

ABSTRACT
The alphaherpesvirus pseudorabies virus (PRV) encodes a single immediate early gene called IE180. The IE180 protein is a potent transcriptional activator of viral genes involved in DNA replication and RNA transcription. A PRV mutant with both copies of IE180 deleted was constructed 20 years ago (S. Yamada and M. Shimizu, Virology 199:366–375, 1994, doi:10.1006/viro.1994.1134), but propagation of the mutant depended on complementing cell lines that expressed the toxic IE180 protein constitutively. Recently, Oyibo et al. constructed a novel set of PRV IE180 mutants and a stable cell line with inducible IE180 expression (H. Oyibo, P. Znamenskiy, H. V. Oviedo, L. W. Enquist, A. Zador, Front. Neuroanat. 8:86, 2014, doi:10.3389/fnana.2014.00086), which we characterized further here. These mutants failed to replicate new viral genomes, synthesize immediate early, early, or late viral proteins, and assemble infectious virions. The PRV IE180-null mutant did not form plaques in epithelial cell monolayers and could not spread from primary infected neurons to second-order neurons in culture. PRV IE180-null mutants lacked the property of superinfection exclusion. When PRV IE180-null mutants infected cells first, subsequent superinfecting viruses were not blocked in cell entry and formed replication compartments in epithelial cells, fibroblasts, and neurons. Cells infected with PRV IE180-null mutants survived as long as uninfected cells in culture while expressing a fluorescent reporter gene. Transcomplementation with IE180 in epithelial cells restored all mutant phenotypes to wild type. The conditional expression of PRV IE180 protein enables the propagation of replication-incompetent PRV IE180-null mutants and will facilitate construction of long-term single-cell-infecting PRV mutants for precise neural circuit tracing and high-capacity gene delivery vectors.
IMPORTANCE
Pseudorabies virus (PRV) is widely used for neural tracing in animal models. The virus replicates and spreads between synaptically connected neurons. Current tracing strains of PRV are cytotoxic and kill infected cells. Infected cells exclude superinfection with a second virus, limiting multiple virus infections in circuit tracing. By removing the only immediate early gene of PRV (called IE180), the mutant virus will not replicate or spread in epithelial cells, fibroblasts, or neurons. The wild-type phenotype can be restored by transcomplementation of infected cells with IE180. The PRV IE180-null mutant can express fluorescent reporters for weeks in cells with no toxicity; infected cells survive as long as uninfected cells. Infection with the mutant virus allows superinfection of the same cell with a second virus that can enter and replicate. The PRV IE180-null mutant will permit conditional long-term tracing in animals and is a high-capacity vector for gene delivery.
INTRODUCTION
Alphaherpesviruses are neuroinvasive and pantropic pathogens that infect a wide range of mammalian hosts (1). Pseudorabies virus (PRV) is a significant infectious agent in agriculture and veterinary medicine, infecting a remarkably wide range of mammals, including pigs, cows, sheep, goats, horses, rabbits, dogs, cats, rats, and mice but not higher-order primates (2, 3). While most adult pigs survive PRV infection, neonatal pigs and all other infected mammals usually die after only 2 to 3 days, often exhibiting symptoms of Aujeszky’s disease, such as intense pruritus (“mad itch”), ataxia, fevers, and seizures (4). In rodents, virulent strains of PRV induce a lethal inflammatory response as well as pathogenic processes such as synchronized firing of peripheral nervous system neurons (5, 6). As a result of the rapid lethal peripheral inflammatory response, wild-type PRV cannot penetrate very far into the mouse nervous system. However, certain replicating vaccine strains of PRV such as PRV Bartha are attenuated and do not induce the lethal inflammatory response and peripheral symptomology (5, 6). These mutants and their derivatives spread faithfully between synaptically connected neurons, revealing the architecture of a neuronal circuit (7).
In this report, we characterize the in vitro properties of a highly attenuated PRV mutant that should be suitable not only for the precise mapping of neuronal connectivity but also for gene delivery. The PRV IE180 gene codes for the master sole immediate early transcriptional activator of viral genes required for DNA replication and RNA transcription (8). IE180 does not require de novo protein synthesis to be transcribed and is a promiscuous activator of both viral and host transcripts (9). The IE180 protein is 1,460 amino acids long and contains a positively charged RRKRR nuclear localization signal and two ICP4-like domains (10, 11). IE180 is similar to other alphaherpesvirus immediate early (IE) proteins, such as p180 of bovine herpesvirus, IE1 of equid herpesvirus 1, IE40 of varicella-zoster virus, and ICP4 of herpes simplex type 1 virus (12). During PRV infection, IE180 suppresses phosphorylation of translation initiation factor eIF2α by increasing activity of phosphatase PP1 (13). Transgenic mouse lines expressing IE180 showed abnormal spermatogenesis (14) and impaired motor coordination, spatial learning, and memory retrieval (15). Deletion of IE180 from the viral genome should therefore reduce toxicity of PRV in infected cells. IE180 activates transcription of early and late viral genes, and no PRV gene products should be synthesized without IE180. A PRV mutant with both copies of IE180 deleted was constructed 20 years ago, but propagation of the mutant depended on a complementing cell line that expressed the toxic IE180 protein constitutively with the aforementioned consequences (9). Cells expressing PRV IE180 protein are unstable and die after only a few passages. Consequently, it has been difficult to propagate PRV IE180 mutants, and little work on these mutants has been published. However, Oyibo et al. (16) recently described the construction of a stable cell line with conditional expression of PRV IE180 and bacterial artificial chromosome (BAC)-derived PRV IE180-null mutants based on the PRV Becker genome. The same group described the in vivo labeling properties of these mutants and their utility for the precise labeling of neuronal connectivity (16). In the present study, we further characterized the properties of the PRV IE180-null mutant, demonstrating that these mutants do not replicate their genome, synthesize viral proteins, or produce infectious progeny. Remarkably, PRV IE180-infected cells survived as long as uninfected cells. In addition, PRV IE180-null mutants did not block infection by a secondary replication-competent PRV strain (no superinfection exclusion). These findings suggest several applications of PRV IE180-null mutants in neuronal tracing and gene delivery.
RESULTS
PRV IE180-null mutants do not replicate.
DNA replication was assessed by EdU incorporation and click chemistry. The EdU nucleoside analogue is incorporated into nascent DNA and is fluorescently detected by the click reaction (see Materials and Methods). As a positive control, nonconfluent dividing RAT-2 fibroblasts were incubated with EdU. The click reaction detected diffuse EdU signal in the nuclei due to S-phase incorporation. Additionally, mitotic cells showed a stronger EdU signal. A cell in telophase exhibiting robust EdU signal is shown in the top right of the second panel of the first row in Fig. 1A. When mock-infected contact-inhibited RAT-2 cells were grown to confluence and serum starved to prevent DNA replication, no signal was detected (second panel of the second row in Fig. 1A). Infection of confluent cells with wild-type PRV 151 but not PRV 128 (the PRV IE180-null mutant) elicited an EdU signal that colocalized with the nucleus (n > 100) (Fig. 1A). The characteristic pattern of nascent DNA during PRV 151 infection was similar to that previously observed by fluorescent in situ hybridization (17) and differed from the pattern of newly synthesized cellular DNA of dividing uninfected cells (compare the second panel of the first row with the second panel of the third row in Fig. 1A). We also observed nuclear remodeling after PRV 151 infection as large, bright foci in the Hoechst stain that corresponded to sites of exclusion in the EdU stain. In contrast, no nuclear remodeling was observed after PRV 128 infection. PRV 128-infected cells more closely resembled the EdU pattern of mock-infected cells, showing no nascent DNA after viral infection.
FIG 1 .
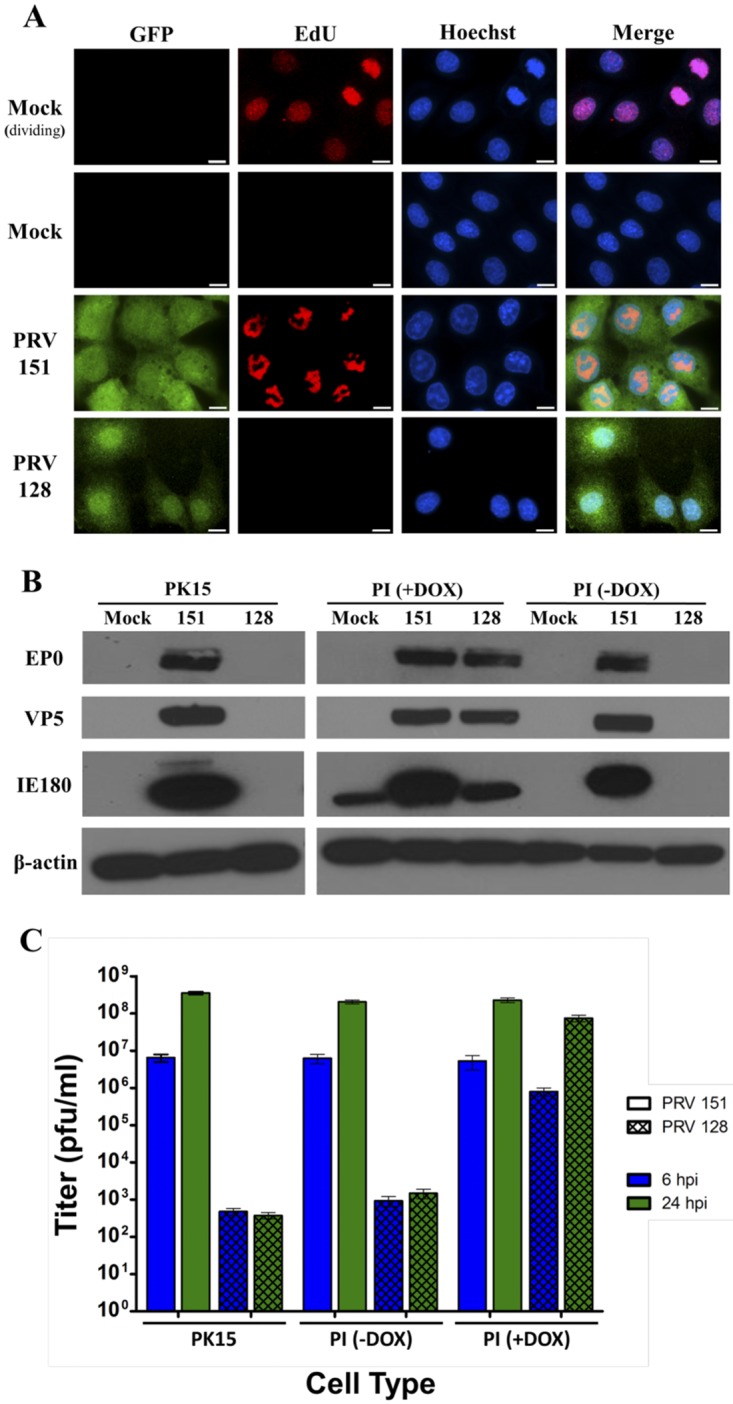
The IE180-null PRV mutant is nonreplicating. (A) DNA-associated click chemistry. Rat-2 fibroblasts were infected with PRV 151 or PRV 128 (green) (MOI = 10) or mock infected and then incubated with EdU. Cells were fixed 6 hours after PRV 151 infection or 16 hours after PRV 128 infection. The click reaction detected incorporation of EdU into newly synthesized DNA (red). The mock (dividing) condition showed S-phase EdU incorporation. For all other conditions, cells were grown to confluence and serum starved to reduce cell division. Nuclei were stained with Hoechst (blue). Scale bars = 10 µm. (B) Western immunoblots. PK15 and PI epithelial cells were infected with PRV 151 or PRV 128 (MOI = 10) or mock infected. PI cells were incubated with or without doxycycline (DOX) during infection. Protein extracts were prepared 16 hours postinfection and blotted with anti-EP0, anti-VP5, anti-IE180 and anti-β-actin antibodies, respectively. (C) Viral titer assay. PK15 and PI epithelial cells were infected with PRV 151 or PRV 128 (MOI = 10). PI cells were incubated with and without doxycycline (DOX) during infection. Infected-cell lysates were harvested 6 and 24 hours postinfection (hpi), and titers were determined. Error bars indicate standard errors of the mean (SEM).
Next, we determined if the loss of the IE180 transcriptional activator abolished PRV early or late gene expression by Western blot analysis. We could not detect any expression of EP0 protein (early transactivator) or the major capsid protein VP5 (late protein) when PK15 and PI (PK15 IE180-expressing) cells were infected with PRV 128 (Fig. 1B), while we saw robust expression after PRV 151 infection. The PI cell line contains the PRV IE180 gene driven by a promoter activated by doxycycline. IE180 protein was detected only when doxycycline was added. IE180 band intensity (Fig. 1B) reflects the amount of exogenous IE180 protein produced in PI cells with doxycycline versus that of replicating PRV 151. As expected, only when PI cells were treated with doxycycline to induce IE180 transcomplementation and then infected with PRV 128 were early EP0 and late VP5 proteins robustly expressed.
We verified that PRV 128 infection of noncomplementing cells produced few, if any, detectable infectious particles (Fig. 1C). Lysates from cells infected with 5 × 106 PFU PRV 128 were collected at 6 and 24 h postinfection (hpi), and titers were determined on PK15 and PI cells. By 24 h, the mean titers measured in PK15 and PI cells without doxycycline were 3.75 × 102 and 1.48 × 103 PFU/ml, respectively. These titers did not increase over time and were significantly lower than the amount of initial infection input (5 × 106 PFU). They probably reflect residual inoculum that adhered to the tissue culture dishes. When PI cells were induced before PRV 128 infection, the resulting titers were comparable to those of PRV 151, and abundant progeny virus was produced (Fig. 1C).
The complemented PRV IE180-null mutant infects noncomplementing cells once and cannot spread.
No plaques formed on PK15 cell lines after low-multiplicity infection with PRV 128 produced on complementing cells. We did see infection of single PK15 and PI cells by expression of the fluorescent viral reporter. However, on induced complementing PI cells, PRV 128 formed fluorescent plaques that were indistinguishable from wild-type virus plaques (Fig. 2A).
FIG 2 .

IE180-null mutant PRV does not exhibit cell-to-cell spread. (A) Assay of PRV spread in epithelial cells. PK15 and PI epithelial cells were infected with PRV 128 (green) (MOI = 0.1). PI cells were incubated with or without doxycycline (DOX) during infection. Infected cells were imaged 48 h postinfection to assess viral spread plaque formation. Scale bars = 25 µm [PK15 and PI (–DOX)] and 250 µm [PI (+DOX)]. (B) Method for assessing viral spread in neurons. The N compartment of a trichamber neuronal culture is treated with green fluorescent virus and red fluorescent lipophilic dye (DiI). The virus and dye travel from the axons to the cell bodies in the S compartment, so first-order infected cell bodies are green and red (yellow). Since not all cell bodies have axons that extend into the N compartment, second-order infected cell bodies from viral spread are green only. Uninfected cell bodies are red only if their axons extend into the N compartment. (C) Assay of PRV spread in primary SCG neurons. Red fluorescent dye (DiI) and green fluorescent virus (PRV 128 or PRV 151; 106 PFU) were added to the N compartment. A field of the S compartment was imaged 48 h postinfection for first-order (red/green) and second-order (green only) infection. Close-ups of cell body clusters are shown for each field. Scale bars = 250 µm (fields) and 25 µm (close-ups).
Spread of PRV 128 between axon-infected primary neurons was assessed using modified Campenot chambers (Fig. 2B). Isolated axons in the N compartment were infected with 106 PFU of PRV 128 or PRV 151. The red fluorescent lipophilic dye DiI was also added to the N compartment, which labeled only cell bodies in the S compartment whose axons penetrated the M compartment and extended into the N compartment. Therefore, first-order infected neurons would be yellow (simultaneous expression of green fluorescent protein [GFP] from PRV 128 or PRV 151 and red DiI). Second-order infected cell bodies in the S compartment would be green, as a result of neuron-to-neuron spread (GFP from viral infection and no red DiI). Uninfected neurons would be colorless or red only (Fig. 2B). We imaged cell bodies in the S compartment 2 days after infection of axons in the N compartment (n = 300 cell bodies). Axonal infection by PRV 151 was efficient; all cell bodies were GFP positive, indicative of viral infection and spread. Cell bodies were either yellow (primary infected neurons) or green (secondary infected neurons). However, after PRV 128 infection, only yellow or colorless cell bodies were detected (Fig. 2C). No cell bodies were green only, confirming that PRV 128 could not spread from neuron to neuron. The data also show that retrograde transport of PRV 128 capsids from axons to cell bodies was efficient, as there were many yellow cell bodies. Taken together, these results suggested that the IE180 gene is necessary for cell-to-cell spread of PRV in both epithelial cells and neurons.
PRV IE180-null mutants do not exclude secondary infection by homologous viruses.
Superinfection exclusion, also known as homologous interference, is a well-documented phenomenon where an established primary viral infection inhibits a subsequent infection with the same or a closely related secondary virus (18). As an example, Banfield et al. showed that the PRV Bartha recombinant PRV 614 establishes superinfection exclusion in dorsal root ganglion neurons (19). Since PRV IE180 mutants deliver their genomes to cells but no further gene expression occurs, it was of interest to determine if the PRV IE180 protein is necessary for superinfection exclusion. We infected SCG, RAT-2, PK15, and PI cells with a primary virus followed by a superinfection with a secondary virus after specific intervals (0, 2, 4, and 8 h). The dual infections consisted of PRV 614 (primary strain) plus PRV 128 (secondary strain) and PRV 128 plus PRV 614. Eight hours after each secondary infection, the cells expressing the primary virus fluorescent reporter were identified and counted (n = 400 cells). Of this population, only cells that also expressed the secondary virus fluorescent reporter were counted. We determined the ratio of the dual-colored cells to the primary-colored cells (Fig. 3A). When cells were simultaneously coinfected with a 1:1 mixture of primary and secondary virus, essentially 100% of cells expressed both reporters. As time between primary and secondary infection increased, the percent of dual-colored cells decreased markedly for PRV 614 plus PRV 128. PRV 614 established complete exclusion of PRV 128 by 8 h in all cell types. In contrast, when PRV 128 was the primary strain, nearly 100% of cells were dual colored even after 8 h postinfection, demonstrating that PRV 128 could not exclude the secondary virus in SCG, RAT-2, PK15, and PI cells (Fig. 3B to E). Remarkably, we observed that 100% of SCG cells that were infected with primary PRV 128 remained permissive to secondary PRV 614 infection 5 weeks later (data not shown). When PI cells were treated with doxycycline to induce IE180 transcomplementation, PRV 128 superinfection exclusion of PRV 614 was restored (Fig. 3F). These data demonstrate that infection by PRV IE180-null mutants does not exclude superinfecting PRV strains.
FIG 3 .

IE180-null mutant PRV does not establish superinfection exclusion. (A) Overview of the superinfection exclusion assay. Cells were infected with fluorescent primary virus and then infected with differently colored fluorescent secondary virus at later time points. Cells were imaged 8 h after secondary infection. Primary infected cells (white circles) were examined for secondary-virus fluorescence. The normalized percentage of dual-colored cells was calculated by dividing the number of dual-colored cells (yellow circles) by the total number of primary-colored cells (white circles). The remaining percentage of non-dual-colored cells therefore represents the extent of exclusion. (B) SCG neurons were infected with PRV 614 (red) or PRV 128 (green) as the primary virus (106 PFU). Zero to eight hours later, cells were infected with PRV 128 or PRV 614, respectively, as the secondary virus (106 PFU). (C to F) Cells were infected with PRV 614 (red) or PRV 128 (green) as the primary virus (MOI = 10). Zero to eight hours later, cells were infected with PRV 128 or PRV 614 as the secondary virus (MOI = 10). (C) RAT-2 fibroblasts; (D) PK15 epithelial cells; (E) PI epithelial cells; (F) PI epithelial cells treated with doxycycline (DOX). (B to F) Cells were imaged 8 h after secondary infection. The percentage of dual-colored cells was calculated for 0, 2, 4, and 8 h between primary and secondary infections. Error bars indicate standard errors of the mean (SEM). Scale bars = 50 µm.
Next, we examined the fate of secondary infection in SCG neurons previously infected with either IE180-null PRV mutants or replicating PRV. SCG neurons were infected with PRV 128 or PRV 151 for 24 h and subsequently superinfected with PRV 180 (PRV Becker with mRFP-VP26 capsid fusion) for 2 h before imaging (Fig. 4A). We examined the localization of PRV 180 red capsids in cell bodies that were mock infected or infected with green PRV 128 as the primary virus. In both cases, there was abundant accumulation of red capsids in the cytoplasm and nuclear membrane, compatible with a productive superinfection (Fig. 4A, first and third rows). The pattern of red capsid distribution was indistinguishable between mock and PRV 128 primary infection. In both cases, there was no superinfection exclusion of PRV 180 and fluorescent capsids reached the nucleus (Fig. 4A; also, see Movies S1 and S3 in the supplemental material). Strikingly, when PRV 151 was used for primary infection, there was complete superinfection exclusion of PRV 180. We could not detect PRV 180 capsids entering the cell body; no capsids were observed in the cytoplasm or perinuclear regions of cell bodies (Fig. 4A, second row; also, see Movie S2 in the supplemental material). These results suggests that superinfection exclusion blocks viral entry into the cell. Secondary virus appears to adsorb to the cell membrane without penetration of capsids into the cell These data are compatible with HSV-1 gD-mediated infection resistance to HSV-1 (20). PRV interference mediated by homologous viral gene products seems to rely on IE180-mediated viral replication.
FIG 4 .

IE180-null mutant PRV allows superinfection with a secondary virus. (A) SCG cell bodies were mock infected or infected with PRV 151 or PRV 180 as the primary virus. In all cases, PRV 180 was added 24 h after primary infection and neurons were imaged after 2 h. GFP signal indicates primary infection, RFP signal corresponds to PRV 180 capsids, and blue signal corresponds to Hoechst nuclear staining. The fate of PRV 180 capsids can be observed with more detail in the magnified panel at the right of each row to determine if there was secondary infection, as evidence by capsid entry and transport to the perinuclear region. Scale bar = 10 µm for the first three panels of each row and 2 µm for the magnified inset of each row. (B) PK15 epithelial cells were infected with PRV 614 (red) or PRV 242 (red, IE180-null PRV) as the primary virus (MOI = 10) or mock infected. Eight hours later, cells were infected with PRV 927 (green, ICP8-EGFP PRV) as the secondary virus (MOI = 10). Cells were imaged 6 h after secondary infection. Scale bars = 5 µm.
To further track the fate of secondary infecting virus, we determined if PRV 128 allowed replication of superinfecting PRV. It was possible that PRV 128 infection enabled translation of the fluorescent transcript produced by the secondary virus rather than permitting a productive secondary viral infection. To address this possibility, we infected PK15 cells with PRV 242 (a PRV IE180-null mutant isogenic with PRV 128 except that the enhanced GFP [EGFP] gene replaced the mCherry gene) followed by infection with PRV 927 (a PRV mutant with ICP8 fused to GFP and expressed from the ICP8 promoter). ICP8 is a viral single-strand DNA-binding protein that localizes to PRV replication compartments in the nucleus and therefore serves as a marker of productive infection (21). PK15 cells infected with PRV 927 alone showed GFP foci in the nucleus 6 h postinfection (Fig. 4B, first row). PK15 cells infected with PRV 242 followed by infection with PRV 927 8 h later showed nuclear GFP foci 6 h after secondary infection (Fig. 4B, third row). As expected, PK15 cells infected first with PRV 614 excluded PRV 927 expression (no ICP8 foci or green diffusible nuclear signal). These observations were consistent with the hypothesis that PRV IE180-null mutants do not exclude entry, gene replication, and expression of a secondary infecting PRV strain. IE180-null-PRV-infected cells behave like mock-infected or uninfected cells.
Neurons survive infection by PRV IE180-null mutants.
Primary cultures of SCG neurons were infected with 106 PFU of PRV 128 or PRV 151. Cytopathic effects were easily observed 10 days after PRV 151 infection: axons disintegrated and cell bodies lifted from the dish. By day 30, all cell bodies had been destroyed, and only debris could be observed. In striking contrast, neurons infected with PRV 128 showed no cytopathic effects. PRV 128-infected neurons were indistinguishable from uninfected neurons even 1 month after infection (Fig. 5).
FIG 5 .

Viability of neurons infected with IE180-null PRV mutant. SCG neurons were infected with 106 PFU of PRV 151 or PRV 128 (green) or mock infected. Neuron morphology and health were monitored over time. Images were taken 2, 10, and 30 days postinfection. Scale bars = 50 µm.
DISCUSSION
Our data showed that PRV IE180-null mutants infect single cells efficiently, do not replicate or spread, and do not kill infected neurons. These properties extend the seminal observations of Yamada and Shimizu (9) and are consistent with the role of IE180 as the sole activator of PRV RNA transcription and DNA replication. Two noteworthy new findings were that infection of cells with PRV IE180-null mutants did not exclude infection by a second PRV strain (no superinfection exclusion) and that infected cells showed no cytopathic effects. The mutant phenotypes were fully restored when IE180 was supplied by complementation in epithelial cells. Construction of replication-incompetent PRV mutants is technically more straightforward than what is possible for the closely related alphaherpesvirus herpes simplex virus 1 (HSV-1), which encodes five immediate early genes. For PRV, deletion of the sole immediate early (IE) gene IE180 is sufficient to render the virus completely replication deficient and nontoxic. Consequently, transcomplementation is possible with a cell line expressing IE180 alone. The HSV-1 IE genes ICP0, ICP4, ICP22, ICP27, and ICP47 have diverse roles in viral infection, and their deletion involves complicated genetics and construction of cell lines expressing multiple IE genes. Several HSV-1 deletion mutants have been developed as gene delivery vectors containing partial deletion of some of the aforementioned IE genes, the neurovirulence factor ICP34.5, VP16, or gH (22).
The lack of superinfection exclusion by PRV IE180-null mutants is noteworthy. One mechanism of superinfection exclusion could be that replication compartments are fully occupied during replication by the primary virus (23). Since PRV IE180-null mutants do not replicate, the replication compartments may be available for occupancy by other infecting viral genomes. Other mechanisms for exclusion rely on the expression of a crucial viral protein that interferes with infection by the second virus. For example, bovine viral diarrhea virus requires E2 glycoprotein to block entry of secondary virus (24), duck hepatitis B virus requires large surface antigen to regulate nucleocapsid trafficking (25), and the closely related HSV-1 requires glycoprotein D (gD) to bind to receptors to block entry of the second infecting virus (20). Our data suggest that excluded secondary PRV adsorbs to the cell surface but is unable to penetrate and enter the cytoplasm. However, when a PRV IE180-null mutant is the primary infection strain, secondary infecting virions can enter and replicate with normal kinetics similar to those of mock primary infection. PRV IE180-null mutants are completely defective in viral protein synthesis (early and late) and must fail to synthesize one or more gene products that block secondary infection. It is possible that HSV-1 and PRV share the gD-mediated exclusion mechanism. Even if a reduced number of capsids enter the cell, the bottleneck at the level of nuclear replication might act as a second exclusion barrier (26).
The lack of cytopathic effects after PRV IE180-null infection is remarkable. Primary neurons infected with PRV 128 survived as long as mock-infected cells, whereas all neurons perished by day 10 after wild-type PRV infection. Strikingly PRV IE180-null mutants are completely avirulent in mice, persisting even after 6 months (16). Cell death is caused by viral gene expression, because PRV gB-null mutants, which produce noninfectious particles, kill neurons as efficiently as wild-type virus infection (27). However, we noticed that a fraction of PRV 128-infected cells showed cytopathic effects if infected at an extremely high multiplicity of infection (MOI) (i.e., MOI > 100; >108 PFU). It may be that binding of an excessive number of virions to host receptors or subsequent delivery of many viral genomes to the nucleus may be sufficient to activate antiviral cell responses such as transcription of interferon-stimulated genes (ISGs). In fact, infection of mice with replication-defective HSV vectors increases the expression of ISGs, albeit to a lower degree than wild-type infection (28). Although neurons infected with 106 PFU of PRV 128 were viable for over a month, the intensity of GFP signal varied among individual neurons and slowly faded with time. By the fifth week of infection, the GFP signal was barely detectable. It may be that expression is being silenced or repressed. Alphaherpesviruses such as PRV can establish episomal latency (29) and express latency-associated transcripts (30). PRV IE180-null mutant genomes may be especially prone to repression, because no viral proteins are produced that would normally counteract host defenses. It will be important to test expression and stability of transgene transcripts from other promoters in the PRV IE180-null mutant background. Similar studies have been done with the HSV-1 replication-deficient mutant d109 (ICP4−, ICP27−, ICP0−, ICP22−, and ICP47−). This mutant included the fluorescent reporter EGFP driven from the cytomegalovirus (CMV) promoter and showed no cytotoxicity in vitro (31). In agreement with our observation of decay of EGFP expression in cells infected with the IE180-null PRV mutant, those authors reported a decay in fluorescence 1 to 2 weeks after infecting Vero cells with d109 mutant. Importantly, they demonstrated that d109 genomes resident in noncomplementing cells persisted in a long-term silent but functional state for weeks after the decay of EGFP expression. Those authors were able to induce EGFP expression from the silent genomes by expression of ICP0. Our hypothesis is that IE180-null PRV mutants also establish a persistent infection and that the viral genomes are in a potentially functional state in noncomplementing cells even after the decay of fluorophore expression.
PRV IE180-null mutants have potential applications for neuroscience research because they infect only the primary cell, do not spread, and do not kill infected cells. For example, it is possible to determine if neuron A is synaptically connected to a distant neuron B (Fig. 6). Using virus microinjection, neuron A could be infected with a replicating attenuated PRV recombinant expressing RFP and neuron B could be infected with PRV 128 expressing GFP. The target neuron B will not die from infection and the green signal will not spread, thus labeling neuron B exclusively. The red signal would spread via synaptically connected neurons throughout neuron A’s circuit. If neuron B is a member of neuron A’s circuit, it would eventually show yellow fluorescence (green and red), because PRV IE180-null mutants do not exclude secondary virus. If neuron B is not a member of neuron A’s circuit, it would remain green. This technique for mapping neuronal networks is more straightforward and efficient than using two well-timed wild-type virus infections (i.e., if and when the viruses meet, they must do so in the narrow time window before superinfection exclusion is established). Additionally, neuron B can be easily identified and monitored over long periods of time simply by examining green fluorescence.
FIG 6 .

Model for detecting synaptic connectivity between distance neurons with IE180-null PRV mutant. Neuron A is injected with red fluorescent wild-type PRV. Neuron B is injected with green fluorescent IE180-null PRV. Due to no viral spread and low toxicity during IE180-null infection, neuron B will be fluorescently labeled over time for easy identification and monitoring over time. Red fluorescence will spread throughout neuron A’s circuit via synaptically connected neurons. Due to the lack of superinfection exclusion during IE180-null PRV infection, neuron B will show both green and red (yellow) fluorescence if it is a member of neuron A’s circuit. If it is not, neuron B will show green fluorescence only.
The properties of PRV IE180-null mutants suggest that they could be used for gene delivery in vivo. After the removal of the two copies of IE180 (9 kb of DNA), the genome of IE180-null PRV will package more foreign DNA than traditional viral vectors, such as adenovirus, adeno-associated virus, rabies virus, or lentivirus. We have already shown that PRV 128 effectively delivers GFP to neurons without significantly affecting cell viability. Furthermore, individual neurons can be precisely targeted due to the absence of viral spread, and subsequent viral gene delivery could occur due to the lack of superinfection exclusion. One transgene of interest that could be delivered by PRV IE180-null recombinants is the fast calcium sensor GCaMP6f (32). Neurons expressing GCaMP6 variants produce large transient fluorescence signals in response to single action potentials (33, 34). Therefore, PRV IE180-null mutants harboring a GCaMP6 transgene would have diverse applications in extended long-term electrophysiological and behavioral experiments.
We have shown that PRV IE180-null mutants can efficiently label a population of cells with fluorescent proteins for long-term in vitro studies. Additionally, the mutant can be useful in studies of the initial steps of PRV infection by removing the background effects of viral replication. For example, the fate of incoming virions can be clearly tracked with capsid-tagged PRV IE180-null mutants, whereas during wild-type infection, incoming and progeny virions must be distinguished. We suggest that PRV IE180-null mutants will have utility in both neuroscience and alphaherpesvirus research.
MATERIALS AND METHODS
Epithelial and neuron cell cultures.
Cells and viruses used in this study are listed in Table 1. Epithelial cell lines were grown at 37°C and 5% CO2 and maintained in Dulbecco’s modified Eagle medium (DMEM) (Thermo Scientific) supplemented with 10% fetal bovine serum (Thermo Scientific) and 1% penicillin-streptomycin (Thermo Scientific). Medium for PK15 IE180-expressing cells (PI cells) was also supplemented with 500 µg/ml Geneticin (Life Technologies) to select for cells possessing the PRV IE180 transgene. When necessary, 2 µg/ml doxycycline (Clontech) was added to cell medium to induce expression of the IE180 gene.
TABLE 1 .
Cells and viral strains
| Cell line or virus | Description |
|---|---|
| Cells | |
| PK15 | Porcine kidney epithelial cells |
| PK15 IE180 (PI) | Transgenic PK15 cells with Tet-On promoter for IE180 gene; addition of doxycycline activates IE180 transcription (16) |
| RAT-2 | Rat embryonic fibroblast-like cells |
| SCG | Primary rat superior cervical ganglion neurons |
| PRV strains | |
| PRV 128 | PRV IE180 null with EF1a-GFP in Us9 (16) |
| PRV 151 | PRV Becker with CMV-EGFP in gG (39) |
| PRV 180 | PRV Becker with mRFP-VP26 (40) |
| PRV 242 | PRV IE180 null with EF1a-mCherry in Us9 (16) |
| PRV 614 | PRV Bartha with CMV-RFP in gG (19) |
| PRV 927a | PRV Becker with ICP8-EGFP (J. B. Bosse, unpublished data) |
PRV 927 was produced by homologous recombination of PRV Becker DNA and plasmid pUC57-mEGFP-ICP8 containing EGFP fused to the C terminus of ICP8 by a 19-amino-acid flexible linker. The plasmid contained 500-bp flanking ends homologous to the upstream and downstream regions of the PRV UL29 gene, coding for ICP8.
Superior cervical ganglia (SCG) were dissected from day 14 embryonic rats and plated on plastic dishes coated with poly-l-ornithine (Sigma-Aldrich) and murine laminin (Invitrogen). Neurons were grown at 37°C and 5% CO2 and maintained in neuronal medium consisting of neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), 1% penicillin-streptomycin-glutamine (Invitrogen), and 50 ng/ml nerve growth factor (Invitrogen). Cultures differentiated for at least 14 days prior to viral infection.
Modified Campenot trichamber neuron cultures were constructed as described previously (35, 36). SCG neurons were plated in the left, proximal compartment (S compartment) and maintained in neuronal medium. Over the course of 14 to 21 days, axons extended along the grooves and penetrated through the middle compartment (M compartment) and into the right, distal compartment (N compartment). To assess the rate of retrograde spread of IE180-null recombinants, 1 µg/ml of red fluorescent lipophilic dye (DiI) (Invitrogen) was added to the N compartment 3 h after N compartment infection. Only a subset of neurons grow axons into the N compartment, and neurons that do not project into the N compartment have synaptic contacts with neurons that do. DiI labels neurons via lateral diffusion in the plasma membrane without transfer to unlabeled cells and does not affect cell viability or physiological properties of parent cell bodies in the S compartment.
DNA-associated click chemistry.
Confluent cultures of contact-inhibited RAT-2 cells were serum-starved and infected at a multiplicity of infection (MOI) of 10 with PRV 151 or PRV 128 or mock infected. The nucleoside analog EdU (10 µM) was added 1 h postinfection. Cells were fixed with 3% paraformaldehyde for 15 min at 37°C 6 h after PRV 151 infection to preserve cell morphology and 16 h after PRV 128 infection to allow stronger green fluorescent signal driven from the EF1a promoter. Cells were then washed with 3% bovine serum albumin (BSA) (Sigma-Aldrich) in phosphate-buffered saline (PBS) twice and permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) for 20 min at room temperature. The click reaction was performed according to the Click-iT EdU imaging kit (Invitrogen) manual with Alexa Fluor 647 azide (Invitrogen). Nuclei were stained with 5 µg/ml Hoechst 33342 (Invitrogen).
Western immunoblotting.
PK15 or PI cells were infected at an MOI of 10 with PRV 151 or PRV 128 or mock infected and then harvested 16 h postinfection. Cell samples were lysed with radioimmunoprecipitation assay (RIPA)-light buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.1% SDS, 0.1% Triton X-100), protease inhibitor (Roche), and 100 mM dithiothreitol (DTT). Protein samples were heated to 70°C for 10 min, run on 4 to 12% SDS-PAGE gels, and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk–Tris-buffered saline supplemented with 0.1% Tween (TBS-T) for 1 h at room temperature and then incubated with a 1:200 dilution of rabbit anti-PRV EP0 (37), 1:1,000 mouse anti-PRV VP5 (38), 1:200 rabbit anti-PRV IE180 (12), and 1:10,000 mouse anti-β-actin (Sigma-Aldrich). Primary antibodies were washed with TBS-T, and membranes were incubated with 1:15,000 anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (KPL) for 1 h at room temperature. Signal was detected on radiographic film using a chemiluminescent kit (Thermo Scientific) according to the manufacturer’s instructions. The IE80 film was exposed longer than that of EP0 or VP5 to obtain comparable signal intensity.
Viral titer.
Infectious virus was titrated by infecting a monolayer of epithelial cells with serial dilutions of the sample. Infected cells were maintained in cell medium supplemented with 1% methylcellulose. PRV 151, PRV 180, PRV 614, and PRV 927 were titrated on PK15 cells. PRV 128 and PRV 242 were titrated on PI cells with 2 µg/ml doxycycline supplementing 1% methylcellulose. Titers were reported as PFU per milliliter.
Superinfection exclusion studies.
PK15, PI, RAT-2, and 293A cells were infected at an MOI of 10, and SGC axons were infected with 106 PFU. Times between primary infection and secondary superinfection were 0, 2, 4, and 8 h. For the time point of 0 h between primary and secondary infections, cells were coinfected with an equal mixture of the two viruses (MOI of 5 each for epithelial cells and fibroblasts or 5 × 105 PFU each for neurons). After 1 h of primary infection, inoculum was aspirated and fresh medium was added. At the time for secondary infection, medium was aspirated and viral inoculum was added. After 1 h of secondary infection, inoculum was aspirated and fresh medium was added. For each condition and time point, the percentage of cells expressing the primary viral reporter gene that also expressed the secondary viral reporter gene was calculated.
Fluorescence microscopy.
Cells were imaged with a Nikon Ti-Eclipse epifluorescent inverted microscope under a heated cell culture chamber (Ibidi). Fluorescent images were captured using an X-Cite series 120 lamp (EXFO) and a Roper Scientific CoolSnap HQ2 camera (Photometrics). Images were processed with NIS Elements software (Nikon).
SUPPLEMENTAL MATERIAL
SCG cell body infection with PRV 180 and mock infection. A z-axis-stack movie of Fig. 4, row 1, shows the localization of PRV 180 capsids. Abundant PRV 180 capsids are observed in the cell body cytoplasm and perinuclear region. The movie was acquired at 4 frames per s. Scale bar = 10 µm. Download
SCG cell body infection with PRV 151 and PRV 180. A z-axis-stack movie of Fig. 4, row 2, shows the localization of PRV 180 capsids. PRV 180 capsids are observed only in the cell body periphery, not entering the cell. Infection with PRV 151 excludes superinfection with PRV 180 and blocks viral entry. The movie was acquired at 4 frames per s. Scale bar = 10 µm. Download
SCG cell body infection with PRV 128 and PRV 180. A z-axis-stack movie of Fig. 4, row 3, shows the localization of PRV 180 capsids. Abundant PRV 180 capsids are observed in the cell body cytoplasm and perinuclear region. Infection with PRV 128 allows superinfection with PRV 180. The movie was acquired at 4 frames per s. Scale bar = 10 µm. Download
ACKNOWLEDGMENTS
This research was supported by NIH grants P40 RR 018604 and R01 NS060699 to L.W.E. and a Pew Latin American Fellowship in the Biomedical Sciences (grant 2010-000225-002) to E.A.E.
We thank Anthony Zador, Hassana Oyibo, and Peter Znamenskiy for building the PRV IE180-null mutants and the cell line expressing IE180, Orkide Koyuncu for assistance with click chemistry and discussion of experiments, Enrique Tabares for sharing the IE180 antibody, Etsuno Ono for sharing the EP0 antibody, Jens Bosse for building the PRV 927 mutant with ICP8-EGFP, and Ren Song for critical reading of the manuscript.
Footnotes
Citation Wu BW, Engel EA, Enquist LW. 2014. Characterization of a replication-incompetent pseudorabies virus mutant lacking the sole immediate early gene IE180. mBio 5(6):e01850-14. doi:10.1128/mBio.01850-14.
REFERENCES
- 1. Goodpasture EW, Teague O. 1923. Transmission of the virus of herpes febrilis along nerves in experimentally infected rabbits. J. Med Res. 44:139–184.137. [PMC free article] [PubMed] [Google Scholar]
- 2. Kimman TG, Binkhorst GJ, van den Ingh TS, Pol JM, Gielkens AL, Roelvink ME. 1991. Aujeszky’s disease in horses fulfils Koch’s postulates. Vet. Rec. 128:103–106. 10.1136/vr.128.5.103. [DOI] [PubMed] [Google Scholar]
- 3. McCracken RM, McFerran JB, Dow C. 1973. The neural spread of pseudorabies virus in calves. J. Gen. Virol. 20:17–28. 10.1099/0022-1317-20-1-17. [DOI] [PubMed] [Google Scholar]
- 4. Aujeszky A. 1902. Über eine neue Infektionskrankheit bei Haustieren. Zbl. Bakt. Abt. Orig. 32:353–373. [Google Scholar]
- 5. Granstedt AE, Bosse JB, Thiberge SY, Enquist LW. 2013. In vivo imaging of alphaherpesvirus infection reveals synchronized activity dependent on axonal sorting of viral proteins. Proc. Natl. Acad. Sci. U. S. A. 110:E3516–E3525. 10.1073/pnas.1311062110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brittle EE, Reynolds AE, Enquist LW. 2004. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J. Virol. 78:12951–12963. 10.1128/JVI.78.23.12951-12963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Card JP, Enquist LW. 2014. Transneuronal circuit analysis with pseudorabies viruses. 68:1.5.1–1.5.39. 10.1002/0471142301.ns0105s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 69:462–500. 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamada S, Shimizu M. 1994. Isolation and characterization of mutants of pseudorabies virus with deletion in the immediate-early regulatory gene. Virology 199:366–375. 10.1006/viro.1994.1134. [DOI] [PubMed] [Google Scholar]
- 10. Cheung AK. 1989. DNA nucleotide sequence analysis of the immediate-early gene of pseudorabies virus. Nucleic Acids Res. 17:4637–4646. 10.1093/nar/17.12.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taharaguchi S, Ono E, Yamada S, Shimizu Y, Kida H. 1995. Mapping of a functional region conferring nuclear localization of pseudorabies virus immediate-early protein. Arch. Virol. 140:1737–1746. 10.1007/BF01384338. [DOI] [PubMed] [Google Scholar]
- 12. Gómez-Sebastián S, Tabarés E. 2004. Negative regulation of herpes simplex virus type 1 ICP4 promoter by IE180 protein of pseudorabies virus. J. Gen. Virol. 85:2125–2130. 10.1099/vir.0.80119-0. [DOI] [PubMed] [Google Scholar]
- 13. Van Opdenbosch N, Van den Broeke C, De Regge N, Tabarés E, Favoreel HW. 2012. The IE180 protein of pseudorabies virus suppresses phosphorylation of translation initiation factor eIF2α. J. Virol. 86:7235–7240. 10.1128/JVI.06929-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomioka Y, Miyazaki T, Taharaguchi S, Yoshino S, Morimatsu M, Uede T, Ono E, Watanabe M. 2008. Cerebellar pathology in transgenic mice expressing the pseudorabies virus immediate-early protein IE180. Eur. J. Neurosci. 27:2115–2132. 10.1111/j.1460-9568.2008.06174.x. [DOI] [PubMed] [Google Scholar]
- 15. López-Ramos JC, Tomioka Y, Morimatsu M, Yamamoto S, Ozaki K, Ono E, Delgado-García JM. 2010. Motor-coordination-dependent learning, more than others, is impaired in transgenic mice expressing pseudorabies virus immediate-early protein IE180. PLoS One 5:e12123. 10.1371/journal.pone.0012123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oyibo H, Znamenskiy P, Oviedo HV, Enquist LW, Zador A. 2014. Long-term Cre-mediated retrograde tagging of neurons using a novel recombinant pseudorabies virus. Front. Neuroanat. 8:86. 10.3389/fnana.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kobiler O, Brodersen P, Taylor MP, Ludmir EB, Enquist LW. 2011. Herpesvirus replication compartments originate with single incoming viral genomes. mBio 2:e00278-11. 10.1128/mBio.00278-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bennett CW. 1953. Interactions between viruses and virus strains. Adv. Virus Res. 1:39–67. 10.1016/S0065-3527(08)60461-3. [DOI] [PubMed] [Google Scholar]
- 19. Banfield BW, Kaufman JD, Randall JA, Pickard GE. 2003. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. J. Virol. 77:10106–10112. 10.1128/JVI.77.18.10106-10112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson RM, Spear PG. 1989. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 63:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao M, Knipe DM. 1991. Potential role for herpes simplex virus ICP8 DNA replication protein in stimulation of late gene expression. J. Virol. 65:2666–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Y, Nemunaitis J. 2006. Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther. 13:975–992. 10.1038/sj.cgt.7700946. [DOI] [PubMed] [Google Scholar]
- 23. Simon KO, Cardamone JJ, Jr, Whitaker-Dowling PA, Youngner JS, Widnell CC. 1990. Cellular mechanisms in the superinfection exclusion of vesicular stomatitis virus. Virology 177:375–379. 10.1016/0042-6822(90)90494-C. [DOI] [PubMed] [Google Scholar]
- 24. Lee YM, Tscherne DM, Yun SI, Frolov I, Rice CM. 2005. Dual mechanisms of pestiviral superinfection exclusion at entry and RNA replication. J. Virol. 79:3231–3242. 10.1128/JVI.79.6.3231-3242.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walters KA, Joyce MA, Addison WR, Fischer KP, Tyrrell DL. 2004. Superinfection exclusion in duck hepatitis B virus infection is mediated by the large surface antigen. J. Virol. 78:7925–7937. 10.1128/JVI.78.15.7925-7937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobiler O, Lipman Y, Therkelsen K, Daubechies I, Enquist LW. 2010. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat. Commun. 1:146. 10.1038/ncomms1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Curanovic D, Enquist LW. 2009. Virion-incorporated glycoprotein B mediates transneuronal spread of pseudorabies virus. J. Virol. 83:7796–7804. 10.1128/JVI.00745-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shayakhmetov DM, Di Paolo NC, Mossman KL. 2010. Recognition of virus infection and innate host responses to viral gene therapy vectors. Mol. Ther. 18:1422–1429. 10.1038/mt.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minarovits J. 2006. Epigenotypes of latent herpesvirus genomes, p 61-80 In Doerfler PDW, Böhm P. (ed), DNA methylation: development, genetic disease and cancer. Springer Verlag, Berlin, Germany. [Google Scholar]
- 30. Jin L, Scherba G. 1999. Expression of the pseudorabies virus latency-associated transcript gene during productive infection of cultured Cells. J. Virol. 73:9781–9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Samaniego LA, Neiderhiser L, DeLuca NA. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shang W, Lu F, Sun T, Xu J, Li LL, Wang Y, Wang G, Chen L, Wang X, Cannell MB, Wang SQ, Cheng H. 2014. Imaging Ca2+ nanosparks in heart with a new targeted biosensor. Circ. Res. 114:412–420. 10.1161/CIRCRESAHA.114.302938. [DOI] [PubMed] [Google Scholar]
- 33. Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL. 2012. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 32:13819–13840. 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. 2013. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wojaczynski GJ, Engel EA, Steren KE, Enquist LW, Card PJ. March 2014. The neuroinvasive profiles of H129 (herpes simplex virus type 1) recombinants with putative anterograde-only transneuronal spread properties. Brain Struct. Funct. 10.1007/s00429-014-0733-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ch’ng TH, Enquist LW. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 79:10875–10889. 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watanabe S, Ono E, Shimizu Y, Kida H. 1995. Pseudorabies virus early protein 0 transactivates the viral gene promoters. J. Gen. Virol. 76:2881–2885. 10.1099/0022-1317-76-11-2881. [DOI] [PubMed] [Google Scholar]
- 38. Lyman MG, Feierbach B, Curanovic D, Bisher M, Enquist LW. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 81:11363–11371. 10.1128/JVI.01281-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Demmin GL, Clase AC, Randall JA, Enquist LW, Banfield BW. 2001. Insertions in the gG gene of pseudorabies virus reduce expression of the upstream Us3 protein and inhibit cell-to-cell spread of virus infection. J. Virol. 75:10856–10869. 10.1128/JVI.75.22.10856-10869.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. del Rio T, Ch’ng TH, Flood EA, Gross SP, Enquist LW. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 79:3903–3919. 10.1128/JVI.79.7.3903-3919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SCG cell body infection with PRV 180 and mock infection. A z-axis-stack movie of Fig. 4, row 1, shows the localization of PRV 180 capsids. Abundant PRV 180 capsids are observed in the cell body cytoplasm and perinuclear region. The movie was acquired at 4 frames per s. Scale bar = 10 µm. Download
SCG cell body infection with PRV 151 and PRV 180. A z-axis-stack movie of Fig. 4, row 2, shows the localization of PRV 180 capsids. PRV 180 capsids are observed only in the cell body periphery, not entering the cell. Infection with PRV 151 excludes superinfection with PRV 180 and blocks viral entry. The movie was acquired at 4 frames per s. Scale bar = 10 µm. Download
SCG cell body infection with PRV 128 and PRV 180. A z-axis-stack movie of Fig. 4, row 3, shows the localization of PRV 180 capsids. Abundant PRV 180 capsids are observed in the cell body cytoplasm and perinuclear region. Infection with PRV 128 allows superinfection with PRV 180. The movie was acquired at 4 frames per s. Scale bar = 10 µm. Download