceptor morphogenesis and the integrity of the external limiting membrane. Mice lacking the two proteins display severe retinal disorganization, abnormal layering, and mislocalized cells within retinal nuclear layers. The removal of Crb2 from mouse photoreceptors has also been linked to retinal degeneration [98,102,103]. In our study, the observed decrease in relative expression of Crb1 and Crb2 may point to a loss of adhesion between photoreceptors and Müller glia, signifying collapse of the external limiting membrane and phase 2 remodeling [92,98]. This breakdown subsequently alters Müller-neuron interactions that span the retina. As a result, Müller cell hypertrophy and migration to sites of injury is said to lead inner neuronal remodeling [92]. However, additional cell-specific studies will be needed to disambiguate downregulation of Crb1 and 2 from changes in the relative abundance of cell types between the wild-type and rd10 retinas.
Given this knowledge, we also analyzed genes associated with the extracellular matrix (ECM), an organized scaffold of proteins and carbohydrates that interact with nearby cells [104]. We discovered increased relative expression of inhibitory ECM genes such as Cd44 and Ncan, which influence cell migration and inhibit axon growth and synapse formation. Paired alongside heightened levels of cell adhesion molecules, this environment of inhibitory ECM molecules becomes a significant barrier to neuronal regeneration, particularly in areas of glial scarring [105]. In response, we discovered significantly increased relative expression of Mmp2 and 9, gelatinase matrix metalloproteinase (Mmp) genes whose products participate in extracellular matrix proteolysis, removal of inhibitory basement membrane molecules, and vascular remodeling [105]. Increased Mmp expression has also been found in the rd1 mouse as well as in patients with macular degeneration [106,107]. MMP2 also breaks down CD44 and NCAN, proving key to neuronal remodeling [105]. However, increased expression of tissue inhibitors of metalloproteinases (Timp) 1, 2, 3, and 4, suggest attempts to tightly regulate ECM degradation. ECM dynamics are also influenced by the expression of intermediate and microfilament-related genes such as plectin (Plec), vimentin (Vim), and stathmin1 (Stmn1), whose expression levels were also increased [59]. Altogether, these data reflect the complex interactions contributing to ECM stability and the importance of the ECM in retinal architecture.
As a whole, gaps in the current understanding of neural-glial interactions and reorganization are of concern since many retinal therapeutics operate under the assumption that inner retinal cells are viable in late stages of degeneration. These assumptions overlook the complex nature of retinal circuitry and retinal cell interdependence from structural and metabolic perspectives. Thus, future studies examining cell migration at varying stages of RP, as well as within specific inner retinal cells, will be crucial to our comprehensive understanding of rod-cone degeneration and RP retinal dystrophies.
Acknowledgments
Funding provided by Burroughs Wellcome Fund CASI award. Dr. Andrew D. Smith (andrewds@usc.edu) and Dr. Alan Horsager (horsager@usc.edu) are co-corresponding authors for this paper.
Appendix 1. Genes found to be differentially expressed between rd10 and wild-type mouse retina.
To access the data, click or select the words “Appendix 1.”
Appendix 2. Gene ontology terms associated with genes found to have increased relative expression in rd10 retina.
To access the data, click or select the words “Appendix 2.”
Appendix 3. Exons found to have changed relative inclusion rates in rd10 retina from wild-type.
To access the data, click or select the words “Appendix 3.”
References
- 1.Ferrari S, Iorio ED, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis Pigmentosa: Genes and Disease Mechanisms. Curr Genomics. 2011;12:238–49. doi: 10.2174/138920211795860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Soest S, Westerveld A, De Jong PTVM, Bleeker-Wagemakers EM, Bergen AAB. Retinitis Pigmentosa: Defined From a Molecular Point of View. Surv Ophthalmol. 1999;43:321–34. doi: 10.1016/s0039-6257(98)00046-0. [DOI] [PubMed] [Google Scholar]
- 3.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–8. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos A, Humayun MS, de Juan E, Jr, Greenburg RJ, Marsh MJ, Klock IB, Milam AH. Preservation of the inner retina in retinitis pigmentosa: A morphometric analysis. Arch Ophthalmol. 1997;115:511–5. doi: 10.1001/archopht.1997.01100150513011. [DOI] [PubMed] [Google Scholar]
- 5.Humayun MS, Prince M, de Juan E, Barron Y, Moskowitz M, Klock IB, Milam AH. Morphometric analysis of the extramacular retina from postmortem eyes with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1999;40:143–8. [PubMed] [Google Scholar]
- 6.Strettoi E, Pignatelli V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2000;97:11020–5. doi: 10.1073/pnas.190291097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marc RE, Jones BW. Retinal remodeling in inherited photoreceptor degenerations. Mol Neurobiol. 2003;28:139–47. doi: 10.1385/MN:28:2:139. [DOI] [PubMed] [Google Scholar]
- 8.Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. J Comp Neurol. 2007;500:222–38. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li ZY, Wong F, Chang JH, Possin DE, Hao Y, Petters RM, Milam AH. Rhodopsin transgenic pigs as a model for human retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1998;39:808–19. [PubMed] [Google Scholar]
- 10.Peng Y-W, Hao Y, Petters RM, Wong F. Ectopic synaptogenesis in the mammalian retina caused by rod photoreceptor-specific mutations. Nat Neurosci. 2000;3:1121–7. doi: 10.1038/80639. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan RKP, WoldeMussie E, Pow DV. Dendritic and Synaptic Plasticity of Neurons in the Human Age-Related Macular Degeneration Retina. Invest Ophthalmol Vis Sci. 2007;48:2782–91. doi: 10.1167/iovs.06-1283. [DOI] [PubMed] [Google Scholar]
- 12.Furukawa T, Morrow EM, Li T, Davis FC, Cepko CL. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet. 1999;23:466–70. doi: 10.1038/70591. [DOI] [PubMed] [Google Scholar]
- 13.Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–25. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- 14.Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and Functional Abnormalities in the Inner Retina of the rd/rd Mouse. J Neurosci. 2002;22:5492–504. doi: 10.1523/JNEUROSCI.22-13-05492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strettoi E, Pignatelli V, Rossi C, Porciatti V, Falsini B. Remodeling of second-order neurons in the retina of rd/rd mutant mice. Vision Res. 2003;43:867–77. doi: 10.1016/s0042-6989(02)00594-1. [DOI] [PubMed] [Google Scholar]
- 16.Stone JL, Barlow WE, Humayun MS, de Juan E, Jr, Milam AH. Morphometric analysis of macular photoreceptors and ganglion cells in retinas with retinitis pigmentosa. Arch Ophthalmol. 1992;110:1634–9. doi: 10.1001/archopht.1992.01080230134038. [DOI] [PubMed] [Google Scholar]
- 17.Margolis DJ, Newkirk G, Euler T, Detwiler PB. Functional Stability of Retinal Ganglion Cells after Degeneration-Induced Changes in Synaptic Input. J Neurosci. 2008;28:6526–36. doi: 10.1523/JNEUROSCI.1533-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varela C, Igartua I, De la Rosa EJ, De la Villa P. Functional modifications in rod bipolar cells in a mouse model of retinitis pigmentosa. Vision Res. 2003;43:879–85. doi: 10.1016/s0042-6989(02)00493-5. [DOI] [PubMed] [Google Scholar]
- 19.Marc RE, Jones BW, Anderson JR, Kinard K, Marshak DW, Wilson JH, Wensel T, Lucas RJ. Neural Reprogramming in Retinal Degeneration. Invest Ophthalmol Vis Sci. 2007;48:3364–71. doi: 10.1167/iovs.07-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhingra A, Jiang M, Wang T-L, Lyubarsky A, Savchenko A, Bar-Yehuda T, Sterling P, Birnbaumer L, Vardi N. Light Response of Retinal ON Bipolar Cells Requires a Specific Splice Variant of Gαo. J Neurosci. 2002;22:4878–84. doi: 10.1523/JNEUROSCI.22-12-04878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stasheff SF. Emergence of Sustained Spontaneous Hyperactivity and Temporary Preservation of off Responses in Ganglion Cells of the Retinal Degeneration (rd1) Mouse. J Neurophysiol. 2008;99:1408–21. doi: 10.1152/jn.00144.2007. [DOI] [PubMed] [Google Scholar]
- 22.Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–55. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 23.Ye JH, Goo YS. The slow wave component of retinal activity in rd / rd mice recorded with a multi-electrode array. Physiol Meas. 2007;28:1079. doi: 10.1088/0967-3334/28/9/009. [DOI] [PubMed] [Google Scholar]
- 24.Lagali PS, Balya D, Awatramani GB, Munch TA, Kim DS, Busskamp V, Cepko CL, Roska B. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci. 2008;11:667–75. doi: 10.1038/nn.2117. [DOI] [PubMed] [Google Scholar]
- 25.Doroudchi MM, Greenberg KP, Liu J, Silka KA, Boyden ES, Lockridge JA, Arman AC, Janani R, Boye SE, Boye SL, Gordon GM, Matteo BC, Sampath AP, Hauswirth WW, Horsager A. Virally delivered Channelrhodopsin-2 Safely and Effectively Restores Visual Function in Multiple Mouse Models of Blindness. Mol Ther. 2011;19:1220–9. doi: 10.1038/mt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humayun MS, de Juan E, Jr, Dagnelie G, Greenberg RJ, Propst RH, Phillips D. VIsual perception elicited by electrical stimulation of retina in blind humans. Arch Ophthalmol. 1996;114:40–6. doi: 10.1001/archopht.1996.01100130038006. [DOI] [PubMed] [Google Scholar]
- 27.Horsager A, Greenwald SH, Weiland JD, Humayun MS, Greenberg RJ, McMahon MJ, Boynton GM, Fine I. Predicting Visual Sensitivity in Retinal Prosthesis Patients. Invest Ophthalmol Vis Sci. 2009;50:1483–91. doi: 10.1167/iovs.08-2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–8. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 29.Cai L, Lyu YL. Analysis of Retinal Development and Diseases Using RNA-Seq. Cell Dev Biol. 2012;1:e113. doi: 10.4172/2168-9296.1000e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks MJ, Rajasimha HK, Swaroop A. Retinal Transcriptome Profiling by Directional Next-Generation Sequencing Using 100 ng of Total RNA number. T Retin Dev. 2012: p. 319–34. [DOI] [PubMed] [Google Scholar]
- 31.Gamsiz ED, Ouyang Q, Schmidt M, Nagpal S, Morrow EM. Genome-wide transcriptome analysis in murine neural retina using high-throughput RNA sequencing. Genomics. 2012;99:44–51. doi: 10.1016/j.ygeno.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholl HPN, Issa PC, Walier M, Janzer S, Pollok-Kopp B, Börncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, Holz FG, Weber BHF, Oppermann M. Systemic Complement Activation in Age-Related Macular Degeneration. PLoS ONE. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT, Nusinowitz S, Rengarajan K, Boyd AP, Starr SS, Chaudhury RC, Nickerson JM, Heckenlively JR, Boatright JH. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007;47:624–33. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis RJ, Tosi J, Janisch KM, Kasanuki JM, Wang N-K, Kong J, Tsui I, Cilluffo M, Woodruff ML, Fain GL, Lin C-S, Tsang SH. Functional Rescue of Degenerating Photoreceptors in Mice Homozygous for a Hypomorphic cGMP Phosphodiesterase 6 b Allele (Pde6bH620Q). Invest Ophthalmol Vis Sci. 2008;49:5067–76. doi: 10.1167/iovs.07-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith AD, Chung W-Y, Hodges E, Kendall J, Hannon G, Hicks J, Xuan Z, Zhang MQ. Updates to the RMAP short-read mapping software. Bioinformatics. 2009;25:2841–2. doi: 10.1093/bioinformatics/btp533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanamori M, Konno H, Osato N, Kawai J, Hayashizaki Y, Suzuki H. A genome-wide and nonredundant mouse transcription factor database. Biochem Biophys Res Commun. 2004;322:787–93. doi: 10.1016/j.bbrc.2004.07.179. [DOI] [PubMed] [Google Scholar]
- 39.Galante PAF, Sandhu D, de Sousa Abreu R, Gradassi M, Slager N, Vogel C, Jose de Souza S, Penalva LOF. A comprehensive in silico expression analysis of RNA binding proteins in normal and tumor tissue; identification of potential players in tumor formation. RNA Biol. 2009;6:426–33. doi: 10.4161/rna.6.4.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Phillips MJ, Otteson DC, Sherry DM. Progression of neuronal and synaptic remodeling in the rd10 mouse model of retinitis pigmentosa. J Comp Neurol. 2010;518:2071–89. doi: 10.1002/cne.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwong JMK, Caprioli J, Piri N. RNA Binding Protein with Multiple Splicing: A New Marker for Retinal Ganglion Cells. Invest Ophthalmol Vis Sci. 2010;51:1052–8. doi: 10.1167/iovs.09-4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Squire J, Zhou A, Hassel BA, Nie H, Silverman RH. Localization of the Interferon-Induced, 2–5A-Dependent RNase Gene (RNS4) to Human Chromosome 1q25. Genomics. 1994;19:174–5. doi: 10.1006/geno.1994.1033. [DOI] [PubMed] [Google Scholar]
- 44.Mehta A, Kinter MT, Sherman NE, Driscoll DM. Molecular Cloning of Apobec-1 Complementation Factor, a Novel RNA-Binding Protein Involved in the Editing of Apolipoprotein B mRNA. Mol Cell Biol. 2000;20:1846–54. doi: 10.1128/mcb.20.5.1846-1854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talmud PJ, Converse C, Krul E, Huq L, McIlwaine GG, Series JJ, Boyd P, Schonfeld G, Dunning A, Humphries S. A novel truncated apolipoprotein B (apo B55) in a patient with familial hypobetalipoproteinemia and atypical retinitis pigmentosa. Clin Genet. 1992;42:62–70. doi: 10.1111/j.1399-0004.1992.tb03141.x. [DOI] [PubMed] [Google Scholar]
- 46.Schippert R, Schaeffel F, Feldkaemper MP. Microarray analysis of retinal gene expression in Egr-1 knockout mice. Mol Vis. 2009;15:2720–39. [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma YV, Cojocaru RI, Ritter LM, Khattree N, Brooks M, Scott A, Swaroop A, Goldberg AFX. Protective Gene Expression Changes Elicited by an Inherited Defect in Photoreceptor Structure. PLoS ONE. 2012;7:e31371. doi: 10.1371/journal.pone.0031371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muto A, Iida A, Satoh S, Watanabe S. The group E Sox genes Sox8 and Sox9 are regulated by Notch signaling and are required for Müller glial cell development in mouse retina. Exp Eye Res. 2009;89:549–58. doi: 10.1016/j.exer.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 49.Shi Z, Trenholm S, Zhu M, Buddingh S, Star EN, Awatramani GB, Chow RL. Vsx1 Regulates Terminal Differentiation of Type 7 ON Bipolar Cells. J Neurosci. 2011;31:13118–27. doi: 10.1523/JNEUROSCI.2331-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valleix S, Nedelec B, Rigaudiere F, Dighiero P, Pouliquen Y, Renard G, Le Gargasson J-F, Delpech M. H244R VSX1 Is Associated with Selective Cone ON Bipolar Cell Dysfunction and Macular Degeneration in a PPCD Family. Invest Ophthalmol Vis Sci. 2006;47:48–54. doi: 10.1167/iovs.05-0479. [DOI] [PubMed] [Google Scholar]
- 51.Barbaro V, Di Iorio E, Ferrari S, Bisceglia L, Ruzza A, De Luca M, Pellegrini G. Expression of VSX1 in Human Corneal Keratocytes during Differentiation into Myofibroblasts in Response to Wound Healing. Invest Ophthalmol Vis Sci. 2006;47:5243–50. doi: 10.1167/iovs.06-0185. [DOI] [PubMed] [Google Scholar]
- 52.Coppieters F, Leroy BP, Beysen D, Hellemans J, De Bosscher K, Haegeman G, Robberecht K, Wuyts W, Coucke PJ, De Baere E. Recurrent Mutation in the First Zinc Finger of the Orphan Nuclear Receptor NR2E3 Causes Autosomal Dominant Retinitis Pigmentosa. Am J Hum Genet. 2007;81:147–57. doi: 10.1086/518426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber S, Rozet JM, Takezawa SI, dos Santos LC, Lopes L, Gribouval O, Penet C, Perrault I, Ducroq D, Souied E, Jeanpierre M, Romana S, Frézal J, Ferraz F, Yu-Umesono R, Munnich A, Kaplan J. The photoreceptor cell-specific nuclear receptor gene (PNR) accounts for retinitis pigmentosa in the Crypto-Jews from Portugal (Marranos), survivors from the Spanish Inquisition. Hum Genet. 2000;107:276–84. doi: 10.1007/s004390000350. [DOI] [PubMed] [Google Scholar]
- 54.Zhang C-L, Zou Y, Yu RT, Gage FH, Evans RM. Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev. 2006;20:1308–20. doi: 10.1101/gad.1413606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramachandran R, Zhao X-F, Goldman D. Insm1a-mediated gene repression is essential for the formation and differentiation of Müller glia-derived progenitors in the injured retina. Nat Cell Biol. 2012;14:1013–23. doi: 10.1038/ncb2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wan J, Ramachandran R, Goldman D. HB-EGF Is Necessary and Sufficient for M¸ller Glia Dedifferentiation and Retina Regeneration. Dev Cell. 2012;22:334–47. doi: 10.1016/j.devcel.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dyer MA, Cepko CL. Control of Müller glial cell proliferation and activation following retinal injury. Nat Neurosci. 2000;3:873–80. doi: 10.1038/78774. [DOI] [PubMed] [Google Scholar]
- 58.Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 59.Roesch K, Stadler MB, Cepko CL. Gene expression changes within Müller glial cells in retinitis pigmentosa. Mol Vis. 2012;18:1197–214. [PMC free article] [PubMed] [Google Scholar]
- 60.Ghai K, Zelinka C, Fischer AJ. Notch Signaling Influences Neuroprotective and Proliferative Properties of Mature Müller Glia. J Neurosci. 2010;30:3101–12. doi: 10.1523/JNEUROSCI.4919-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischer AJ, Scott MA, Tuten W. Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia. 2009;57:166–81. doi: 10.1002/glia.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang Y, Ding Q, Xie X, Libby RT, Lefebvre V, Gan L. Transcription Factors SOX4 and SOX11 Function Redundantly to Regulate the Development of Mouse Retinal Ganglion Cells. J Biol Chem. 2013;288:18429–38. doi: 10.1074/jbc.M113.478503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Surzenko N, Crowl T, Bachleda A, Langer L, Pevny L. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Müller glia. Development. 2013;140:1445–56. doi: 10.1242/dev.071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mizeracka K, DeMaso CR, Cepko CL. Notch1 is required in newly postmitotic cells to inhibit the rod photoreceptor fate. Development. 2013;140:3188–97. doi: 10.1242/dev.090696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swaroop A, Kim D, Forrest D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci. 2010;11:563–76. doi: 10.1038/nrn2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mullins RF, Kuehn MH, Radu RA, Enriquez GS, East JS, Schindler EI, Travis GH, Stone EM. Autosomal Recessive Retinitis Pigmentosa Due To ABCA4 Mutations: Clinical, Pathologic, and Molecular Characterization. Invest Ophthalmol Vis Sci. 2012;53:1883–94. doi: 10.1167/iovs.12-9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006;83:380–9. doi: 10.1016/j.exer.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 68.Guimarães CA, Benchimol M, Amarante-Mendes GP, Linden R. Alternative Programs of Cell Death in Developing Retinal Tissue. J Biol Chem. 2003;278:41938–46. doi: 10.1074/jbc.M306547200. [DOI] [PubMed] [Google Scholar]
- 69.Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2012;1824:22–33. doi: 10.1016/j.bbapap.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 70.Huang C-C, Chen K-L, Cheung CHA, Chang J-Y. Autophagy induced by cathepsin S inhibition induces early ROS production, oxidative DNA damage, and cell death via xanthine oxidase. Free Radic Biol Med. 2013;65:1473–86. doi: 10.1016/j.freeradbiomed.2013.07.020. [DOI] [PubMed] [Google Scholar]
- 71.Komeima K, Rogers BS, Lu L, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2006;103:11300–5. doi: 10.1073/pnas.0604056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen J, Yang X, Dong A, Petters RM, Peng Y-W, Wong F, Campochiaro PA. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. 2005;203:457–64. doi: 10.1002/jcp.20346. [DOI] [PubMed] [Google Scholar]
- 73.Copello M, Menéndez S, Hernández F. Ozone Therapy in Retinitis Pigmentosa Patients: Clinical Evolution and Oxidative Stress Behavior in Retinitis Pigmentosa Patients Treated with Ozone Therapy over 20 Years. Ozone Sci Eng. 2012;34:476–83. [Google Scholar]
- 74.Kim I, Moon S-O, Hoon Kim S, Jin Kim H, Soon Koh Y, Young Koh G. Vascular Endothelial Growth Factor Expression of Intercellular Adhesion Molecule 1 (ICAM-1), Vascular Cell Adhesion Molecule 1 (VCAM-1), and E-selectin through Nuclear Factor-κB Activation in Endothelial Cells. J Biol Chem. 2001;276:7614–20. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 75.Hughes JM, Groot AJ, van der Groep P, Sersansie R, Vooijs M, van Diest PJ, Van Noorden CJF, Schlingemann RO, Klaassen I. Active HIF-1 in the Normal Human Retina. J Histochem Cytochem. 2010;58:247–54. doi: 10.1369/jhc.2009.953786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sedeek M, Nasrallah R, Touyz RM, Hébert RL. NADPH Oxidases, Reactive Oxygen Species, and the Kidney: Friend and Foe. J Am Soc Nephrol. 2013;24:1512–8. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim JH, Kim JH, Jun HO, Yu YS, Min BH, Park KH, Kim K-W. Protective Effect of Clusterin from Oxidative Stress–Induced Apoptosis in Human Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci. 2010;51:561–6. doi: 10.1167/iovs.09-3774. [DOI] [PubMed] [Google Scholar]
- 78.Harned J, Ferrell J, Nagar S, Goralska M, Fleisher LN, McGahan MC. Ceruloplasmin alters intracellular iron regulated proteins and pathways: Ferritin, transferrin receptor, glutamate and hypoxia-inducible factor-1α. Exp Eye Res. 2012;97:90–7. doi: 10.1016/j.exer.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tapryal N, Mukhopadhyay C, Das D, Fox PL, Mukhopadhyay CK. Reactive Oxygen Species Regulate Ceruloplasmin by a Novel mRNA Decay Mechanism Involving Its 3′-Untranslated Region: IMPLICATIONS IN NEURODEGENERATIVE DISEASES. J Biol Chem. 2009;284:1873–83. doi: 10.1074/jbc.M804079200. [DOI] [PubMed] [Google Scholar]
- 80.Lu M, Perez VL, Ma N, Miyamoto K, Peng HB, Liao JK, Adamis AP. VEGF Increases Retinal Vascular ICAM-1 Expression In Vivo. Invest Ophthalmol Vis Sci. 1999;40:1808–12. [PubMed] [Google Scholar]
- 81.Dong A, Shen J, Zeng M, Campochiaro PA. Vascular cell-adhesion molecule-1 plays a central role in the proangiogenic effects of oxidative stress. Proc Natl Acad Sci USA. 2011;108:14614–9. doi: 10.1073/pnas.1012859108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krishnamoorthy R, Agarwal N, Chaitin MH. Upregulation of CD44 expression in the retina during the rds degeneration. Brain Res Mol Brain Res. 2000;77:125–30. doi: 10.1016/s0169-328x(00)00035-8. [DOI] [PubMed] [Google Scholar]
- 83.Cao G, Savani RC, Fehrenbach M, Lyons C, Zhang L, Coukos G, DeLisser HM. Involvement of Endothelial CD44 during in Vivo Angiogenesis. Am J Pathol. 2006;169:325–36. doi: 10.2353/ajpath.2006.060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.El-Amraoui A, Petit C. Cadherins as Targets for Genetic Diseases. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER. Mutations of the Protocadherin Gene PCDH15 Cause Usher Syndrome Type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kohno H, Chen Y, Kevany BM, Pearlman E, Miyagi M, Maeda T, Palczewski K, Maeda A. Photoreceptor Proteins Initiate Microglial Activation via Toll-like Receptor 4 in Retinal Degeneration Mediated by All-trans-retinal. J Biol Chem. 2013;288:15326–41. doi: 10.1074/jbc.M112.448712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arthur JSC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–92. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 88.Egwuagu C, Yu C-H, Mahdi R, Mameza M, Eseonu C, Takase H, Ebong S. Cytokine-Induced Retinal Degeneration: Role of Suppressors of Cytokine Signaling (SOCS) Proteins in Protection of the Neuroretina. In: Hollyfield J, Anderson R, LaVail M, editors. Retin Degenerative Dis. Vol 572: Springer US; 2006. p. 275–81. [DOI] [PubMed] [Google Scholar]
- 89.Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13:649–65. doi: 10.1038/nri3499. [DOI] [PubMed] [Google Scholar]
- 90.Jacobson SG, Sumaroka A, Aleman TS, Cideciyan AV, Danciger M, Farber DB. Evidence for retinal remodelling in retinitis pigmentosa caused by PDE6B mutation. Br J Ophthalmol. 2007;91:699–701. doi: 10.1136/bjo.2006.104463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O'Brien EE, Fletcher EL, Meffin H, Burkitt AN, Grayden DB, Greferath U. Viability of the inner retina in a novel mouse model of retinitis pigmentosa. Eng Med Biol Soc Annu Int Conf; 2010 Aug. 31 2010-Sept. 4 2010. p. 553–6. [DOI] [PubMed] [Google Scholar]
- 92.Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–806. [PMC free article] [PubMed] [Google Scholar]
- 93.Sakagami K, Chen B, Nusinowitz S, Wu H, Yang X-J. PTEN regulates retinal interneuron morphogenesis and synaptic layer formation. Mol Cell Neurosci. 2012;49:171–83. doi: 10.1016/j.mcn.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hilgert N, Kahrizi K, Dieltjens N, Bazazzadegan N, Najmabadi H, Smith RJH, Van Camp G. A large deletion in GPR98 causes type IIC Usher syndrome in male and female members of an Iranian family. J Med Genet. 2009;46:272–6. doi: 10.1136/jmg.2008.060947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hmani-Aifa M, Benzina Z, Zulfiqar F, Dhouib H, Shahzadi A, Ghorbel A, Rebai A, Soderkvist P, Riazuddin S, Kimberling WJ, Ayadi H. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur J Hum Genet. 2009;17:474–82. doi: 10.1038/ejhg.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lotery AJ, Malik A, Shami SA, Sindhi M, Chohan B, Maqbool C, Moore PA, Denton MJ, Stone EM. CRB1 mutations may result in retinitis pigmentosa without para-arteriolar RPE preservation. Ophthalmic Genet. 2001;22:163–9. doi: 10.1076/opge.22.3.163.2222. [DOI] [PubMed] [Google Scholar]
- 97.den Hollander AI, ten Brink JB, de Kok YJM, van Soest S, van den Born LI, van Driel MA, van de Pol DJR, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FPM, Bergen AAB. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23:217–21. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 98.Alves CH, Pellissier LP, Wijnholds J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog Retin Eye Res. 2014;40:35–52. doi: 10.1016/j.preteyeres.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 99.Schwartz SB, Aleman TS, Cideciyan AV, Windsor EAM, Sumaroka A, Roman AJ, Rane T, Smilko EE, Bennett J, Stone EM, Kimberling WJ, Liu X-Z, Jacobson SG. Disease Expression in Usher Syndrome Caused by VLGR1 Gene Mutation (USH2C) and Comparison with USH2A Phenotype. Invest Ophthalmol Vis Sci. 2005;46:734–43. doi: 10.1167/iovs.04-1136. [DOI] [PubMed] [Google Scholar]
- 100.Cantrup R, Dixit R, Palmesino E, Bonfield S, Shaker T, Tachibana N, Zinyk D, Dalesman S, Yamakawa K, Stell WK, Wong RO, Reese BE, Kania A, Sauvé Y, Schuurmans C. Cell-Type Specific Roles for PTEN in Establishing a Functional Retinal Architecture. PLoS ONE. 2012;7:e32795. doi: 10.1371/journal.pone.0032795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McMillan DR, Kayes-Wandover KM, Richardson JA, White PC. Very Large G Protein-coupled Receptor-1, the Largest Known Cell Surface Protein, Is Highly Expressed in the Developing Central Nervous System. J Biol Chem. 2002;277:785–92. doi: 10.1074/jbc.M108929200. [DOI] [PubMed] [Google Scholar]
- 102.Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, Bechtold L, Haider NB, Tepass U, Heckenlively JR, Chang B, Naggert JK, Nishina PM. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003;12:2179–89. doi: 10.1093/hmg/ddg232. [DOI] [PubMed] [Google Scholar]
- 103.Alves CH, Pellissier LP, Vos RM, Garcia Garrido M, Sothilingam V, Seide C, Beck SC, Klooster J, Furukawa T, Flannery JG, Verhaagen J, Seeliger MW, Wijnholds J. Targeted ablation of Crb2 in photoreceptor cells induces retinitis pigmentosa. Hum Mol Genet. 2014;23:3384–401. doi: 10.1093/hmg/ddu048. [DOI] [PubMed] [Google Scholar]
- 104.Al-Ubaidi MR, Naash MI, Conley SM. A Perspective on the Role of the Extracellular Matrix in Progressive Retinal Degenerative Disorders. Invest Ophthalmol Vis Sci. 2013;54:8119–24. doi: 10.1167/iovs.13-13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tucker B, Klassen H, Yang L, Chen DF, Young MJ. Elevated MMP Expression in the MRL Mouse Retina Creates a Permissive Environment for Retinal Regeneration. Invest Ophthalmol Vis Sci. 2008;49:1686–95. doi: 10.1167/iovs.07-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meighan PC, Meighan SE, Rich ED, Brown RL, Varnum MD. Matrix metalloproteinase-9 and −2 enhance the ligand sensitivity of photoreceptor cyclic nucleotide-gated channels. Channels. 2012;6:181–96. doi: 10.4161/chan.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Plantner JJ, Jiang C, Smine A. Increase in Interphotoreceptor Matrix Gelatinase A (MMP-2) Associated with Age-related Macular Degeneration. Exp Eye Res. 1998;67:637–45. doi: 10.1006/exer.1998.0552. [DOI] [PubMed] [Google Scholar]
- 108.Fakin A, Zupan A, Glavač D, Hawlina M. Combination of retinitis pigmentosa and hearing loss caused by a novel mutation in PRPH2 and a known mutation in GJB2: Importance for differential diagnosis of Usher syndrome. Vision Res. 2012;75:71–6. doi: 10.1016/j.visres.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 109.Wada Y, Abe T, Takeshita T, Sato H, Yanashima K, Tamai M. Mutation of Human Retinal Fascin Gene (FSCN2) Causes Autosomal Dominant Retinitis Pigmentosa. Invest Ophthalmol Vis Sci. 2001;42:2395–400. [PubMed] [Google Scholar]
- 110.Makino CL, Peshenko IV, Wen X-H, Olshevskaya EV, Barrett R, Dizhoor AM. A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J Biol Chem. 2008;283:29135–43. doi: 10.1074/jbc.M804445200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sato M, Nakazawa M, Usui T, Tanimoto N, Abe H, Ohguro H. Mutations in the gene coding for guanylate cyclase-activating protein 2 (GUCA1B gene) in patients with autosomal dominant retinal dystrophies. Graefes Arch Clin Exp Ophthalmol. 2005;243:235–42. doi: 10.1007/s00417-004-1015-7. [DOI] [PubMed] [Google Scholar]
- 112.Roger JE, Ranganath K, Zhao L, Cojocaru RI, Brooks M, Gotoh N, Veleri S, Hiriyanna A, Rachel RA, Campos MM, Fariss RN, Wong WT, Swaroop A. Preservation of Cone Photoreceptors after a Rapid yet Transient Degeneration and Remodeling in Cone-Only Nrl−/− Mouse Retina. J Neurosci. 2012;32:528–41. doi: 10.1523/JNEUROSCI.3591-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dvir L, Srour G, Abu-Ras R, Miller B, Shalev SA, Ben-Yosef T. Autosomal-Recessive Early-Onset Retinitis Pigmentosa Caused by a Mutation in PDE6G, the Gene Encoding the Gamma Subunit of Rod cGMP Phosphodiesterase. Am J Hum Genet. 2010;87:258–64. doi: 10.1016/j.ajhg.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nakazawa M, Wada Y, Tamai M. ARrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch Ophthalmol. 1998;116:498–501. doi: 10.1001/archopht.116.4.498. [DOI] [PubMed] [Google Scholar]
- 115.Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–6. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 116.Beetz N, Hein L. The physiological roles of phosducin: from retinal function to stress-dependent hypertension. Cell Mol Life Sci. 2011;68:599–612. doi: 10.1007/s00018-010-0550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nishiguchi KM, Berson EL, Dryja TP. Mutation screening of the phosducin gene PDC in patients with retinitis pigmentosa and allied diseases. Mol Vis. 2004;10:62–4. [PubMed] [Google Scholar]