

Abstract
Background
Pathogenic mutations range from single nucleotide changes to deletions or duplications that encompass a single exon to several genes. The use of gene-centric high-density array comparative genomic hybridization (aCGH) has revolutionized the detection of intragenic copy number variations. We implemented an exon-centric design of high-resolution aCGH to detect single- and multi-exon deletions and duplications in a large set of genes using the OGT 60 K and 180 K arrays. Here we describe the molecular characterization and breakpoint mapping of deletions at the smaller end of the detectable range in several genes using aCGH.
Results
The method initially implemented to detect single to multiple exon deletions, was able to detect deletions much smaller than anticipated. The selected deletions we describe vary in size, ranging from over 2 kb to as small as 12 base pairs. The smallest of these deletions are only detectable after careful manual review during data analysis. Suspected deletions smaller than the detection size for which the method was optimized, were rigorously followed up and confirmed with PCR-based investigations to uncover the true detection size limit of intragenic deletions with this technology. False-positive deletion calls often demonstrated single nucleotide changes or an insertion causing lower hybridization of probes demonstrating the sensitivity of aCGH.
Conclusions
With optimizing aCGH design and careful review process, aCGH can uncover intragenic deletions as small as dozen bases. These data provide insight that will help optimize probe coverage in array design and illustrate the true assay sensitivity. Mapping of the breakpoints confirms smaller deletions and contributes to the understanding of the mechanism behind these events. Our knowledge of the mutation spectra of several genes can be expected to change as previously unrecognized intragenic deletions are uncovered.
Keywords: aCGH, Intragenic deletions, Breakpoint analysis, Molecular characterization
Background
Laboratories that offer diagnostic mutation testing use a number of methodologies to detect pathogenic chromosomal rearrangements, coding sequence aberrations, abnormal methylation patterns, and other biochemical indicators of genetic disease. These analyses help with diagnoses, management, carrier testing, and counseling for families affected by an inherited genetic disease. The mutation spectrum of a particular gene guides clinical test development, so that the adapted method promises the highest yield in detection without compromising sensitivity, specificity, and cost effectiveness. Small mutations, such as nucleotide changes predicted to cause missense, nonsense, or altered splicing, as well as frameshifts due to small deletions and duplications of a few bases, can be detected by sequence analysis. Larger pathogenic copy number variations (CNVs) are efficiently detected by high-resolution G-banding, fluorescence in situ hybridization (FISH), and cytogenomic array comparative genomic hybridization (aCGH); however, the size limitation of these methods is approximately 200–500 kb or larger.
Recurrent microdeletions and microduplications that occur between repeat sequences via nonallelic homologous recombination (NAHR) are a class of large pathogenic CNVs that can easily be detected in diagnostic tests, as the known breakpoints are amenable to the development of targeted methods [1,2]. On the other hand, there are CNVs that primarily represent private non-recurrent familial mutations encompassing several to a single gene. Nonhomologous end-joining (NHEJ) and microhomology-mediated break-induced replication (MMBIR) are two mechanisms responsible for these mutations [3-5]. Chromosomal microarray is the recommended technique to screen the entire genome for CNVs, when there is no specific locus clinically suspected [6].
Gene-targeted diagnostic testing methods can be developed to screen a specific genomic locus for CNVs, which is best illustrated by the diagnostic testing for Duchenne muscular dystrophy [7]. Pathogenic deletions and duplications within the DMD gene account for approximately 65 percent of mutations. Clinical testing for these mutations has been performed by multiplex standard PCR (males only) [8,9], quantitative PCR (q-PCR) [10], and Southern blotting [11], as well as multiple ligation-dependent probe amplification (MLPA) [12]. These methodologies are laborious and lack sensitivity, particularly for females, often requiring confirmation testing by a second method. To date, the most cost-effective and sensitive method for the detection of mutations in Duchenne muscular dystrophy is array-based comparative genomic hybridization (aCGH) [7,13,14].
Several gene-targeted arrays have been developed with probes concentrated within loci of interest. Examples include an aCGH that targets regions with segmental duplications and arrays that target only 5–8 specific genes of interest [15-18]. To be useful in a diagnostic laboratory, the design of aCGH has to be optimized to yield coverage of as many disease-associated genes as possible without compromising resolution and sensitivity to detect small intragenic pathogenic CNVs. Roughly, the detection criteria can be considered single or multiple exonic CNVs. Detection of pathogenic CNVs at sub-kilobase resolution have been reported by our laboratory and Boone et al. (2010), illustrating the ability of this technology to identify mutations in patients with various diseases and syndromes [19,20]. We have previously reported the development of a custom exon-centric array designed and implemented at Emory Genetics Laboratory (EGL) [20]. We now report aCGH data from a set of representative deletions identified with the use of these arrays during routine laboratory testing that demonstrate the power and sensitivity of this technology and illustrate the limit of detection in terms of deletion size (Table 1).
Table 1.
Table lists all the cases with intragenic deletions discussed in this manuscript
| Case | Gene | Disease: Inheritance | Mutation detected with sequencing | Mutation detected with aCGH | Deletion size |
|---|---|---|---|---|---|
|
1 |
BCKDHB |
Maple syrup urine disease: AR |
c.596_597delGT |
Exon 9 deletion |
~58 kb |
|
2 |
FH |
Hereditary leiomyomatosis and renal cell cancer: AD |
|
Exons 2–9 deletion |
~19 kb |
|
3 |
DBT |
Maple syrup urine disease: AR |
c.871C > T (p.R291X) |
Exon 5 deletion |
~3.7 kb |
|
4 |
HPRT1 |
Lesch-Nyhan syndrome: XL |
|
Exon 5 deletion |
2,319 bp |
|
5 |
STK11 |
Peutz-Jeghers syndrome: AD |
|
Exon 8 deletion |
1,325 bp |
|
6 |
STK11 |
Peutz-Jeghers syndrome: AD |
|
Exon 3 deletion |
971 bp |
|
7 |
PAH |
Phenylketonuria: AR |
c.838G > A (p.E280K) |
Partial exon 6 deletion |
801 bp |
|
8 |
EMD |
Emery-Dreifuss muscular dystrophy: XL |
|
Exon 2 deletion |
267 bp |
|
9 |
DBT |
Maple syrup urine disease: AR |
|
Partial exon 11 deletion |
> 3.5 kb |
| |
|
|
|
c.344-4del12 |
12 bp |
|
10 |
POMT1 |
Walker-Warburg syndrome AR |
c.2167dupG |
No deletion: c.160_161ins349 |
False positive: Alu insertion |
|
11 |
SLC9A6 |
X-linked intellectual disability: XL |
|
No deletion |
False positive: hemizygous missense |
| 12 | GALT | Galactosemia: AR | c.855G > T (p.K285N) mutation & c.844C > G (p.L282V) variant | No deletion | False positive: compound heterozygous missense |
AD: Autosomal Dominant; AR: Autosomal Recessive; XL: X-linked.
Results
Univocal detection of deletions larger than 2 kb
Custom-designed gene-targeted aCGH, manufactured on an Agilent aCGH platform developed by OGT’s Genefficiency service (Oxford Gene Technology, Oxford, UK), was used for deletion and duplication mutational analysis for genes that are part of our laboratory’s sequence analysis repertoire [20]. The Circular Binary Segmentation (CBS) algorithm generated deletion calls using a log2 ratio of each segment that has a minimum of four probes [21]. Threshold factor for deletions was set as a log2 ratio of -0.6. Figure 1 shows several examples of aCGH data with intragenic deletions larger than 2 kb that were easily detected with CBS calls that crossed the -0.6 log2 ratio threshold. Sequence analysis of the three maple syrup urine disease (MSUD) genes, BCKDHA, BCKDHB, or DBT, in two unrelated cases of known biochemical diagnosis of MSUD detected only one familial mutation each (Table 1) [22]. One had a known pathogenic mutation in the BCKDHB gene (c.596_597delGT), whereas the other family had a DBT gene nonsense mutation (c.871C > T (p.R291X). Reflex testing for with aCGH yielded the corresponding second familial mutation; an approximately 58-kb deletion encompassing exon 9 of the BCKDHB gene (Figure 1a top) and an approximately 3.7-kb deletion encompassing exon 5 of the DBT gene were detected in the two above mentioned MSUD cases (Figure 1a bottom). These examples demonstrate how conclusive a single exon deletion can be when several probes target the deleted sequence within the breakpoints and the CBS call is well below the log2 ratio threshold. Parental samples were tested to confirm biallelic inheritance in the probands of both families. Finding two familial mutations in this autosomal recessive disorder made carrier-testing possible for other at-risk family members.
Figure 1.
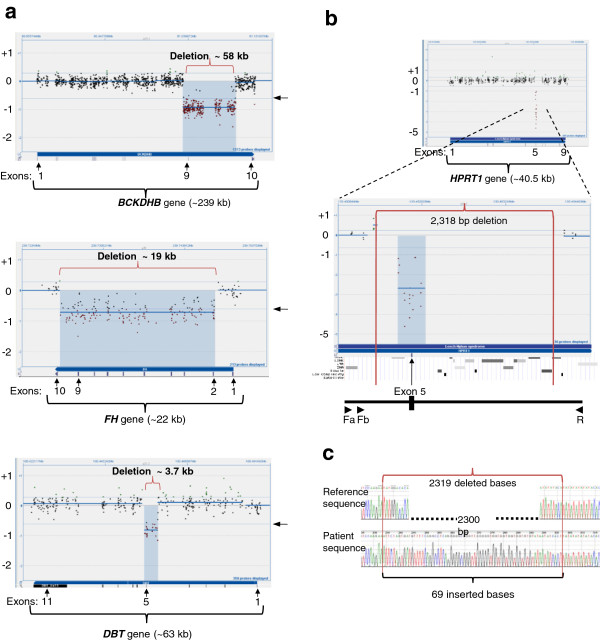
Examples of large deletions (>2 kb). CytoSure display of aCGH data across genes, with exon locations marked below. The patient versus reference Cy3/Cy5 ratio is plotted for each sample. Shown are the mean of the log2 ratio (thick blue horizontal line) and thresholds for deletion and duplication calls at log2 ratios of +0.4 and -0.6, respectively (thin blue horizontal lines), derived from the CBS algorithm. Arrows on the right of the CytoSure display mark the -0.6 log2 threshold for deletions. Below the CytoSure display are the corresponding exon tracks. 1a top) ~58-kb deletion encompassing exon 9 of the BCKDHB gene. 1a middle) ~19-kb deletion encompassing exons 2–9 of the FH gene. 1a bottom) ~3.7-kb deletion encompassing exon 5 of the DBT gene. 1b) 2,318-bp deletion encompassing exon 5 of the HPRT1 gene. The breakpoints of the deletion are shown with vertical red lines. Below the CytoSure display are the UCSC RepeatMasker track showing repeat elements at the deletion locus, followed by an illustration of the breakpoint PCR design with location of primers (Fa, Fb, and R) shown as arrows. 1c) Electropherogram of sequenced HPRT1 deleted allele with 69 inserted bases shown. Image in Figure 1c was reproduced, with kind permission of Springer Science + Business Media, as previously published in Molecular Genetics and Personalized Medicine, edited by Best DH and Swensen JJ, Jan 2012, Chapter 2: Array Comparative Genomic Hybridization in Cytogenetics and Molecular Genetics, Askree SH and Hegde M, Figure 2.5, Page 32.
An additional example presented here is where aCGH analysis of the FH gene detected a heterozygous deletion of an intermediate size compared to the two single exon deletions detected in MSUD cases described above. However, this 19-kb deletion resulted in a loss of 8 out of 10 exons of the FH gene. This testing was triggered due to strong clinical suspicion, in an adult male with a personal and family history that was highly suggestive of the autosomal dominant disorder, hereditary leiomyomatosis and renal cell cancer. Sequencing of the relevant FH gene did not detect a mutation, and aCGH analysis confirmed the familial (autosomal dominant) FH deletion mutation (Figure 1a middle).
In contrast to the deletions detected in autosomal genes, deletions in X-linked diseases show high sensitivity in male probands due to lack of an interfering normal allele. Lesch-Nyhan syndrome (LNS) is an X-linked recessive disorder caused by deficiency of the enzyme hypoxanthine guanine phosphoribosyltransferase (HPRT). A mutation in the single copy of the HPRT1 gene in a male causes LNS.[23] A 20-year-old male proband was found to carry a 2.3-kb hemizygous deletion mutation encompassing exon 5 in the HPRT1 gene (Figure 1b). Subsequently, his sister was found to carry the familial mutation. We tested amniocytes from the sister’s pregnancy and determined that the fetus did not inherit the familial deletion mutation. Allele-specific PCR was developed that amplified the deleted allele in the proband and his sister. Sequence analysis confirmed a 2319-bp deletion encompassing exon 5 with breakpoints at the exon 5 splice site boundary (Figure 1b, 1c). There was an insertion of 69 bp with no homology to any flanking sequence. Upon BLAT query, the inserted bases mapped to chr5p13.1 (Chr5:40,844,202-40,844,270/hg18) [24]. Data included in additional information shows the aCGH analyses on all three family members, the fragment analysis of the breakpoint PCR, as well as the complete sequence of the deletion locus (see Additional file 1).
1325-bp and 971-bp deletions in the STK11 gene
Peutz-Jeghers syndrome (PJS) is an autosomal dominant disorder characterized by intestinal hamartomatous polyps, an increased risk of certain malignancies, and hyperpigmented cutaneous lesions. Mutations in the STK11 gene cause PJS. In two unrelated Caucasian patients who had clinical presentations consistent with PJS, sequence analysis of the STK11 gene did not detect a mutation.[25] Figure 2 shows aCGH data where a deletion call encompassing exon 8 did not cross the -0.6 log2 ratio threshold set. However, low hybridization of 15 probes leading to a deletion call of a >1-kb segment triggered PCR confirmation. Using primers flanking the breakpoint, the deleted allele was preferentially amplified over the larger normal allele. Sequencing data confirmed a 1325-bp deletion with a four-base microhomology at the breakpoints in intron 7 and intron 8 (Figure 2a). Fragment analysis of breakpoint PCR, and the complete sequence of the deletion locus is included in Additional file 2.
Figure 2.
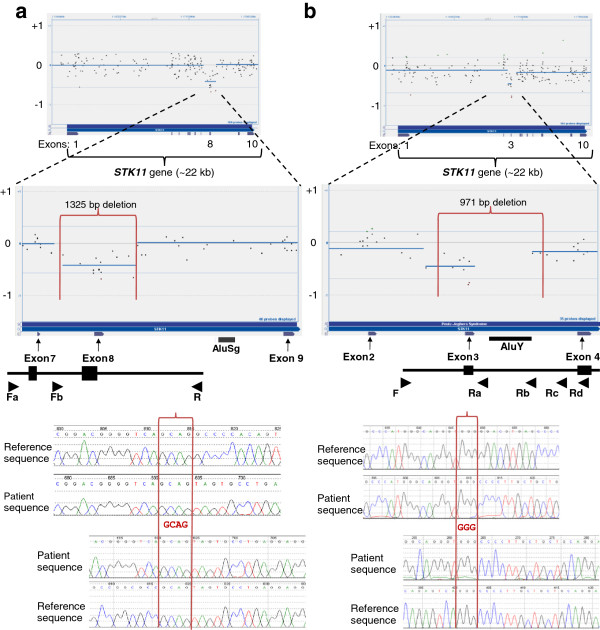
Single exon deletions in the STK11 gene. Figure 2 shows the data from two independent aCGH analyses with probes targeting the entire STK11 gene, and the zoomed-in view of where the deletion was present. CBS-generated deletion call did not cross the -0.6 threshold in either analysis. The breakpoints of the actual deletion are shown with vertical red lines. Below the CytoSure display are the corresponding exon tracks. Locations of Alu elements in the region of the deletion are marked. At the bottom is an illustration of the breakpoint PCR design, with the location of respective primers shown as arrows. Electropherograms of bidirectionally sequenced deleted alleles are shown with sequencing in the forward direction on top and reverse sequencing below. The four- and three-base pair microhomology at the breakpoints are shown within the two vertical red lines that demarcate the breakpoints. 2a) 1325-bp deletion encompassing exon 8 of the STK11 gene, with electropherogram of sequenced STK11 across deleted exon 8. 2b) 971-bp deletion encompassing exon 3 of the STK11 gene, with electropherogram of sequenced STK11 across deleted exon 3.
In the second PJS patient, a deletion call encompassing exon 3 did not cross the -0.6 log2 ratio threshold set, but was appreciated in manual review (Figure 2b). In contrast to the previous case, the call was based on 9 probes. However, the patient’s clinical presentation, as reported by the referring physician, was highly suggestive of PJS syndrome. A 971-bp deletion encompassing exon 3 of the STK11 gene was subsequently confirmed and breakpoints mapped with allele-specific PCR and sequencing (see additional file 2). Several probes that map within the deletion did not show hybridization ratios, as would be expected with the deletion in one allele.
801-bp deletion resulting in partial deletion in the PAH gene
A sample from a biochemically diagnosed 6-year-old phenylketonuria (PKU) patient was received for PAH gene sequencing. One copy of a previously reported missense mutation, c.838G > A (p.E280K), was detected.[26] Since a second mutation was not detected, deletion/duplication analysis was ordered. A deletion was identified with four probes partially covering exon 6 and six probes covering a few hundred bases of immediately flanking intron 6 sequence (Figure 3a). Four probes over the 5’ end of exon 6 showed a normal hybridization signal. To confirm the partial deletion of exon 6, breakpoint PCR was conducted; an 801-bp deletion, with an insertion of 11 bp between the breakpoints in intron 6 and exon 6, was confirmed. The inserted bases corresponded to the reverse compliment of the intron 6 breakpoint (figure 3a bottom). The fragment analysis of the breakpoint PCR, and the complete sequence of the deletion locus is included in Additional file 3. A SINE (Short Interspersed Element) of the MIRb subfamily maps within the deleted sequence, 32 bases from the intron 6 breakpoint.
Figure 3.
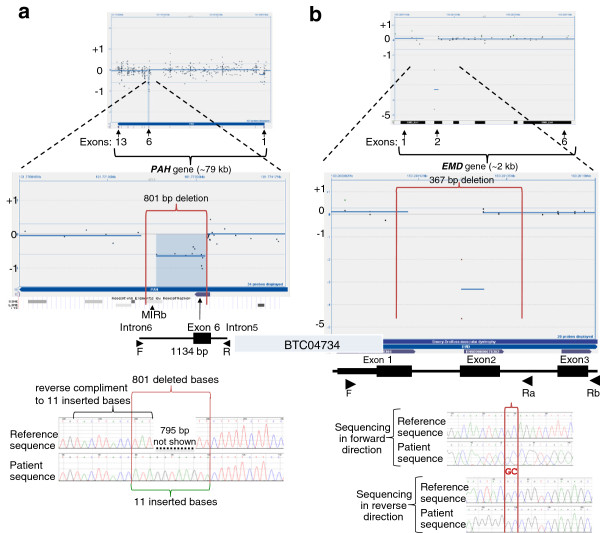
Deletions with a breakpoint in exons. Figure 3 shows CytoSure display of aCGH data with probes targeting the PAH gene and EMD. The top panel displays the entire gene view, and the lower panel zooms into the region showing a deletion. Below the CytoSure display are the corresponding exon tracks, and the UCSC RepeatMasker track showing repeat elements at the deletion locus. Vertical red lines mark the breakpoints. At the bottom is an illustration of the PCR design. Primers are shown as arrows. 3a) 801-bp deletion encompassing part of exon 6 of the PAH gene, with electropherogram of sequenced PAH gene across deletion. The breakpoints are shown with vertical red lines. The inserted 11 bases, shown with a green bracket, correspond to the reverse compliment of the bases labeled with a black bracket in the reference sequence across one of the breakpoints. 3b) 367-bp deletion encompassing part of exon 1 and all of exon 2 of the EMD gene, with electropherogram of sequenced EMD gene across deletion. The two-base pair microhomology at the breakpoints is shown within the two vertical red lines that demarcate the breakpoints.
Subsequently, we detected one copy of the same PAH indel mutation allele, in a presumably unrelated, four-month-old who was diagnosed via newborn screening (NBS) to have elevated phenylalanine. PAH gene sequencing identified a c.168 + 1G > A splice donor site mutation (data not shown). Deletion/duplication analysis with aCGH detected intron6/exon6 deletion that was confirmed with the allele-specific PCR and sequencing developed for the previous patient.
267-bp deletion mutation encompassing exon 2 of the EMD gene
Emery-Dreifuss muscular dystrophy can be inherited in an autosomal recessive or an X-linked pattern, depending upon the gene that carries the mutation. Mutations in EMD cause X-linked Emery-Dreifuss muscular dystrophy [27]. We detected a 267-bp deletion encompassing exon 2 of the EMD gene in a 46-year-old male (Figure 3b). Despite the general criteria set for a minimum of four probes to determine a deletion call, two probes in a hemizygous condition were sufficient to prompt further investigation. Breakpoint mapping with allele-specific PCR revealed that certain probes within the deleted regions showed normal hybridization (Figure 3b). The fragment analysis of the breakpoint PCR, and the complete sequence of the deletion locus is included in Additional file 3.
12-bp intronic deletion in intron 5 of the BCKDHB gene
No mutation was detected by sequence analysis of the BCKDHA, BCKDHB, and DBT genes in a sample from a 13-year-old patient with a biochemical diagnosis of MSUD. However, aCGH analysis resulted in one CBS deletion call within the BCKDHB gene encompassing the 5’ end of exon 11. In addition, manual review picked up a possible small deletion at the 5’ end of exon 5 based on only two probes that map to the sense and antisense of the same 60-bp sequence. Breakpoint PCR and sequencing of amplicons across this region revealed a 12-bp deletion (Figure 4). This c.344-4del12 change is only three bp from the splice acceptor site at the 5’ end of exon 5 and was not detected in the original sequence analysis, since the original amplification primers hybridized to the deleted region, resulting in allelic dropout. The sequencing primer had to be placed very close to the splice site to avoid amplification through an intronic AT-rich simple repeat region distally. The fragment analysis of the breakpoint PCR, and the complete sequence of the deletion locus is included in Additional file 4.
Figure 4.
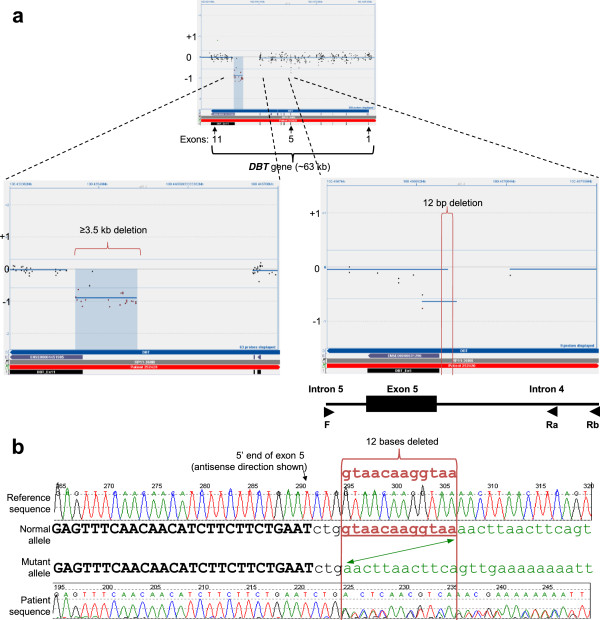
Compound heterozygous deletions in the DBT gene. Figure 4 shows CytoSure display of aCGH data with probes targeting the entire DBT gene, and zoomed-in views of two deletions: a large, >3.5-kb deletion encompassing the 5’ end of exon 11 is on the left, and a small 12-bp deletion in intron 4, three bases from the intron 4/exon 5 boundary, is on the right. The breakpoints of the small deletion are shown with vertical red lines. At the bottom is an illustration of the breakpoint PCR design, with the location of primers shown as arrows. Figure 4b shows the electropherogram indicating the deletion of 12 bases in one allele of the patient. The breakpoints are shown with two vertical red lines.
False deletion call due to an insertion mutation in the POMT1 gene
In some suspected intragenic deletions, further investigation leads to the discovery of an insertion, SNP, or point mutation causing poor hybridization. For example, sequence analysis of the POMT1 gene identified a copy of a c.2167dupG mutation in exon 20 in a patient with Walker-Warburg syndrome (WWS) (data not shown) [28]. Finding one definite mutation triggered reflex deletion duplication testing. Analysis by aCGH showed four overlapping probes over exon 3, suggesting a possible deletion (Figure 5a). Breakpoint PCR and sequence analysis identified an Alu insertion (c.160_161ins349). The c.160_161ins349 Alu insertion has been previously reported in a patient with WWS in cis with a c.2203C > T nucleotide change in exon 20 that was also found in our patient [29]. By placing the forward primer within the Alu sequence, the presence of the Alu was confirmed. The fragment analysis of the breakpoint PCR is included in Additional file 5.
Figure 5.
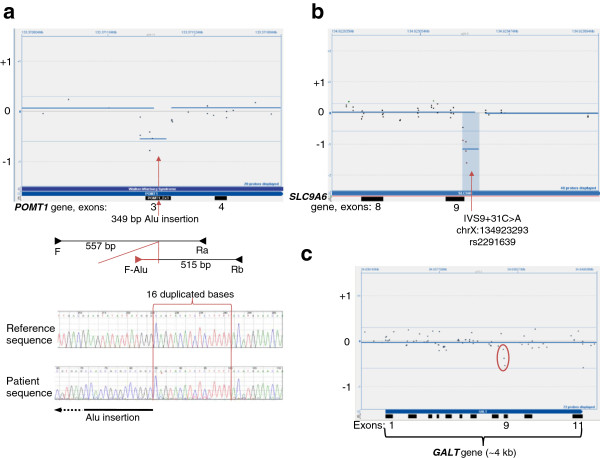
False deletion calls. Figure5 shows CytoSure display of three independent sets of aCGH data, where probe hybridizations on three separate genes resulted in false deletion calls. 5a) Zoomed-in view of the POMT1 gene, where red arrow marks the breakpoint in exon 3, where an Alu insertion interrupts probe hybridization. Below the CytoSure view is an illustration of the breakpoint PCR design, with the location of primers shown as arrows. At the bottom is an electropherogram of the allele with the Alu inserted. The 16 bases, duplicated in the two ends of the inserted element, are shown within two vertical red lines. 5b) Zoomed-in view of the SLC9A6 gene, where red arrow marks the location of a SNP in intron 9 that is targeted by all the probes that triggered the false deletion call. 5c) The entire GALT gene, with three probes showing slightly poor hybridization due to compound heterozygous missense changes.
False-positive deletion call due to hemizygous SNP in the SLC9A6 gene
A mutation in the X-linked SLC9A6 gene in males results in intellectual disability, epilepsy, and ataxia, a phenotype similar to Angelman syndrome [30]. A possible deletion at the 5’ end of exon 9 of the SLC9A6 gene in a three-year-old male patient was found to be a false-positive call. Since this is an X-linked gene, a true deletion call is expected to cross well below the threshold. The deletion call here did fall below the -1, but not to the degree expected in a hemizygous deletion (Figure 5b). Sequencing of exon 9 and flanking intronic sequences revealed a hemizygous single nucleotide polymorphism SNP (c.1140 + 31C > A; rs2291639). All probes suggesting a deletion encompass this SNP and result in poor hybridization on the aCGH. An electropherogram trace encompassing this SNP is included in Additional file 5.
Low probe hybridization due to compound heterozygous missense changes in the GALT gene
A five-year-old patient with galactosemia was referred for GALT gene sequencing [31]. Sequence analysis of exon 9 of the GALT gene identified one copy of the c.855G > T (p.K285N) mutation and one copy of a c.844C > G (p.L282V) nucleotide change of unknown significance, in this individual. Parental testing showed that these two missense changes (a mutation and a variant of unknown significance) were in trans. Since the c.844C > G (p.L282V) nucleotide change has not been previously reported in a patient with galactosemia, aCGH was ordered to rule out the possibility of a deletion or duplication (Figure 5c). The three probes highlighted with a red circle overlap both the missense changes, demonstrating low hybridization that can be appreciated upon manual review. An electropherogram trace encompassing these nucleotide changes is included in Additional file 5.
Discussion
Custom-designed high-density oligonucleotide arrays for molecular diagnostics are used to target specific disease-associated genes and are designed to detect single and multiple exon deletions and duplications [7,13,19,20]. The limit of detection in terms of the size of pathogenic deletions has improved immensely with the implementation of high-resolution, gene-targeted aCGH in diagnostic genetics [32,33]. The smallest size of deletion that analysis software can detect depends upon the density of probes targeting that sequence and the criteria set for software-generated calls. For example, if four consecutive probes targeted an overlapping sequence, and all four crossed the threshold set to detect deletions in our method (-0.6 log2 ratio), then a call could be generated even for deletions smaller than the length of the probes. As the size of a deletion gets closer to the limit of detection, the confidence in a call becomes weaker, and an alternate confirmation is necessary. Investigating suspicious events with breakpoint mapping helped us elucidate the true detection limit of our gene-targeted aCGH design. Several cases where software-generated calls did not cross the threshold nevertheless aroused suspicion upon manual review and warranted further investigation.
Detection of deletions is highly sensitive in the hemizygous genotype of males with X-linked disease. It is important to obtain relevant clinical information, family history, and any biochemical findings to help interpret the results of molecular testing; identification of a single copy of one mutation by gene sequencing for a patient suspected of an autosomal recessive disorder is also an indication to investigate any suspicious microarray data.
The smallest deletion we detected with aCGH was the 12-bp intronic deletion in the DBT gene of a child with a biochemical diagnosis of MSUD (Figure 4). The call made due to this deletion was only due to the hybridization of two probes targeting the same 60 bp and was only appreciated upon manual review. The location of these probes mapped to the sequence that the primer used in sequencing. Therefore, this deletion was not detected by sequencing due to allelic dropout, highlighting the fact that it was ultimately detected based on high clinical suspicion, the presence of one copy of another mutation in the same gene, and keen manual review.
Selection of probes and the density and redundancy in the coverage in the array design, are critical in the detection of intragenic CNVs. Not all probes perform equally well. Both deletions in the STK11 gene described here failed to generate a call that crossed the threshold set for deletions (-0.6 log2 ratio). Several probes within the deletions had discrepant array and breakpoint PCR data (Figure 2 and 3). These data may indicate that the probes may be prone to non-specific signal and should be redesigned or removed. However, it is important to recognize the possibility that sequences within a deleted region may have translocated to another location within the gene, or elsewhere in the genome, and consequently may carry the potential for clinical implications. However in the STK11 deletions presented, the clinical findings are consistent with the deletions alone.
Most deletions reported here had microhomology of at least a few bases at the breakpoints. This is consistent with the replication-based mechanism and break-induced repair (BIR) mechanism hypothesis for such events [4,34,35]. Interestingly, in the 801-bp deletion encompassing part of exon 6 of the PAH gene, there was an 11-bp insertion that corresponds to the reverse compliment of bases along the intron 6 breakpoint, demonstrating the involvement of at least two double-strand breaks in the mechanism resulting in this deletion (Figure 3). The familial HPRT1 deletion also had an insertion of 69 bp (Figure 1). In this mutation, the inserted bases aligned to a region on chromosome 5 (chr5p13.1:40,844,202-40,844,270/hg18) that has no homology to the locus on the X-linked gene. A second recurrent theme at breakpoints is close proximity of SINEs (Short INterspersed Elements) or other repeat tracks, suggestive of non-allelic homologous recombination.
In spite of the fear that higher probe density generates more noise in aCGH data, in our experience, the greater the number of probes within a deleted area generally helped in its detection. This is true even when there is redundancy in the probes; for example, the EMD gene deletion, where the same 60 bp were targeted by probes complimentary to the two strands. There was a definite call made by CBS software for the 801-bp PAH gene deletion. In contrast, the larger STK11 deletions did not cross the threshold for the software to generate a call. This difference is due entirely to probe density, which is higher for the PAH gene in our array. With sufficient data, probe performance can be evaluated and array design modified for optimal sensitivity. Possible deletions that were deemed false positive did demonstrate how single nucleotide changes could decrease the hybridization of probes, highlighting the sensitivity of this technology (Figure 5). In one case, the call generated did lead to the identification of a pathogenic Alu insertion. We have found that familiarity with specific probe performance within a gene helps differentiate between informative variation and noise.
It is important to remember that oligonucleotide arrays have the same limitations as any method that relies on hybridization to unique sequence probes. Therefore, repeat sequences are not targeted, and pseudogenes and homologs will interfere with assessments. Also, the information on copy number variation gives no insight on the orientation or location of insertions, duplications or rearrangements.
Gene targeted aCGH technology described here is complementary to diagnostic analyses utilizing next generation sequencing (NGS) that have been rapidly adopted in clinical laboratories, especially for genetically heterogenous diseases where more than one gene can contribute to a disorder. Several gene panels are being offered by clinical laboratories, for example gene panels for X linked intellectual disability, cardiomyopathy, neuromuscular disorders and congenital disorders of glysosylation. Detection of small indels from NGS data is still not optimal, and detection of CNVs via NGS cannot be easily adopted in clinical laboratories since the required lowered stringency would introduce a high false positive variant call rate. Gene targeted can easily fill these gaps and make the gene panels complete by offering combination of NGS based test and gene targeted arrays to detect the near complete range of mutations detected in genes.
Conclusion
We present the examples of pathogenic intragenic deletions ranging from several kilobases to as small as 12 bases, to highlight the limit of detection with high-density gene-targeted aCGH. Although probe coverage and performance are critical parameters to consider, however, there is not a minimum criteria of probe density that can be applied across all genomic sequences of interest. Based on our experiences, rigorous efforts to detect the smallest of these intragenic CNVs extend beyond simple aCGH analysis algorithms. As the size of deletion gets smaller, the cumulative data from all encompassing probes is insufficient to make a confident call. CBS software does allow identification of these events during manual review, even when the call does not cross the threshold set for the detection. For detection of these smaller CNVs, we routinely investigate further if one or more of the following criteria are met: a) the call was generated with at least two entirely non-overlapping probes, b) the location of the call overlaps with a primer used in sequencing that may have caused allelic dropout, c) the disease gene is recessive, with one mutation within the gene identified, or d) the disease gene is dominant, with a strong clinical suspicion in the patient. Ultimately, these data can be used to track individual probe performance across samples to improve the sensitivity of the array. In conclusion, high-density targeted aCGH is a very powerful tool for detection of intragenic deletions, and the identification of novel intragenic deletions and duplications will help expand the known spectrum of disease-associated genes.
Methods
Array design
All array data discussed in this manuscript were generated using the custom-designed EGL_NMD_NBSplus_v1 array. This 4X180K array was developed on the Agilent Technologies (Santa Clara, CA) aCGH platform using the Genefficiency service (Oxford Gene Technology (OGT), Yarnton, Oxford OX5 1PF UK). OGT uses proprietary ink-jet in situ printer technology (IJISS) developed by Rosetta InPharmatics (Kirkland, WA) and Agilent Technologies that allows in situ synthesis of long oligonucleotides. The probes are ~60 bp in length and annotated against NCBI build 36.1 (UCSC hg18, March 2006). This array has 207 control probes and 15,028 backbone probes spread in between regions of interest. 157,448 probes are targeting 261 genes (see Additional file 6).
aCGH protocol
DNA was extracted from whole blood collected in EDTA (purple-top) collection tubes and from amniocytes received for prenatal testing using the Puregene DNA extraction kit (Qiagen, Valencia, CA) according to the manufacturer’s recommendation. aCGH was performed following the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA). Each patient’s DNA was spiked with a combination of PCR products (spike-in) unique to each sample per array. The reference DNA was used from two pools (male and female) from normal individuals, run as a same-sex control. DNA was sonicated using a Branson Sonifier 450 with cup horn (Danbury, CT) and visualized on a two-percent agarose gel prior to labeling, as a quality control measure. Each patient and reference DNA was labeled with Cy3 and Cy5 9mer primers, respectively. Purification of labeled products, hybridization, and post-wash of the array was carried out according to Agilent’s recommendation and with their proprietary solutions. Array slides were scanned with Agilent’s High-Resolution C Scanner and extraction software.
aCGH analysis
CytoSure Interpret software 02002 (OGT) was used for analysis of array data (referred to as CytoSure). The program uses the Circular Binary Segmentation (CBS) algorithm to generate segments along the chromosomes that have similar copy number relative to reference chromosome [21]. Averaging of the segments is with median value of all segments on a chromosome as the baseline. Deletion or duplication calls are made using the log2 ratio of each segment that has a minimum of four probes. Threshold factor for deletions was set as a log2 ratio of -0.6 that is less stringent than the theoretical log2 score of -1 (heterozygous deletion log2(1/2) = -1; No change in allele number log2(2/2) = 0; heterozygous duplication log2(3/2) = 0.59). The software uses the standard deviation of the log2 ratio to calculate a deviation log ratio (DLR), which is used as a quality control check. A DLR of 0.08-0.19 is accepted, 0.20-0.29 is borderline, and ≥0.30 is rejected. The DLR for all arrays shown was scored by this scale. Data is analyzed only for the gene ordered for testing. The data for others genes is masked and not analyzed.
Breakpoint mapping design
CytoSure segment calls were used to generate minimum and maximum genomic coordinates of possible aberrations using NCBI build 36.1 (UCSC hg18, March 2006). The UCSC Genome Browser was used to determine the composition of the involved DNA [36,37]. We assessed repeat tracks and segmental duplications, as well as all annotated SNPs [24,38-43]. Breakpoint mapping by PCR was used to confirm deletion calls encompassing all or part of at least one exon. Primers for breakpoint PCR were designed using Light Scanner Primer Design software (Idaho Technologies Inc, Salt Lake City, UT). Several primer sets were designed by walking along the DNA sequence proximal and distal to the possible CNV.
PCR
All primers were ordered from Integrated DNA Technologies, Inc. (Coralville, IA). In all PCRs, 50 ng of genomic DNA was amplified in a 50-μl reaction that had a final composition of 2 U FastStart Taq, 1X FastStart Taq buffer with MgCl2, and 0.2 mmol/L dNTPs (Roche Applied Science, Indianapolis, IN), as well as 10 pmol forward and reverse primers (for primers sequences see Table 2). The PCR cycling had an initial melting at 95°C for 3 min followed by 40 three-temperature cycles (60 s at 95°C, 60 s at the lower of the two primer Tms, and 72°C for 1 min). The 40 cycles were followed by a final extension at 72°C for 7 min, and then held at 4°C. 15 μl of each PCR product was visualized on a 2% agarose gel (Fisher Scientific, Waltham, MA). Primers that successfully amplified across the breakpoints for cases described are listed in table 1.
Table 2.
Table lists primers used in confirming breakpoint mapping for the cases listed in this manuscript
| Primer name | Primer sequence |
|---|---|
| PAH_bkpt_ex6_F |
GCTAAATTAACATCCTCTTGACAGAA |
| PAH_bkpt_ex6_R |
ACCCTTTCATGTGGGAAATC |
| STK11_bkpt_ex8_Fa |
CCAGTGGCCTTGGGAGAA |
| STK11_bkpt_ex8_Fb |
GAGATGCGCCAGGAAGG |
| STK11_bkpt_ex8_R |
GGCTGGCTGCCAATGTG |
| STK11_bkpt_ex3_F |
GTTGTGGGCCATTTTGGT |
| STK11_bkpt_ex3_Ra |
TGGCCTCACGGAAAGGA |
| STK11_bkpt_ex3_Rb |
CAGCAAAGATGGAGGCG |
| STK11_bkpt_ex3_Rc |
ATTTTCCTGTGGGCCACAGG |
| STK11_bkpt_ex3_Rd |
AATCAGCTGACAGAAGT |
| HPRT1_bkpt_ex5_Fa |
TATATGACAGAGTATGATGAGAGCTACA |
| HPRT1_bkpt_ex5_Fb |
GCCTCATTCTTATAACTAGCATAAGAAC |
| HPRT1_bkpt_ex5_R |
ACAGTGGCTCATGCCTAT |
| EMD_bkpt_ex2_F |
CTCGGCCGGTTTTGGTA |
| EMD_bkpt_ex2_Ra |
CAGACTTCCCTCCCCTTTCT |
| EMD_bkpt_ex2_Rb |
AGGTCTCAGGTCCTCCCTGT |
| DBT_bkpt_int4_F |
CAGAGATACAAATGTACACTTCCTA |
| DBT_bkpt_int4_Ra |
AGTTGTGTTTTCCTATTCTGAAGTAGTT |
| DBT_bkpt_int4_Rb |
TGACATATCCACCAGGTACTAATAATTAAA |
| POMT1_bkpt_ex3_F |
CAGGATTAGCCTTGCGTC |
| POMT1_bkpt_ex3_Ra |
GGCAAGCAATAAACAAGATGC |
| POMT1_bkpt_ex3_Rb |
TCTGTGGGACTAGGTATGAAAGG |
| F-Alu_sense |
GTCTCGATCTCCTGACCTCG |
| SLC9A6_ex9_F |
TCTACTGTGAAGAAAGAACCTCAG |
| SLC9A6_ex9_R |
GGAAGAGGAGCCAAAATGAG |
| GALT_ex9_F |
CTAGGCACTGGATGGAGGTT |
| GALT_ex9_R | TCACTAGGCTGAGCCCCAGG |
Sequencing
PCR products were purified using the Millipore Ultrafiltration PCR purification kit (Millipore, Billerica, MA). Sequencing reactions (15 μl total) were prepared with the BD v3.1 sequencing kit (Applied Biosystems, Foster City, CA). Each PCR product was sequenced bidirectionally using the amplification primers. The sequencing reaction products were purified using a Sephadex cleanup plate (Edge Biosystems, Gaithersburg, MD) according to the manufacturer’s instructions. Products were heat-denatured (5 min at 95°C) and sequenced on a 3730xl capillary sequencer (Applied Biosystems, Carlsbad, CA). Sequence analysis was performed using Mutation Surveyor v2.61 software (SoftGenetics, State College, PA).
Abbreviations
PCR: Polymerase chain reaction; aCGH: array Comparative Genomic Hybridization; MLPA: Multiple ligation-dependent probe amplification; CBS: Circular binary segmentation; CNV: Copy number variation; SNP: Single nucleotide polymorphism; SINE: Short interspersed element; NHEJ: Non-homologous end-joining; MMBIR: Microhomology-mediated break-induced replication; DMD: Duchenne muscular dystrophy; MSUD: Maple syrup urine disease; LNS: Lesch-nyhan syndrome; HPRT: Hypoxanthine guanine phospho-ribosyl-transferase; PJS: Peutz-jeghers syndrome; PKU: Phenylketonuria; EMD: Emery-dreifuss muscular dystrophy; WWS: Walker-warburg syndrome.
Competing interests
All work presented was performed at Emory Genetics Laboratory. Since the completion of the work, author SHA has been employed by Medical Neurogenetics Inc, and author EC has been employed by Oxford Gene Technology. All other authors declare no conflict of interest.
Author’s contributions
SHA designed and carried out the breakpoint mapping for confirmation, and drafted the manuscript. Targeted aCGH was developed, validated and implemented under the leadership of MH and EC. All authors were involved with aCGH data review. All authors read and approved the final manuscript.
Authors’ information
This work was part of SHA’s project assignment during ABMG (American Board of Medical Genetics) fellowship at Emory University, under the guidance of MH.
Supplementary Material
2319-bp deletion in the HPRT1 gene.
1325-bp deletion in the STK11 gene.
801-bp deletion in the PAH gene and 267-bp deletion in the EMD gene.
12-bp deletion in the DBT gene.
False deletion calls in the POMT1 , SLC9A6 and GALT genes.
List of genes included in the custom-designed gene-targeted array.
Contributor Information
S Hussain Askree, Email: askree@gmail.com.
Ephrem LH Chin, Email: ephrem.chin@ogt.com.
Lora H Bean, Email: ljbean@emory.edu.
Bradford Coffee, Email: bcoffee@emory.edu.
Alice Tanner, Email: aktanne@emory.edu.
Madhuri Hegde, Email: mhegde@emory.edu.
Acknowledgements
We thank Oxford Gene Technology in their help in optimizing probes on this custom build aCGH.
References
- Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1(6):e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10(8):551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5(1):e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131(7):1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde MR, Chin EL, Mulle JG, Okou DT, Warren ST, Zwick ME. Microarray-based mutation detection in the dystrophin gene. Hum Mutat. 2008;29(9):1091–1099. doi: 10.1002/humu.20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16(23):11141–11156. doi: 10.1093/nar/16.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs AH, Hoffman EP, Snyder JR, Arahata K, Specht L, Shapiro F, Angelini C, Sugita H, Kunkel LM. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet. 1991;49(1):54–67. [PMC free article] [PubMed] [Google Scholar]
- Abbs S, Bobrow M. Analysis of quantitative PCR for the diagnosis of deletion and duplication carriers in the dystrophin gene. J Med Genet. 1992;29(3):191–196. doi: 10.1136/jmg.29.3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraishi Y, Kato S, Ishihara T, Takano T. Quantitative Southern blot analysis in the dystrophin gene of Japanese patients with Duchenne or Becker muscular dystrophy: a high frequency of duplications. J Med Genet. 1992;29(12):897–901. doi: 10.1136/jmg.29.12.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics. 2005;6(1):29–35. doi: 10.1007/s10048-004-0204-1. [DOI] [PubMed] [Google Scholar]
- del Gaudio D, Yang Y, Boggs BA, Schmitt ES, Lee JA, Sahoo T, Pham HT, Wiszniewska J, Chinault AC, Beaudet AL. et al. Molecular diagnosis of Duchenne/Becker muscular dystrophy: enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative genomic hybridization. Hum Mutat. 2008;29(9):1100–1107. doi: 10.1002/humu.20841. [DOI] [PubMed] [Google Scholar]
- Bovolenta M, Neri M, Fini S, Fabris M, Trabanelli C, Venturoli A, Martoni E, Bassi E, Spitali P, Brioschi S. et al. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC Genomics. 2008;9:572. doi: 10.1186/1471-2164-9-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Kidd JM, Eichler EE. Human copy number polymorphic genes. Cytogenet Genome Res. 2008;123(1–4):234–243. doi: 10.1159/000184713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami P, Coffey AJ, Abbs S, Vermeesch JR, Dumanski JP, Woodward KJ, Andrews RM, Langford C, Vetrie D. Exon array CGH: detection of copy-number changes at the resolution of individual exons in the human genome. Am J Hum Genet. 2005;76(5):750–762. doi: 10.1086/429588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saillour Y, Cossee M, Leturcq F, Vasson A, Beugnet C, Poirier K, Commere V, Sublemontier S, Viel M, Letourneur F. et al. Detection of exonic copy-number changes using a highly efficient oligonucleotide-based comparative genomic hybridization-array method. Hum Mutat. 2008;29(9):1083–1090. doi: 10.1002/humu.20829. [DOI] [PubMed] [Google Scholar]
- Staaf J, Torngren T, Rambech E, Johansson U, Persson C, Sellberg G, Tellhed L, Nilbert M, Borg A. Detection and precise mapping of germline rearrangements in BRCA1, BRCA2, MSH2, and MLH1 using zoom-in array comparative genomic hybridization (aCGH) Hum Mutat. 2008;29(4):555–564. doi: 10.1002/humu.20678. [DOI] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA. et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31(12):1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayeh MK, Chin EL, Miller VR, Bean LJ, Coffee B, Hegde M. Targeted comparative genomic hybridization array for the detection of single- and multiexon gene deletions and duplications. Genet Med. 2009;11(4):232–240. doi: 10.1097/GIM.0b013e318195e191. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5(4):557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Morton DH. Maple Syrup Urine Disease. Seattle (WA): GeneReviews™; 1993-2013. [updated 2013 May 09] [Google Scholar]
- Nyhan WL, O’Neill JP, Jinnah HA, Harris JC. Lesch-Nyhan Syndrome. Seattle (WA): GeneReviews™; 1993-2013. [updated 2010 Jun 10] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarrity TJ, Kulin HE, Zaino RJ. Peutz-Jeghers syndrome. Am J Gastroenterol. 2000;95(3):596–604. doi: 10.1111/j.1572-0241.2000.01831.x. [DOI] [PubMed] [Google Scholar]
- Mitchell JJ. Phenylalanine Hydroxylase Deficiency. Seattle (WA): GeneReviews™; 1993-2013. [updated 2013 Jan 31] [Google Scholar]
- Bonne G, Leturcq F, Ben Yaou R. Emery-Dreifuss Muscular Dystrophy. Seattle (WA): GeneReviews™; 1993-2013. [PubMed] [Google Scholar]
- Sparks S, Quijano-Roy S, Harper A, Rutkowski A, Gordon E, Hoffman EP, Pegoraro E. Congenital Muscular Dystrophy Overview. Seattle (WA): GeneReviews™; 1993-2013. [updated 2012 Aug 23] [Google Scholar]
- Bouchet C, Vuillaumier-Barrot S, Gonzales M, Boukari S, Bizec CL, Fallet C, Delezoide AL, Moirot H, Laquerriere A, Encha-Razavi F. et al. Detection of an Alu insertion in the POMT1 gene from three French Walker Warburg syndrome families. Mol Genet Metab. 2007;90(1):93–96. doi: 10.1016/j.ymgme.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H. et al. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet. 2008;82(4):1003–1010. doi: 10.1016/j.ajhg.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsas LJ. Galactosemia. Seattle (WA): GeneReviews™; 1993-2013. [updated 2010 Oct 26] [Google Scholar]
- Shaffer LG, Bejjani BA, Torchia B, Kirkpatrick S, Coppinger J, Ballif BC. The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am J Med Genet C Semin Med Genet. 2007;145C(4):335–345. doi: 10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S. et al. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9(9):607–616. doi: 10.1097/GIM.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42(5):385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P. et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A. et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297(5583):1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32:D23–26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22(9):1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
2319-bp deletion in the HPRT1 gene.
1325-bp deletion in the STK11 gene.
801-bp deletion in the PAH gene and 267-bp deletion in the EMD gene.
12-bp deletion in the DBT gene.
False deletion calls in the POMT1 , SLC9A6 and GALT genes.
List of genes included in the custom-designed gene-targeted array.