

Summary
Fragile X syndrome (FXS) is the most common heritable form of cognitive impairment. It results from epigenetic silencing of the X-linked FMR1 gene by a CGG expansion in its 5′-untranslated region. Taking advantage of a large set of FXS-affected human embryonic stem cell (HESC) lines and isogenic subclones derived from them, we show that FMR1 hypermethylation commonly occurs in the undifferentiated state (six of nine lines, ranging from 24% to 65%). In addition, we demonstrate that hypermethylation is tightly linked with FMR1 transcriptional inactivation in undifferentiated cells, coincides with loss of H3K4me2 and gain of H3K9me3, and is unrelated to CTCF binding. Taken together, these results demonstrate that FMR1 epigenetic gene silencing takes place in FXS HESCs and clearly highlights the importance of examining multiple cell lines when investigating FXS and most likely other epigenetically regulated diseases.
Highlights
-
•
FMR1 epigenetic gene silencing commonly occurs in the undifferentiated FXS cells
-
•
FXS HESCs are heterogeneous for repeat size and methylation levels
-
•
This study underscores the importance of multiple HESC lines in disease modeling
Fragile X syndrome (FXS) results from epigenetic silencing of the X-linked FMR1 gene. Using a large set of fragile X-affected human embryonic stem cell lines, Eiges and colleagues show that FMR1 epigenetic gene inactivation commonly occurs prior to embryonic tissue differentiation. This study underscores the importance of examining multiple cell lines in disease modeling, especially in epigenetically regulated disorders like FXS.
Introduction
Fragile X syndrome (FXS; OMIM #300624) is the most common heritable form of cognitive impairment. It is inherited as an X-linked trait and is caused by a deficiency in the fragile X mental retardation protein (FMRP) (Santoro et al., 2012). Most patients lack FMRP due to an unstable expansion of a CGG trinucleotide repeat sequence in the 5′-untranslated region of the FMR1 gene (Kremer et al., 1991; Oberlé et al., 1991; Verkerk et al., 1991; Yu et al., 1991). The number of CGG repeats varies so that normal individuals carry 5 to 55 repeat copies, while affected patients carry over 200 copies (full mutation). Expansion of repeat copy number to over 200 CGGs (Nolin et al., 1996) coincides with local acquirement of abnormal DNA methylation and the gain of repressive histone modifications typical to densely packed chromatin (like H3K9 and H3K27 trimethylation) (Coffee et al., 1999, 2002; Kumari and Usdin, 2010; Oberlé et al., 1991; Pietrobono et al., 2005; Tabolacci et al., 2005, 2008). These epigenetic modifications, which are presumed to be acquired in a developmentally regulated process, are responsible for FMRP deficiency and disease manifestation through transcriptional silencing of the FMR1 gene in affected fetuses as early as 6–13 weeks of age (Devys et al., 1992; Sutcliffe et al., 1992; Suzumori et al., 1993).
Formerly, we established a human embryonic stem cell (HESC) line from a fragile X-affected embryo, which was obtained through a preimplantation genetic diagnostic (PGD) procedure (Eiges et al., 2007). This cell line, termed HEFX, transcribes FMR1 mRNA levels that are comparable to the levels in wild-type (WT) HESCs. In addition, it is completely unmethylated, despite the presence of a full expansion. These findings have led us to propose that epigenetic gene silencing is conditioned by differentiation and that DNA methylation is a relatively late event in the silencing process. To further substantiate the notion that FMR1 is transcriptionally active in the undifferentiated state, we derived and fully characterized eight additional HESC lines established from fragile X-affected embryos. These and the former cell line were used to better define the timing and nature of FMR1 epigenetic gene silencing during early embryo development.
Results
FXS HESC Line Derivation
Twelve different mutant HESC lines were established from embryos with a CGG expansion greater than 55 repeats at the FMR1 gene (Table 1). The embryos, which were obtained through PGD, were donated by seven unrelated couples in which the mothers had a premutation at the FMR1 gene. All newly established cell lines display key features of pluripotent cells, namely unrestricted growth in culture, expression of undifferentiated cell-specific markers, and the potential to differentiate into a wide range of cell types (Figure S1 available online).
Table 1.
FXS HESC Line Collection
| HESC Line | Sex | Expansion Size | Family | Maternal Expansion Size |
|---|---|---|---|---|
| SZ-FX1 | XX | FM | B | 75 |
| SZ-FX3 | XX | FM | A | 100–300 |
| SZ-FX4 | XY | PM | B | 75 |
| SZ-FX5 | XX | PM | B | 75 |
| SZ-FX6 | XY | FM | C | 170 |
| SZ-FX7 | XX | FM (mosaic) | B | 75 |
| SZ-FX8 | XY | FM | D | 80 |
| LS-FX9a | XY | FM (mosaic) | G | NA |
| SZ-FX10 | XX | PM | B | 75 |
| SZ-FX12 | XX | FM (mosaic) | B | 75 |
| SZ-FX14 | XY | FM | E | 75 |
| HEFXb | XY | FM | F | NA |
Twelve different HESC lines were established from embryos with a CGG expansion in the FMR1 gene. The embryos, which were obtained through PGD, were donated by seven unrelated couples in which the mothers had a premutation at the FMR1 gene. Of the 12 available cell lines, 9 (including the former HEFX cell line) were found to carry a CGG expansion greater than 200 repeats. FM, full mutation (>200 repeats); NA, not available; PM, premutation (55–200 repeats).
Kindly provided by Racine IVF Unit, Tel Aviv Sourasky Medical Center.
Analysis of CGG Expansion by Southern Blot Assay
To determine the number of CGG repeats in each cell line and to generally assess the methylation state of FMR1 at the promoter, we employed a commonly used methylation-sensitive Southern blot assay that relies on DNA restriction with a methylation-sensitive enzyme (Rousseau et al., 1991). Employing this potent test has facilitated the identification of full mutations (>200 CGGs) in eight different cell lines: four females and four males, three of which are repeat size mosaics, meaning that they carry both premutation and full mutation alleles concurrently (LS-FX9, SZ-FX7, and SZ-FX12) (Figure 1A). All apart from two cell lines (SZ-FX7 and SZ-FX12) display aberrant methylation. We roughly estimate the length of the CGG expansion in the XY FXS HESC lines as 200–650 (HEFX), 200–330 (SZ-FX6), 200–300 (SZ-FX8), and 50–300 (LS-FX9) repeats when unmethylated, and ranging from 290–600 repeats when methylated (SZ-FX6, SZ-FX8, LS-FX9, and SZ-FX14). In a similar way, we estimate expansion size in XX FXS HESCs as 200–300 (SZ-FX7), 150–300 (SZ-FX12) when unmethylated, and ranging from 300 to 600 repeats when methylated (SZ-FX1 and SZ-FX3). While full mutations consistently show a smear of bands when unmethylated, discrete band patterns are observed when expansions are methylated. These results neatly illustrate the heterogeneous nature of our cell lines.
Figure 1.
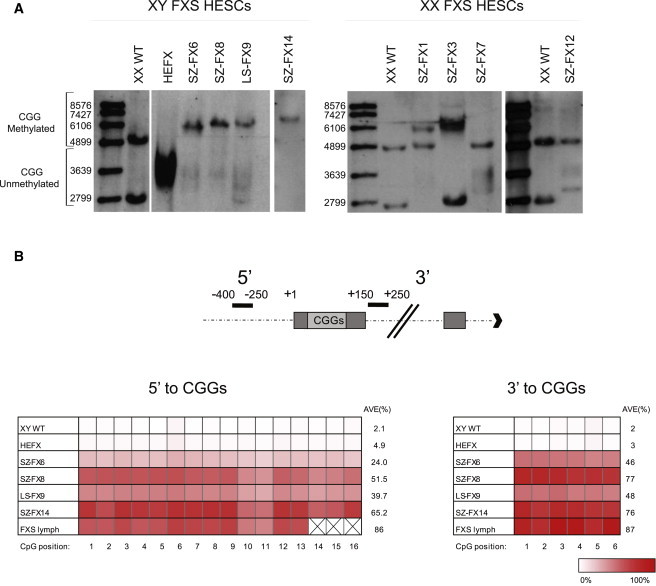
CGG Expansion Size and Methylation State in FXS HESC Lines
(A) CGG expansion size and methylation state were determined by a methylation-sensitive Southern blot assay. This test distinguishes between unmethylated normal (2.8 kb), premutation (2.9–3.4 kb), and full-mutation alleles (3.4–5.8 kb) and their methylated equivalents, as indicated by 5.2 kb, 5.3–5.8 kb, and fragments larger than 5.8 kb, respectively. Full mutations were identified in nine different cell lines (including HEFX): four females and five males. Note that SZ-FX7, SZ-FX12 and LS-FX9 are mosaic for the full mutation (carry both full and premutations) and that most cell lines are aberrantly methylated (SZ-FX1,3,6,8,14 and LS-FX9). Xi patterns are observed within XX FXS cells.
(B) Schematic illustration of the regions analyzed next to the repeats (top) and the bisulfite pyrosequencing results for DNA methylation levels at CpG sites localized 5′ (16 CpGs) and 3′ (6 CpGs) to the CGGs (bottom).
Given that FMR1 is liable to hypermethylation by X inactivation (Xi) and is polymorphic for CGG repeat number in all FXS XX HESCs, we can gain additional information regarding Xi patterns within the XX cells by the Southern blot test. We find that like many XX HESC lines, Xi has already occurred in all of the female cell lines (Shen et al., 2008) and is consistently skewed. The nature of the skewing, which was validated by a methylation-sensitive PCR-based assay (Kiedrowski et al., 2011), demonstrates that in three of four lines the normal X was inactivated (Figure S2A).
Analysis of CpG Methylation by Bisulfite Sequencing and Pyrosequencing
To better define abnormal methylation in FXS HESCs and finely characterize the extent of methylation close to the repeats, we applied bisulfite pyrosequencing, which relies on massive parallel sequencing, at regions that were previously shown by others to be differentially methylated in patients’ somatic cells (Godler et al., 2010). Methylation analysis was limited to XY cells, as this assay does not distinguish between Xi methylation and CGG full-expansion abnormal methylation in XX cells. We find that methylation levels widely vary among our XY FXS HESC lines, ranging from 24% (SZ-FX6) to 65% (SZ-FX14) upstream and 46%–77% downstream of the CGGs (Figure 1B). These levels are generally lower than in somatic cells of patients, which range between 73% and 95% (as determined by the analysis of primary cultures from five different individuals; Figure S2B), and are steady over time in culture, based on the analysis of HEFX and two other partly methylated FXS HESC lines (SZ-FX6 and SZ-FX14) (Figure S2C). In addition, we analyzed methylation levels by bisulfite single colony sequencing to determine methylation patterns at the resolution of single DNA molecules. Using this approach, we find a complete dichotomy of hypomethylated and hypermethylated alleles, reflecting two distinct FMR1 active and inactive states (Figure S2D).
Analysis of Histone Modifications and CTCF Binding in XY FXS HESCs by Chromatin Immunoprecipitation Analysis
To further substantiate the notion of epigenetic silencing being frequently elicited prior to differentiation, two histone modifications were examined in three of our XY FXS HESCs representing low (<5%, HEFX), intermediate (40%–48%, LS-FX9), and high (65%–76%, SZ-FX14) methylation states, by chromatin immunoprecipitation (ChIP). We show that methylation levels strongly correlate with local gain of repressive (H3K9me3) and loss of active (H3K4me2) histone modifications (Figure 2A). At the same time, to explore a potential mechanistic link between hypermethylation and CTCF binding, we examined whether methylation is associated with the binding loss of CTCF protein next to the repeats, as formerly suggested (Ladd et al., 2007). For this purpose, we assessed CTCF enrichment along the 5′ end of FMR1 in four different HESC lines by ChIP analysis. However, no enrichment for CTCF could be detected in both WT and affected HESCs (Figure 2D).
Figure 2.
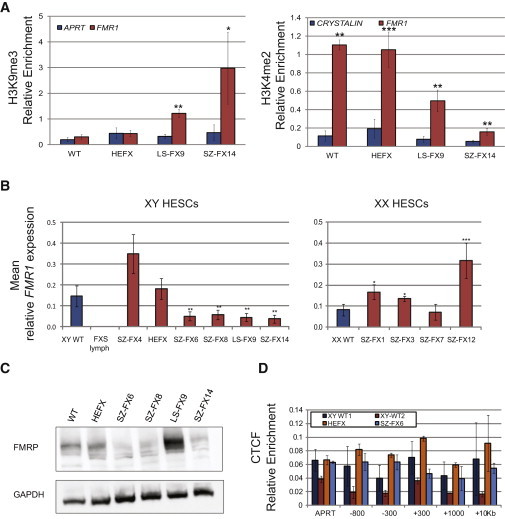
Histone Modifications, FMR1 Expression and CTCF Binding in FXS HESCs
(A) Real-time PCR ChIP analysis for H3K9me3 and H3K4me2 in WT and FXS-affected HESCs. APRT and HOXA9 were used as negative and positive controls for H3K9me3, respectively, while CRYSTALIN and APRT were used as negative and positive controls for H3K4me2, respectively (both positive controls were set to one and are not presented). The data in each panel represents an average of three to five independent ChIP experiments. Error bars represent standard error (paired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(B) Mean real-time RT-PCR analysis of FMR1 transcription in XY (left) and XX (right) WT, FXS affected, and premutation (SZ-FX4) HESC lines. The expression level of each cell line represents an average of three to seven independent experiments. Cycle threshold (Ct) values were normalized to the corresponding Ct value of GAPDH. XY WT cell lines are B200, and the XX WT cell line is B123. Error bars represent standard error (t test for unequal variances, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(C) Western blot analysis of FXS HESCs confirming the expressions of FMRP (∼72 kD) in WT and XY FXS HESCs. GAPDH (40 kD) was used as a loading control.
(D) ChIP analysis of CTCF relative enrichment along the FMR1 locus (−300 to +10 Kb relative to the transcription start site) in WT and FXS-affected HESCs. APRT was used as a negative control, and the average of DMPK (DM1) and FRATAXIN (FXN) genes was used as a positive control (the average of positive controls was set to one). The data in each panel represent an average of three independent ChIP experiments. Error bars represent standard error.
FMR1 Expression in FXS HESCs
To explore whether FMR1 is transcriptionally active in FXS HESCs, real-time RT-PCR was carried out in both XX and XY FXS and control (WT) HESC lines (Figure 2B). For each cell line, the level of transcription was determined by the analysis of at least two unrelated cell cultures. Based on quantitative RT-PCR (qRT-PCR) analysis, we show that FMR1 expression in HEFX cells (0% methylation) is indistinguishable from WT HESCs and is equally reduced in partly methylated FXS cell lines (t test for unequal variances, p ≤ 0.01). This is in contrast to SZ-FX4, a XY premutation cell line that exhibits a marked increase in FMR1 levels as observed in premutation carriers (Tassone et al., 2001). FMR1 transcript levels largely correlate with the extent of FMRP expression, as determined by western blot analysis (Figure 2C). This is true for all examined XY cell lines apart from LS-FX9, which demonstrates marked increase in FMRP, as compared with WT control (two biological replicas), despite reduced FMR1 mRNA levels.
Comparison of Histone Modifications and mRNA Levels between Isogenic Hypermethylated versus Hypomethylated FXS HESC Lines
To better establish the relationship between epigenetic modifications and gene expression at the cellular level, we subcloned undifferentiated SZ-FX6 and SZ-FX14 cells. Single-cell colonies were manually isolated following transfection with a GFP-NEO-resistant gene under the continuous selection of G418. We first screened for FMR1 expression by real-time RT-PCR (Figure 3A) to distinguish between FXS HESC clones that are entirely unmethylated/methylated. FMR1-expressing/nonexpressing clones were verified for methylation state by bisulfite pyrosequencing (Figure 3B). Indeed, FMR1-expressing clones were consistently unmethylated, while FMR1-nonexpressing clones were at all times heavily methylated. Full mutations were validated in all clones by Southern blot and/or PCR analysis (data not shown). In addition, we compared the enrichments of H3K4me2 and H3K9me3 between SZ-FX6 hypermethylated (SZ-FX6 cl.15B) and hypomethylated (SZ-FX6 cl.12B) clones and found a strong correlation between hypermethylation and extensive loss of H3K4me2, coinciding with the acquisition of H3K9me3 (Figure 3C), and vice versa for hypomethylated expansions. Taken together, based on the analysis of FXS HESC clones, we show that aberrant methylation is tightly linked with transcriptional silencing of FMR1 and cannot be separated from the change in histone modifications in HESCs.
Figure 3.
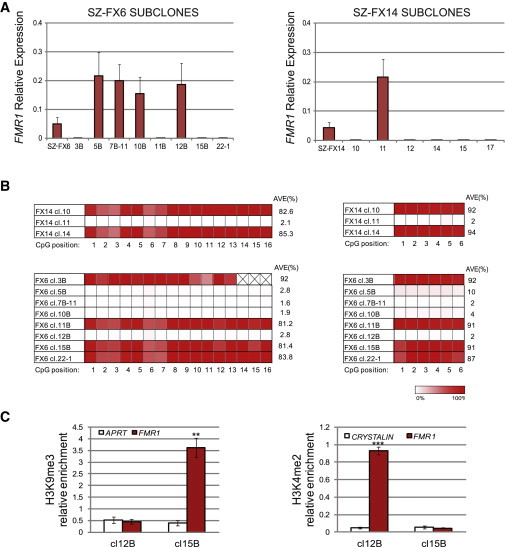
FMR1 Transcription, Methylation Levels and Histone Modifications in Subclones of FXS HESCs
(A) Mean relative FMR1 transcription levels of subclones from SZ-FX6 (left) and SZ-FX14 (right). Two independent qRT-PCR experiments were performed on a single passage culture. Ct values were normalized to the corresponding Ct value of GAPDH. Error bars represent standard error.
(B) Bisufite pyrosequencing for methylation levels near the CGGs (5′ left and 3′ right) in several of FXS HESC (SZ-FX6 and SZ-FX14) subclones.
(C) Real-time PCR ChIP analysis of H3K9me3 and H3K4me2 immediately upstream to the CGGs, in hypomethylated (SZ-FX6 cl.12B) versus hypermethylated (SZ-FX6 cl.15B) isogenic subclones. APRT and CRYSTALIN were used as negative controls for H3K9me3 and H3K4me2, respectively. The data in each panel represent an average of three to five independent ChIP experiments (paired t test, ∗∗p < 0.01, ∗∗∗p < 0.001). Error bars represent standard error.
Discussion
Here we report on altogether nine different HESC lines bearing pathologic expansions larger than 200 copies at the FMR1 gene. This collection of undifferentiated FXS cells, which is the largest to the best of our knowledge, comprises five male and four female cell lines. Taking advantage of this set of HESCs, we show here that FMR1 hypermethylation is frequently present in FXS HESCs (six of nine lines), ranging from 24% to 65% in the FMR1 promoter, unlike what was previously described. The inconsistency between this and former studies regarding the timing by which FMR1 is initially methylated stems from the different cell types/cell lines employed. While we focus on cell lines that are equivalent to the inner cell mass (ICM) of embryos prior to implantation, most other studies relied on the analysis of FMR1 epigenetic gene silencing in chorion villi of FXS fetuses, which often does not reflect that of embryonic tissues (Sutherland et al., 1991; Sutcliffe et al., 1992; Castellví-Bel et al., 1995). In addition, formerly we examined only one XY FXS HESC line (HEFX; Eiges et al., 2007), while here we extended this study and characterized eight additional XX and XY cell lines.
We demonstrate that the CGG expansions are either completely methylated or entirely hypomethylated, reflecting two distinct epigenetic states. Methylation levels remain unchanged over time in culture and are tightly coupled with a change in histone modifications: loss in H3K4me2 (active mark) and gain in H3K9me3 (repressive mark). This cannot be attributed to failed CTCF binding at the FMR1 locus, as previously proposed, since no enrichment for CTCF was found in FMR1 in both WT and affected HESCs. These results are different from the reports by Ladd et al. (2007) and Lanni et al., (2013) and may stem from the different cell types employed.
Regarding FMR1 transcription by FXS HESCs, we show that FMR1 expression in unmethylated FXS HESCs (HEFX, 0% methylation) is indistinguishable from WT HESCs and is significantly reduced in all methylated FXS cell lines. Nevertheless, the relation between the extent of methylation and FMR1 expression levels is not a simple correlation, as all methylated cell lines express FMR1 at similar levels. FMR1 transcription levels often (but not always) reflect FMRP expression. To better establish the association between methylation and FMR1 transcription, we compared undifferentiated isogenic clones that are hypermethylated versus hypomethylated expansions. While all of the unmethylated clones expressed FMR1, their methylated counterparts were completely deficient of FMR1 mRNA. These results further substantiate the relevance of DNA methylation to the silencing process in undifferentiated FXS HESCs. In addition, by comparing the enrichment of H3K4me2 and H3K9me3 between isogenic hypermethylated/hypomethylated clones, we show that aberrant histone modifications are at all times coupled and cannot be separated from DNA hypermethylation in HESCs.
Previous studies involving FXS HESCs, including some of our cell lines, point to the role of cell differentiation in the silencing of the FMR1 gene (Telias et al., 2013). In line with this view is the recent report of Colak and colleagues, who employed two FXS HESC lines to uncover an mRNA-mediated mechanism that drives epigenetic gene inactivation in a way that relies on neuronal differentiation (Colak et al., 2014). It remains to be determined whether this mechanism can act also at earlier developmental stages or if silencing is achieved in undifferentiated cells by a distinct mechanism. Furthermore, it also remains to be determined whether the loss of FMR1 mRNA and FMRP protein in the FXS HESC-derived neurons involves hypermethylation.
Our FXS HESCs widely vary in methylation levels (0%–65%, upstream to the CGGs by pyrosequencing). We show at the resolution of individual DNA molecules by bisulfite single colony sequencing that the methylation states of full mutations are binary, either completely methylated or entirely unmethylated. As FMR1 methylation levels remain unchanged over time in culture, it seems that unmethylated full expansions most likely arise due to imperfect de novo methylation rather than from an inability to reproducibly maintain aberrant methylation patterns. This may suggest that not all the cells in the ICM are evenly FMR1 methylated before or at the time of cell line derivation. We argue that the wide variability in methylation levels among the cell lines reflects a widespread event within mutant FMR1 embryos.
The failure to methylate expansions greater than 200 CGGs may point to a “window of opportunity” within which aberrant methylation can occur so that expansions that have coincidentally escaped de novo methylation persist. We propose a model that relates to the timing and nature of FMR1 hypermethylation, which suggests that abnormal methylation is first gained on full expansions in FXS at a restricted time point before/during blastocyst formation. Once established, it is irreversible and is clonally maintained. Expansions that fail to acquire abnormal methylation during this limited time frame remain unmethylated and under various differentiation conditions become silenced. In line with this model is the existence of methylation mosaicism among affected individuals (which is not that uncommon), the majority of whom express significant levels of FMR1 mRNA (Tassone et al., 2001). In addition, the proposed model also provides an appropriate explanation for the independent results of Urbach and Sheridan, who demonstrate that FMR1 hypermethylation, once established, is irreversible and cannot be erased by cell reprogramming through the formation of FXS-iPS cells (Sheridan et al., 2011; Urbach et al., 2010). In line with this perception, it should be feasible to obtain FXS-iPS clones that are FMR1 active from somatic cells with a full mutation provided that they carry an unmethylated full expansion at least in some of their cells. Indeed, we successfully established a fully reprogrammed (Figures S3A–S3D) and FMR1-active iPS clone with an unmethylated full mutation from patient fibroblasts with intermediate methylation levels (77%) (Figures S3E–S3G).
To summarize, this study demonstrates that FMR1 epigenetic gene silencing can take place in undifferentiated FXS cells and underscores the importance of examining multiple cell lines when investigating epigenetically regulated disorders.
Experimental Procedures
HESC Line Derivation
All cell lines were established in Shaare Zedek Medical Center (Institutional Review Board [IRB] 87/07) apart from LS-FX9, which was kindly provided by the Racine IVF Unit, Tel-Aviv Sourasky Medical Center (IRB 7/04-043). Cell line derivation and characterization were carried out as previously described (Eiges et al., 2007).
RT-PCR
Total RNA was isolated from the cells by TRI reagent extraction. RNA (1 μg) was reverse transcribed (Multi Scribe RT, ABI) with random hexamer primers. Real-time PCR was performed using Power SYBR Green Master Mix (ABI) on an ABI 7900HT instrument. Primers are listed in Table S1.
Western Blot Analysis
Cell pellets were collected in RIPA buffer (50 mM TrisHCl, 0.1% SDS, 0.5% Na Deoxycholate, 1% NP40, 150 mM NaCl, 1 mM phenylmethanesulfonylfluoride, and complete protease inhibitor [Roche]). Resulting lysates were then electrophoretically resolved and transferred onto a polyvinylidene fluoride membrane. Following blocking with 1% skim milk in Tris-buffered saline with Tween 20 (TBST), the membrane was probed either with a rabbit polyclonal antibody against FMRP (Abcam, ab17722, 1:1,000 dilution) followed by goat anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (1:5,000 dilution) or with a monoclonal antibody against GAPDH (Abcam, ab8245 1:2,000) followed by rabbit anti-mouse HRP-conjugated antibody (1:50,000). Detection was carried out using an enhanced chemiluminescence detection kit according to the manufacturer’s protocol (Biological Industries).
Southern Blot Analysis
Genomic DNAs (10–25 μg) were digested with EcoRI and EagI (NEB), separated on 0.8% agarose gels, blotted onto Hybond N+ membranes (Amersham), and hybridized with a PCR Dig-labeled probe (primers: 5′-GCTAGCAGGGCTGAAGAGAA-3′ and 5′-CAGTGGAGCTCTCCGAAGTC-3′).
Bisulfite Sequencing
Genomic DNA (2 μg) was modified by bisulfite treatment (EZ DNA Methylation-Direct Kit, Zymo Research) and amplified by FastStart DNA polymerase (Roche) using primers listed in Table S2. For pyrosequencing, additional internal sequencing primers were used as listed in Table S2. The PCR products were analyzed using PyroMak Q24 (QIAGEN).
ChIP
ChIP was performed according to the Upstate EZ ChIP kit protocol. Immunoprecipitation was performed using an anti-H3K4me2 (Upstate 07-030), anti-H3K9me3 (Abcam 8898), or anti-CTCF (Upstate 07-729) antibody. Real-time PCR was carried out using primers listed in Table S3. ΔΔCt values were normalized according to positive controls.
Transfection of HESCs
Cells were transfected with CMV-EGFP-N1 plasmid using LT1-TransIT transfection reagent (MIRUS) according to the manufacturer’s protocol.
Author Contributions
M.A., H.M.S., S.Y.D., S.A., and S.E.L. contributed to the conception and design of the study, the collection and assembly of data, data analysis and interpretation, and manuscript writing. G.A., P.R., T.E.G., O.S., and E.L.L. contributed to the collection of data. R.E. contributed to the conception and design of the study, financial support, data analysis and interpretation, and manuscript writing.
Acknowledgments
We thank Dr. David Zeevi for critical reading of the manuscript, the embryologists Aharon Peretz for his skillful assistance, the PGD molecular biologists Dr. Rachel Beeri from SZMC, Gidon Toperoff for his kind assistance with pyrosequencing, Dr. Dalit Ben-Yosef for the kind provision of the LS-FX9 affected HESC line, and Professor Douglas Melton and Professor Nissim Benvenisty for the provision of WT HESC lines (HES13, HES-B123, and HES-B200). This work was supported by the March of Dimes Foundation (1-FY09-474), the Chief Scientist Office of the Israel Ministry of Health (300000-5112), and the Marc Rich Foundation.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Castellví-Bel S., Milà M., Soler A., Carrió A., Sánchez A., Villa M., Jiménez M.D., Estivill X. Prenatal diagnosis of fragile X syndrome: (CGG)n expansion and methylation of chorionic villus samples. Prenat. Diagn. 1995;15:801–807. doi: 10.1002/pd.1970150903. [DOI] [PubMed] [Google Scholar]
- Coffee B., Zhang F., Warren S.T., Reines D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999;22:98–101. doi: 10.1038/8807. [DOI] [PubMed] [Google Scholar]
- Coffee B., Zhang F., Ceman S., Warren S.T., Reines D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 2002;71:923–932. doi: 10.1086/342931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D., Zaninovic N., Cohen M.S., Rosenwaks Z., Yang W.Y., Gerhardt J., Disney M.D., Jaffrey S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343:1002–1005. doi: 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D., Biancalana V., Rousseau F., Boué J., Mandel J.L., Oberlé I. Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am. J. Med. Genet. 1992;43:208–216. doi: 10.1002/ajmg.1320430134. [DOI] [PubMed] [Google Scholar]
- Eiges R., Urbach A., Malcov M., Frumkin T., Schwartz T., Amit A., Yaron Y., Eden A., Yanuka O., Benvenisty N., Ben-Yosef D. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Godler D.E., Tassone F., Loesch D.Z., Taylor A.K., Gehling F., Hagerman R.J., Burgess T., Ganesamoorthy D., Hennerich D., Gordon L. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010;19:1618–1632. doi: 10.1093/hmg/ddq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L.A., Raca G., Laffin J.J., Nisler B.S., Leonhard K., McIntire E., Mongomery K.D. DNA methylation assay for X-chromosome inactivation in female human iPS cells. Stem Cell Rev. 2011;7:969–975. doi: 10.1007/s12015-011-9238-6. [DOI] [PubMed] [Google Scholar]
- Kremer E.J., Pritchard M., Lynch M., Yu S., Holman K., Baker E., Warren S.T., Schlessinger D., Sutherland G.R., Richards R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- Kumari D., Usdin K. The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 2010;19:4634–4642. doi: 10.1093/hmg/ddq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd P.D., Smith L.E., Rabaia N.A., Moore J.M., Georges S.A., Hansen R.S., Hagerman R.J., Tassone F., Tapscott S.J., Filippova G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007;16:3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- Lanni S., Goracci M., Borrelli L., Mancano G., Chiurazzi P., Moscato U., Ferrè F., Helmer-Citterich M., Tabolacci E., Neri G. Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet. 2013;9:e1003601. doi: 10.1371/journal.pgen.1003601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolin S.L., Lewis F.A., 3rd, Ye L.L., Houck G.E., Jr., Glicksman A.E., Limprasert P., Li S.Y., Zhong N., Ashley A.E., Feingold E. Familial transmission of the FMR1 CGG repeat. Am. J. Hum. Genet. 1996;59:1252–1261. [PMC free article] [PubMed] [Google Scholar]
- Oberlé I., Rousseau F., Heitz D., Kretz C., Devys D., Hanauer A., Boué J., Bertheas M.F., Mandel J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Pietrobono R., Tabolacci E., Zalfa F., Zito I., Terracciano A., Moscato U., Bagni C., Oostra B., Chiurazzi P., Neri G. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 2005;14:267–277. doi: 10.1093/hmg/ddi024. [DOI] [PubMed] [Google Scholar]
- Rousseau F., Heitz D., Biancalana V., Blumenfeld S., Kretz C., Boué J., Tommerup N., Van Der Hagen C., DeLozier-Blanchet C., Croquette M.F. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 1991;325:1673–1681. doi: 10.1056/NEJM199112123252401. [DOI] [PubMed] [Google Scholar]
- Santoro M.R., Bray S.M., Warren S.T. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- Shen Y., Matsuno Y., Fouse S.D., Rao N., Root S., Xu R., Pellegrini M., Riggs A.D., Fan G. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc. Natl. Acad. Sci. USA. 2008;105:4709–4714. doi: 10.1073/pnas.0712018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan S.D., Theriault K.M., Reis S.A., Zhou F., Madison J.M., Daheron L., Loring J.F., Haggarty S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe J.S., Nelson D.L., Zhang F., Pieretti M., Caskey C.T., Saxe D., Warren S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- Sutherland G.R., Gedeon A., Kornman L., Donnelly A., Byard R.W., Mulley J.C., Kremer E., Lynch M., Pritchard M., Yu S., Richards R.I. Prenatal diagnosis of fragile X syndrome by direct detection of the unstable DNA sequence. N. Engl. J. Med. 1991;325:1720–1722. doi: 10.1056/NEJM199112123252407. [DOI] [PubMed] [Google Scholar]
- Suzumori K., Yamauchi M., Seki N., Kondo I., Hori T. Prenatal diagnosis of a hypermethylated full fragile X mutation in chorionic villi of a male fetus. J. Med. Genet. 1993;30:785–787. doi: 10.1136/jmg.30.9.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabolacci E., Pietrobono R., Moscato U., Oostra B.A., Chiurazzi P., Neri G. Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 2005;13:641–648. doi: 10.1038/sj.ejhg.5201393. [DOI] [PubMed] [Google Scholar]
- Tabolacci E., Moscato U., Zalfa F., Bagni C., Chiurazzi P., Neri G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008;16:1487–1498. doi: 10.1038/ejhg.2008.130. [DOI] [PubMed] [Google Scholar]
- Tassone F., Hagerman R.J., Taylor A.K., Hagerman P.J. A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J. Med. Genet. 2001;38:453–456. doi: 10.1136/jmg.38.7.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telias M., Segal M., Ben-Yosef D. Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013;374:32–45. doi: 10.1016/j.ydbio.2012.11.031. [DOI] [PubMed] [Google Scholar]
- Urbach A., Bar-Nur O., Daley G.Q., Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Yu S., Pritchard M., Kremer E., Lynch M., Nancarrow J., Baker E., Holman K., Mulley J.C., Warren S.T., Schlessinger D. Fragile X genotype characterized by an unstable region of DNA. Science. 1991;252:1179–1181. doi: 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.