

Abstract
Glioblastoma is a deadly malignant brain tumor and one of the most incurable forms of cancer in need of new therapeutic targets. As some cancers are known to be caused by a virus, the discovery of viruses could open the possibility to treat, and perhaps prevent, such a disease. Although an association with viruses such as cytomegalovirus or Simian virus 40 has been strongly suggested, involvement of these and other viruses in the initiation and/or propagation of glioblastoma remains vague, controversial and warrants elucidation. To exhaustively address the association of virus and glioblastoma, we developed and validated a robust metagenomic approach to analyze patient biopsies via high-throughput sequencing, a sensitive tool for virus screening. In addition to traditional clinical diagnostics, glioblastoma biopsies were deep-sequenced and analyzed with a multistage computational pipeline to identify known or potentially discover unknown viruses. In contrast to the studies reporting the presence of viral signatures in glioblastoma, no common or recurring active viruses were detected, despite finding an antiviral-like type I interferon response in some specimens. Our findings highlight a discrete and non-specific viral signature and uncharacterized short RNA sequences in glioblastoma. This study provides new insights into glioblastoma pathogenesis and defines a general methodology that can be used for high-resolution virus screening and discovery in human cancers.
Keywords: glioblastoma multiforme, virus discovery, high-throughput sequencing, metagenomic analysis, antiviral type I interferon response
The World Health Organization (WHO) classifies glial tumors as low and high grade according to their malignancy.1 Glioblastoma multiforme (GBM), representing astrocytoma grade IV, is a deadly malignant brain tumor and one of the most incurable forms of cancer. Despite major advances in clinical medicine, the median survival time is generally 15 months after diagnosis.2 The etiology of GBM remains obscure and only approximately 5% of gliomas represent familial aggregations, with some appearing in known syndromes.3 To date, the only proven environmental factor associated with an increased risk of glioma is exposure to ionizing radiation.4 Yet there is currently much debate over the potential involvement of viruses in GBM.
What’s new? —
Glioblastoma remains frustratingly difficult to cure. There is some evidence to suggest viruses might contribute to glioblastoma, a very tempting possibility, as the involvement of a virus could open doors to formulating novel treatments. However, the role of viruses is still vague and controversial. In this study the authors have developed a robust megagenomic approach to search tumor tissue for the presence of viruses, the first of its kind. They found no common or recurring active viruses, although they did detect a non-specific interferon pattern resembling an antiviral response.
Certain GBM specimens have been found to contain DNA sequences corresponding to the Simian virus 40 (SV40) large tumor antigen5,6 or human cytomegalovirus (CMV).7–11 CMV proteins have been suggested to promote tumor aggressiveness through increased tumorigenicity, invasion and angiogenesis.12 Furthermore, an antiviral response such as the upregulation of type I IFN/STAT1 genes correlates with poor survival outcome in a specific subtype of GBM patients.12 In line with these findings, anti-CMV treatment in GBM patients appears to extend survival rate.13 Yet contrary to these reports, other groups using similar methods did not detect CMV nucleic acids or proteins in GBM samples.14,15 Of note, such discrepancies have little correlation with the type of experimental methodology used in each of these studies. Investigations based on immunohistochemistry, polymerase chain reaction (PCR) or even short-term cultures on brain tumors lead to mixed results.7–9,11,14,15 The inconsistencies can be party attributed to the lack of positive infection controls (e.g., CMV positive glioma tissue) and the variable sensitivity of the end-point PCR used in these analyses. Certain physiological features, such as the epidemiological variation among specimens and the inherent heterogeneity of the tumors, may also lead to variation in the results. Furthermore, numerous technical aspects can be implicated; the use of RNA probes versus biotinylated DNA probes, the uniformity of the Bouin solution used for histological fixation and the differences in working with fixed versus frozen tissues. Yet despite these confounding results, a recent consensus report argues there is sufficient evidence to conclude that CMV sequences and viral gene expression exist in most GBM.12 However, the same report highlights that: (i) to date, no study has demonstrated the production of infectious CMV virions by glioma and, (ii) the existence of CMV in glioma does not appear to fit classic definitions of active or latent infection, casting doubt on any direct influence on GBM.
In contrast to the molecular studies targeting particular viruses, here we present results from a broad unbiased survey of viruses using experimentally characterized glioblastoma biopsies. Next-generation high-throughput sequencing (HTS) is emerging as an improved means to survey human disease origins and progression,16,17 as decreasing cost and increasing throughput make this approach an attractive compliment to traditional diagnostics. For example, one investigation used HTS to analyze human skin lesions and found that 97% of the virus sequences corresponded to human papillomavirus (HPV).16 These results are consistent with the biological context of the study, as HPV is known to infect keratinocytes in mucosa or skin and can elicit a wide range of diseases from benign lesions to invasive tumors. The sensitivity of this approach was somewhat limited, however, as some sample pools determined to be HPV-positive by PCR were found to be HPV-negative in the sequence analysis. Another recent study focused on the analysis of DNA viruses using public HTS data (The Cancer Genome Atlas) from 3,775 malignant neoplasms.17 While many tumor-associated DNA viruses (mostly hepatitis B virus [HBV] and HPV) were detected and catalogued, the approach was limited to the analysis of poly(A)-mRNA data and lacked validation in corresponding cancer tissue samples. Although these studies demonstrate the value of using HTS to detect virus signatures in human biological samples, the methodologies have limited sensitivity and lack corresponding experimental controls. Accordingly, while the generation of large volumes of sequencing data is a relatively straightforward task, thorough and biologically relevant interpretation of the data remains a significant challenge.
In order to provide novel insights into a potential virus–GBM association, we combined traditional clinical diagnostics with HTS. In this study, the availability of human GBM and epileptic biopsies provided a unique opportunity to experimentally validate the computational analysis of the sequence data. In light of the related work, we chose to deep-sequence total RNA from a smaller set of human glioblastoma samples to enable a comprehensive analysis that includes positive and negative controls, using known RNA and DNA viruses, in order to validate the results from the HTS analysis pipeline. To this end, in conjunction with traditional clinical diagnostic analyses, we deep-sequenced GBM biopsies, epileptic control biopsies and in vitro human tissue controls experimentally infected by viruses. As there is not yet any robust method for virus detection in clinical HTS data, we developed a novel sequence analysis pipeline that is able to both identify known viruses and distinguish potential virus-like sequences. In contrast to previous studies reporting the presence of viral signatures in GBM, our results show that despite finding an antiviral-like type I IFN response in human glioblastoma biopsies, no common or recurring active viruses were detected.
Material and Methods
Antibodies
The following primary antibodies against human antigens and human CMV antigens were used: rabbit anti-nestin, rabbit anti-glial fibrillary acidic protein (anti-GFAP) (all from Dako, Glostrup, Denmark, http://www.dako.com), mouse anti-βIII-tubulin (Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com) and mouse anti-Human CMV Immediate-Early antigens (Argene, Varilhes, France, http://www.argene.com).
Culture of undifferentiated ESC
The Embryonic Stem Cell (ESC) line H1 (WiCell Research Institute, Madison, WI, http://www.wicell.org) was maintained as previously described.18
RNA sequencing
Total RNA was extracted from patient biopsies using the RNeasy Mini Kit (Qiagen). Each sample was divided into two libraries to produce RNA-SEQ and RNA-SEQ N libraries. For the RNA-SEQ libraries, total RNA was fragmented using divalent cations. Fragments were reverse transcribed to obtain double stranded cDNA. Adapters were then ligated following the manufacturer’s instructions (Illumina Inc.). Fragments of size 220–300 nt (corresponding to inserts of size 160–240) were purified by gel acrylamide and PCR-amplified. HTS was performed on an Illumina HiSeq 2000 (1 × 100 cycles) (FASTERIS SA, Switzerland).
ENT infection with CMV
Neural differentiation of human ESC in air–liquid interface cell culture system was performed as previously described.18 Briefly, after 3–4 weeks of differentiation, ESC-derived neural tissue was infected with CMV at ±1 MOI per cell. The medium was changed every 2 days: 1 mL of differentiation medium was added underneath the membrane insert. Tissue infection was maintained for 7–10 days and IFN expression analysis and CMV detection were performed at 3, 5 and 7–10 days post-infection.
Virus isolation and analyses
Engineered nervous tissue (ENT) and tumor tissues were homogenized in 1 mL of viral transport medium, centrifuged and supernatant was used to inoculate human fibroblasts, A549, Rita, HeLa, Vero, LLCMK2 and MDCK cells, followed by incubation at 37°C for 21 days in a 5% CO2-containing atmosphere. Cells were monitored daily by light microscopy for the presence of cytopathic effect.
CMV serology
Patient’s plasma were screened for the presence of IgM and IgG antibodies specific to CMV using the Abbott Architect System CMV IgM and IgG assays, respectively, on an Architect i2000SR instrument (Abbott).
Immunofluorescence
Analyses were performed as previously described.18
Statistical analysis
All quantitative data presented are the mean ± SEM. Samples used and the respective n values refer to the number of independent experiments and are listed in the figure legends. Statistical analyses were performed using the Students t-test and ANOVA where p < 0.05 was considered significant.
Bioinformatics
Sample RNA libraries were bar-coded and sequenced in a multiplexed reaction on an illumina HiSeq 2000. Resulting 100 nt single-end reads were demultiplexed (libraries separated by their index) and reads passing quality standards (≥Q30) were used for further analysis. Per sample, reads were first mapped (bwa)19 to the human genome (NCBI GRCh37) and human transcriptome (UniGene Hs.seq.all) to screen human sequences. In an initial discovery phase, remaining reads were mapped to a database of known microbe genomes (EMBL-EBI bacteria, http://www.ebi.ac.uk/genomes/bacteria.html), with remaining reads subsequently mapped to known virus genomes (EMBL-EBI virus, http://www.ebi.ac.uk/genomes/virus.html). All remaining unmapped reads were assembled (SOAPdenovo trans),20 then assemblies generated from each sample were compared (NCBI Blastclust) to identify sequences shared among samples. Assembled sequences found in glioblastoma, but not in control sets, were subsequently analyzed in-depth. Each assembly was Blasted against three databases consisting of: virus (tblastx, translated assemblies blasted against translated virus genomes from EMBL-EBI virus), NR (blastx, translated assemblies blasted against NCBI Non-redundant GenBank CDS translations, PDB, SwissProt, PIR and PRF) and the human genome (blastn, NCBI GRCh37). For each sample, for each assembly, blast results for each database were summarized including blast statistics and read coverage.
Real-time (RT) quantitative PCR
Human reference RNA corresponding to a pool of 20 healthy donors provided from Ambion as well as melanoma, kidney, ovary, lung, breast, liver and colon RNA were used. RNA was extracted using RNeasy Mini Kit or RNeasy Midi kit (for tumor biopsies) and was primed with oligo(dt) and random primers for cDNA synthesis and reverse transcribed with a Takara Kit. RT-PCR was performed using SYBRGreen reagent and an ABI Prism 7000 sequence detection system (Applied Biosystems) according the manufacturer’s instructions. Eleven housekeeping genes were evaluated (Supporting Information Table 6) and Genorm method was used to choose the best housekeeping genes. Finally, TBP, ALAS1, GAPDH and EEF1 transcripts were quantified in all samples and used as housekeeping genes. The results were analyzed by the 2−ΔΔCt method and presented as the ratio between the selected genes and housekeeping gene transcripts. The selected gene/housekeeping ratio was then normalized to the mean ratio of the selected genes in the human reference RNA and/or the epileptic 1 or in ENTs alone to calculate tumor versus normal ratio. In the least, all experiments were performed in triplicates.
Nested touchdown PCR
PCR on CMV was performed essentially as previously described.7,14 Briefly, nested CMV PCR was carried out for 20 cycles with external CMV primers E1 and E2 (Supporting Information Table 6) which amplified a 268 bp fragment within the coding region of gB. About 5 μL of each reaction mix was transferred from external to internal CMV PCR and samples were reamplified for 30 cycles with primers I1 and I2. The PCR setup and post PCR work were performed in separate laboratories in order to reduce the possibility of contamination. DNA derived from ENTs infected with CMV was used as positive control. Amplified DNA products from all samples were visualized on agarose gels with ethidium bromide, bands were excised and DNA was extracted and sequenced (DNA Genetic Analyzer 3130XL Applied Biosystems). Correct CMV sequences were confirmed by NCBI BLAST.
RT-PCR screening for the presence of infectious agents
Tumor tissues were screened for the presence of the following viruses by specific RT-PCR: CMV, human herpes virus 6 (HHV6), varicella zoster virus (VZV), Epstein-Barr virus (EBV), herpes simplex virus types 1 and 2 (HSV-1, HSV-2), JC virus, parechovirus (PeV), enterovirus (EV) and measles virus (MeV) (Supporting Information Table 6). Briefly, tumor tissues were homogenized as described in the “Virus Isolation and Analyses” section. About 400 µL of the supernatant was used for nucleic acid extraction with easyMAG (bioMérieux). For the screening of PeV, MeV and EV viruses, cDNA was performed with both random hexamers (Roche) using the reverse transcriptase Superscript II (Invitrogen) according to the manufacturer’s instructions. RT-PCR screening was then performed with Taqman Universal Mastermix (Applied Biosystems) using the StepOne thermocycler (Applied Biosystems). The screens have been rigorously validated with a limit of detection (LOD) of 100 viral genome copies per milliliter (the same range as commercial quantitative PCR [qPCR] assays) and are quality controlled on an annual basis.
Results and Discussion
Type I interferon (IFN) signaling response is found in a subset of GBM specimens
The initial stage of an antiviral response in non-immune cells involves induction of type I IFN and IFN-related genes including RnaseL, OAS, ISG15, PKR and Mx1.21 Therefore, we first characterized the type I IFN signaling response in the GBM specimens. These IFN-related genes (14 in total) were analyzed by qPCR in a collection of 33 biopsies from various tumors including GBM, tumors localized to the central nervous system, tumors in other peripheral organs and brain metastases from other primary cancer types. Samples with elevated expression of IFN-related genes included three GBM, one squamous cell carcinoma, one invasive ductal carcinoma, one prostate adenocarcinoma, one breast carcinoma and one melanoma brain metastasis (Fig. 1; Supporting Information Table 1). Other GBM samples were distributed among subpopulations and exhibited low to moderate IFN-related gene expression levels. While we did indeed confirm increased gene expression levels in a substantial fraction of cancer biopsies (10/33), the expression patterns were not specific to GBM, and some biopsies did not express any IFN-associated genes (Fig. 1).
Figure 1.

IFN-related gene expression in GBM patient biopsies and other cancers. Expression levels of ISG20, ISG15, Mx1, OAS1, 2 and 3, IFITM3, IFIT1, IFI44, IFI44L, MDA-5, IRF7, STAT1 and RIG1 were detected by quantitative RT-PCR. The heat map depicts values of the fold increase expression of each gene normalized to two housekeeping genes (TBP and GAPDH). Hierarchical clustering illustrates the heterogeneity of gene expression profiles for each type of cancer. GBM biopsies that were deep-sequenced are marked with a red dot.
Only a subpopulation of GBM samples exhibits an increase in expression of type I IFN-related genes, an observation supported by other reports.22 Accordingly, components of the IFN-related signaling pathway have previously been associated with acquired tumor radioresistance in a preclinical model of head and neck cell squamous cancer,23 and in breast, prostate and glioma cells.24 Some previous studies showed that IFN/STAT1 signaling controls antitumorigenic effects through upregulation of caspases,25–28 cyclin-dependent kinase inhibitor 1A (CDKN1A),29 the IFN-regulatory factor 1 (IRF1)/p53 pathway30 and downregulation of the Bcl2 (B-cell CLL/Lymphoma 2) family.31 While no known viruses were found in any of the glioblastoma biopsies, the antiviral-like type I IFN gene activation signatures found in some samples could be explained by the fact that tumor infiltrating leukocytes produce IFNs, or by the sole effect of an antitumor immune response that produces IFNs.
Type I IFN signaling response in GBM is similar to antivirus response
The observed type I IFN-response could potentially be the consequence of a virus infection in the nervous tumor tissue. Therefore, brain-like engineered human nervous tissue, a system that we previously developed,18,32 was used to address this point. Human ENTs,18 derived from human pluripotent embryonic stem cells, were experimentally infected with CMV and Sendai virus. Immunofluorescence analysis clearly showed that neural cells (delineated by βIII-Tubulin immunoreactivity) in the ENT expressed IEA, a commonly used CMV marker (Fig. 2a). Upon infection, the virus propagated into the nervous tissue so that nearly all the tissue was infected 7 days post-infection. A weak CMV signal 3 days post-infection was observed via qPCR, then the virus propagated throughout the ENT by 7–10 days post-infection, followed by increased expression of type I IFN-related genes that include Mx1, OAS1, 2, 3 and MDA5 (Fig. 2b). Furthermore, IRF3, known to play a key role in the induction of type I IFN-related genes following virus infection, was significantly upregulated 5 days post-infection. The same gene expression patterns were also observed when ENTs were infected with Sendai virus (data not shown).
Figure 2.
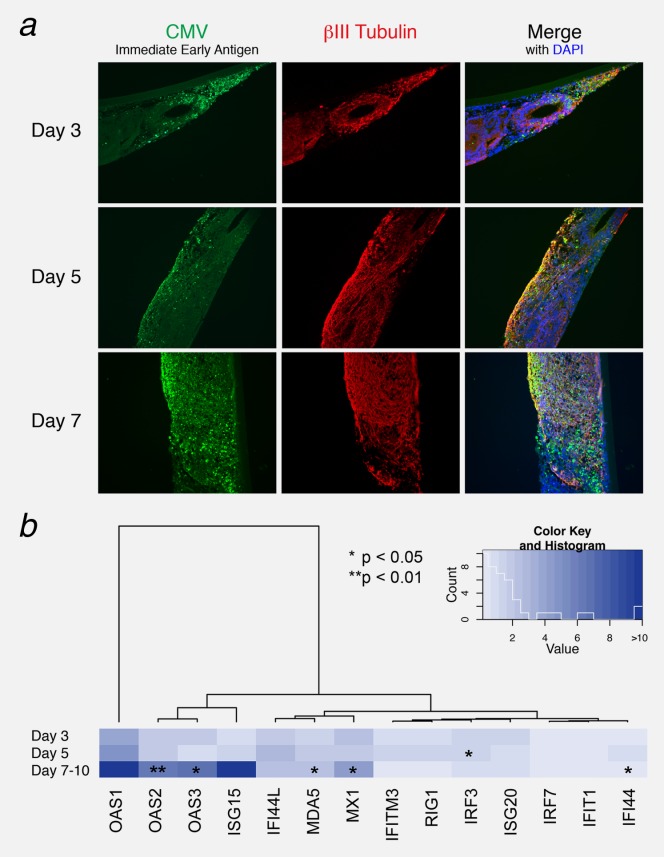
Infection of human brain-like tissue with CMV induces IFN-related gene expression. ENTs were infected with CMV and IFN-related gene expression was analyzed 3, 5 and 7–10 days post-infection. (a) CMV immediate early antigen (IEA) immunofluorescence detection. While little fluorescence was observed at 3 days, the entirety of the neural tissue (marked by beta3-Tubulin) was infected by 7–10 days post-infection. (b) Expression levels of type I IFN-related genes were analyzed by quantitative RT-PCR. The fold increase in expression of each gene was normalized to three housekeeping genes (TBP, ALAS1 and EEF1). Heatmap colors depict the fold increase in expression of each ENT + CMV relative to uninfected ENT at different times post-infection. Data are represented as mean (n = 3) ± SEM (*p < 0.05; **p < 0.01).
Of note, similar gene expression responses are observed both when ENTs are infected by viruses and when ENTs interact with glioblastoma cells.32 Indeed, this previous study demonstrates that GBM cells can develop within ENT and recapitulate the main features of GBM disease. Altogether, these in vitro and in vivo observations reinforce the concept that GBM development could be compatible with the presence of latent or active viruses.
No known active virus detected in GBM
Virus screening: Molecular diagnostic approaches
Traditional clinical diagnostics and HTS were subsequently used to exhaustively analyze the potential association between virus and GBM. Given the type I IFN gene expression profiles observed in some specimens, 20 GBM biopsies including the corresponding patient serum (upon availability), were screened via standard clinical diagnostic methods (semi-qPCR) for the presence of the following common neurotropic viruses: CMV, EBV, HSV, HHV6, MeV, PeV, JC virus, EV and VZV. Although some biopsies were associated with a type I IFN-response (based on Mx1 and OAS1, 2 and 3 expression), none of the above-mentioned viruses were detected in any sample (Supporting Information Table 2). Likewise, biopsies from three low-grade astrocytomas, one oligodendroglioma, two meningiomas, one ependymoma and one oligoastrocytoma were also found to be negative for these viruses. Furthermore, brain metastases from tumors of a different histological origin (breast cancer and melanoma) also tested negative. Only one meningioma biopsy gave a signal for EV at the LOD (Supporting Information Table 2). CMV was not detected in any tumor, although some patients had an IgG positive serology, most likely from a past infection (absence of IgM). As there is currently no common operational definition of CMV positivity in tumor samples, we also screened for CMV using other previously described methods7,14 such as nested touchdown PCR followed by semi-quantitative PCR (Fig. 3). While CMV was detected in the infected ENT, CMV was not found in any GBM biopsy, regardless of the experimental approach (Fig. 3). To vastly increase the scope of detection, and to complement these routine clinical diagnostic molecular techniques, a selection of biopsies and controls were then sequenced via HTS.
Figure 3.
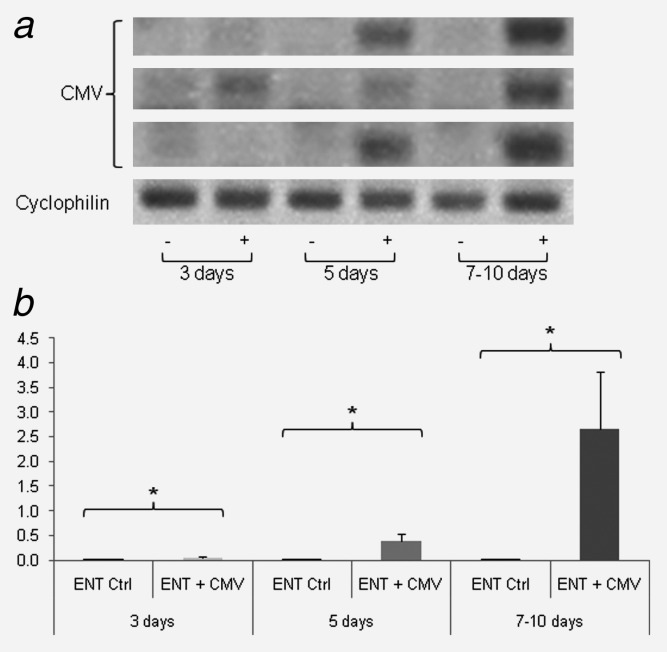
Nested PCR and qPCR of ENTs infected with CMV. ENTs were infected with CMV 3, 5 and 7 days post-infection. CMV was detected only in ENTs infected with the virus by Nested PCR (a) and semi-qPCR (b). Graphs depict the fold increase of CMV expression. Data are represented as mean (n = 4) ± SEM (*p < 0.05, “−” = non-infected ENT, “+” = infected ENT).
Virus screening: Metagenomic deep-sequence analysis
As next-generation HTS is a highly sensitive method capable of detecting specific molecular sequences, and has the potential to exhaustively screen all known virus genomes, we deep-sequenced five of the GBM biopsies, three epileptic control biopsies, and three in vitro control samples (ENT, ENT infected with CMV, and ENT infected with Sendai virus) to search for evidence of a virus signature. Total RNA isolated from the samples was used to generate libraries for single-end sequencing, generating millions of 100 nucleotide-long sequences (reads) per sample. Per glioblastoma sample, approximately 75–85% of the sequence reads mapped to human sequences, resulting in 147,821 annotated human genes (in terms of normalized mapped reads). In contrast to other recent work on HTS data analysis,17,33 the computational methodology presented here was validated by various experimental methods. As a first-pass validation, RT-qPCR results demonstrating a type I-IFN response were compared with the normalized human mRNA expression profiles generated from HTS data for each GBM sample (Supporting Information Table 3). The computational results are concordant with the experimental results, and reveal a type I IFN-related gene induction (IRFs, OASs, Mx1, MDA-5, RIG-1, etc.) (Supporting Information Tables 1 and 3).
To increase sensitivity and specificity of virus discovery in the HTS data, the computational pipeline (Fig. 4a) includes two discovery phases: (1) a search for high-identity nucleotide-level matches to known viral genomes followed by (2) a search for more distant relationships on an amino-acid level. The second phase captures sequences that may not align directly to known virus genomes (e.g., a novel strain not in the database, a distant relative of known strains, etc.) on the nucleotide level. Searching for similarities to known virus coding regions on the level of amino acids increases the likelihood of identifying matches to virus sequences. The raw sequence reads were first quality controlled by trimming and filtering according to standard protocols.34 To remove human sequences and hence reduce data complexity, reads were then mapped to the human genome (NCBI GRCh37) and transcriptome (to capture reads surrounding splice junctions) (UniGene Hs.seq.all) using algorithms designed for aligning short reads to long genomes (bwa).19 This human sequence filter stage effectively removed approximately 80–90% of the total reads from each sample (Figs. 4a and 4b). Sequences of microbial origin were filtered out by mapping the remaining reads against a database of approximately 2,150 genomes (EMBL-EBI bacteria). Any remaining reads with no database matches were subsequently mapped to a database of over 3,250 virus genomes (EMBL-EBI virus) to find high-identity matches to known viruses. To characterize GBM sample similarity in terms of sequence identity, every (non-human) read in each sample was compared with all reads in all other samples (GBM and controls) (Fig. 4c). Overall, only a small (∼2–5%) proportion of the reads were found to be specific to GBM, revealing no prominent sequence difference between GBM and controls.
Figure 4.
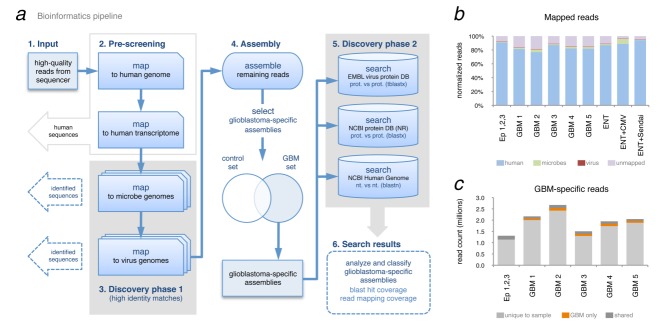
Metagenomic analysis of GBM deep-sequence data. (a) Bioinformatics pipeline developed to filter sequencing reads and discover known viruses (high identity matches step 3), assemble remaining reads and search for more distant similarities to virus (low-identity search step 5). (b) Proportion of mapped reads per sample, per organism. (c) Comparison of read-identity between GBM samples. Per sample, each sequence read with 100% identity to a read found in another sample is labeled as shared (among all samples including control), shared only in GBM samples or not shared (unique to that sample).
The pipeline was initially validated by deep-sequencing three control samples: ENT alone, ENT infected with CMV and ENT infected with Sendai virus. The CMV and Sendai virus signatures are detected in the corresponding controls, and the pipeline captures information along the entire length of each genome (Fig. 5a). Four different viruses were identified in these samples (parainfluenza virus 5 [PIV-5], phage phiX-174, CMV and Sendai virus). The phage phiX-174 was added to each sample preparation as a quality control for the sequencing. In the control samples, CMV and Sendai signals were recovered, with reads distributed along the length of their genomes (Fig. 5a). The PIV-5 is found in GBM and epileptic controls, and likely reflects the presence of this virus in the laboratory setting.
Figure 5.

Virus detection in GBM deep-sequence data. (a) Pipeline validation and positive controls. Discovery phase 1 can capture genome-wide signatures of both DNA (CMV, maximum 110 X-fold coverage) and RNA (Sendai maximum 2,308 X-fold coverage) viruses. (b) Detected virus genomes shown as percentage of total non-human reads mapped to each genome. (c) Virus genome percent coverage and average depth of coverage from discovery phase 1 for the four detected viruses (PIV-5, phage phiX 174, CMV and Sendai virus). Refer to guide on right side for interpreting chart regions (I) low % genome coverage with little depth (number of reads mapped to genome), (II) low % coverage—great depth, (III) high % coverage—little depth and (IV) high % coverage—great depth. (d) Identity of assembled sequences found in two or more GBM specimens. After finding common sequences in GBM-specific assemblies (100% nucleotide sequence identity found in 2, 3, 4 or 5 GBM specimens), each assembly was Blasted against virus, nr and human sequence databases. Assemblies were classified according to databases in which they had significant matches; often an assembly had significant (e-value ≤0.001) matches in more than one database. Sample sets with no common sequences are not listed (e.g., [1,2,4], [2,3,4], etc.).
To compare viral signatures among samples, both the percent genome coverage (regions of the genome covered by the reads) and the normalized X-fold coverage depth (amount of mapped reads at each genome position) were computed for all mapped reads in every sample (Fig. 5c). Typically, the detection of a strain or species is reported as the total count of mapped reads to the genome normalized to the genome length, referred to as genome coverage. However, with such a value, it is not possible to differentiate between a genome that is well covered with little depth and a genome that is partially covered with great depth (see Fig. 5c guide for more detail). Here we present, for the first time, a two-dimensional representation (percent coverage vs. depth of coverage) that provides a more detailed view of the detected genomes (Fig. 5c). With these plots, it is straightforward to observe if HTS reads are evenly distributed across the genome (which may reflect overall virus copy number in the sample), or if the reads originate from particular regions of the genome (corresponding to virus gene activation in the original sample), all while simultaneously representing the “signal strength” of a particular virus (as the X-fold depth).
Very low levels of PIV-5 are detected in most samples (less than half of the genome is covered by only one or two reads) and represent the limit of background signal. The control DNA (phiX 174) is detected in all samples, where the large majority (80–100% genome coverage) of the genome is found with good signal strength (∼2–14 fold depth). Furthermore, both DNA (CMV) and RNA (Sendai) viruses are detected with strong signal (4-fold and 180-fold depth, respectively) in the corresponding control samples (Fig. 5c). Note that the percent genome coverage values for these two positive controls (∼30–40%) indicates active virus, as most reads correspond to transcribed regions of the genome.
No particular differences were found among GBM biopsies and epileptic controls (Figs. 5b and 5c). As an additional control, given the debate over the involvement of CMV in GBM, total reads (without any filtering stages) from all samples were mapped to a select subset of virus genomes including CMV (even though the pipeline would automatically detect these genomes), in addition to the other eight neurotropic viruses tested above, in all GBM biopsies and in the pool of epileptic donors. No sample (GBM or controls) had any substantial number of reads mapped to these genomes (Supporting Information Table 4). However, while no strong viral signatures were detected, we cannot rule out the possibility that viruses such as CMV may play a role in GBM tumorigenesis and then become latent because they are not required for tumor maintenance.
In order to expand the search for virus signatures, any remaining reads with no match to known virus (or human or bacterial genomes) were assembled (SOAPdenovo)20 into longer contigs for further analysis in discovery phase II (Fig. 4a). In order to find GBM-specific sequences, any contigs not shared with the epileptic control set were further analyzed (Fig. 4a, Step 4). Each contig (∼600–800 contigs per GBM specimen) was Blasted against three different databases: a virus protein database (tblastx on translated EMBL genomes), the NCBI NR protein database (blastx) and the NCBI human genome database (blastn). Once Blast results for each contig were agglomerated (Supporting Information Table 5), contigs were then compared (all-against-all) to find those present in at least two GBM specimens (Fig. 5d) in search of sequences enriched in glioblastoma. The contigs were sorted by (i) their blast results from each database, (ii) their “signal strength” (quantity of reads mapped to that contig) and (iii) overall length. While some GBM specimens had contigs with significant matches to sequences only in the virus databases, these matches were generally short (9–12 amino acids long), ambiguous, and relatively uninformative (see Supporting Information Table 5 for more detail). Results from discovery phase 2 (Figs. 4a and 5d) revealed no noteworthy virus signatures. This computational assembly process was validated by PCR using primers designed from the sequences of the contigs with the strongest signals (number of mapped reads). PCR performed on the original GBM samples yielded products of correct sequence and size (Fig. 6). Using this two-stage virus discovery pipeline, aside from controls, no known viruses were detected in any of the GBM biopsies.
Figure 6.
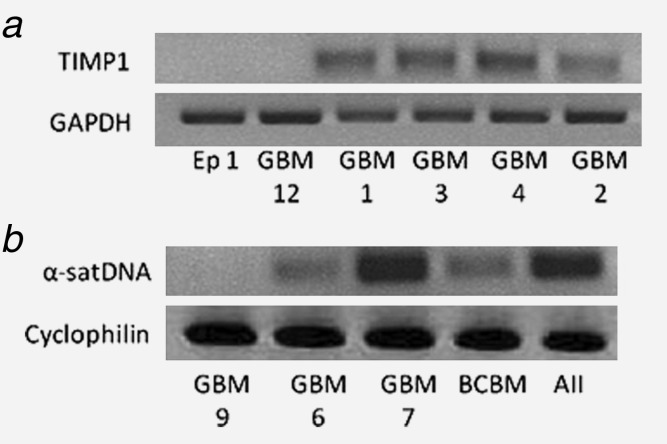
PCR validation of assembled contigs. The assembly process was verified by PCR followed by sequencing. The PCRs were performed on several biopsies: epileptic (Ep), Glioblastoma multiforme (GBM), breast carcinoma brain metastasis (BCBM) and astrocytoma grade II (AII). Amplified DNA products from all samples were visualized on agarose gels with ethidium bromide. PCR amplicons were then extracted and sequenced (DNA Genetic Analyzer 3130XL Applied Biosystems). (a) TIMP1 and (b) α-satellite DNA (α-satDNA) sequence identities were confirmed by NCBI BLAST.
Conclusion
This study is the first to harness HTS to comprehensively survey the virus landscape of GBM with corresponding experimental validation, and is designed to address the questions surrounding a possible virus–GBM association. Previously, these questions were difficult to answer due to the inherent technological limitations of molecular analysis. Here we provide, for the first time, a complete answer to this question by developing a robust virus discovery pipeline with an accurate and elegant means of representation. Moreover, we provide new insights reinforced by experimental validation and appropriate controls. We conclude that no known active human viruses, and notably no CMV signatures, were present in the GBM specimens analyzed, despite an antivirus-like, but non-specific, IFN response. Altogether, our findings illustrate how a metagenomic HTS approach can provide a means of high-resolution virus screening and discovery in cancer, and highlight the urgency of reassessing the significance of ongoing clinical studies targeting active viruses in GBM.
Acknowledgments
The authors thank Lorena Sacco and Aline Mamin (Virology laboratory, Geneva University Hospitals), Géraldine Philipin and Valérie Widmer (Oncology, Geneva University Hospitals), for their help in biopsy preparation. We thank Dominic Garcin for supplying the Sendai virus.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Schwartzbaum JA, Fisher JL, Aldape KD, et al. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503; quiz 1 p following 16. doi: 10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109:93–108. doi: 10.1007/s00401-005-0991-y. [DOI] [PubMed] [Google Scholar]
- Kouhata T, Fukuyama K, Hagihara N, et al. Detection of simian virus 40 DNA sequence in human primary glioblastomas multiforme. J Neurosurg. 2001;95:96–101. doi: 10.3171/jns.2001.95.1.0096. [DOI] [PubMed] [Google Scholar]
- Zhen HN, Zhang X, Bu XY, et al. Expression of the simian virus 40 large tumor antigen (Tag) and formation of Tag-p53 and Tag-pRb complexes in human brain tumors. Cancer. 1999;86:2124–2132. [PubMed] [Google Scholar]
- Cobbs CS, Harkins L, Samanta M, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62:3347–3350. [PubMed] [Google Scholar]
- Mitchell DA, Xie W, Schmittling R, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008;10:10–18. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier J, Uro-Coste E, Pommepuy I, et al. Detection of human cytomegalovirus genome and gene products in central nervous system tumours. Br J Cancer. 2005;92:747–750. doi: 10.1038/sj.bjc.6602339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheurer ME, Bondy ML, Aldape KD, et al. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008;116:79–86. doi: 10.1007/s00401-008-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas KG, Bao L, Bruggeman R, et al. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol. 2011;103:231–238. doi: 10.1007/s11060-010-0383-6. [DOI] [PubMed] [Google Scholar]
- Dziurzynski K, Chang SM, Heimberger AB, et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012;14:246–255. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg-Naucler C, Rahbar A, Stragliotto G. Survival in patients with glioblastoma receiving valganciclovir. N Engl J Med. 2013;369:985–986. doi: 10.1056/NEJMc1302145. [DOI] [PubMed] [Google Scholar]
- Lau SK, Chen YY, Chen WG, et al. Lack of association of cytomegalovirus with human brain tumors. Mod Pathol. 2005;18:838–843. doi: 10.1038/modpathol.3800352. [DOI] [PubMed] [Google Scholar]
- Poltermann S, Schlehofer B, Steindorf K, et al. Lack of association of herpesviruses with brain tumors. J Neurovirol. 2006;12:90–99. doi: 10.1080/13550280600654573. [DOI] [PubMed] [Google Scholar]
- Bzhalava D, Johansson H, Ekstrom J, et al. Unbiased approach for virus detection in skin lesions. PLoS One. 2013;8:e65953. doi: 10.1371/journal.pone.0065953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury JD, Tannir NM, Williams MD, et al. Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA-Seq. J Virol. 2013;87:8916–8926. doi: 10.1128/JVI.00340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preynat-Seauve O, Suter DM, Tirefort D, et al. Development of human nervous tissue upon differentiation of embryonic stem cells in three-dimensional culture. Stem Cells. 2009;27:509–520. doi: 10.1634/stemcells.2008-0600. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte CW, Willey CD, Zhi D, et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS One. 2012;7:e29653. doi: 10.1371/journal.pone.0029653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodarev NN, Beckett M, Labay E, et al. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101:1714–1719. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MH, Cook JA, Chandramouli GV, et al. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007;67:3845–3852. doi: 10.1158/0008-5472.CAN-06-4250. [DOI] [PubMed] [Google Scholar]
- Bhanoori M, Yellaturu CR, Ghosh SK, et al. Thiol alkylation inhibits the mitogenic effects of platelet-derived growth factor and renders it proapoptotic via activation of STATs and p53 and induction of expression of caspase1 and p21(waf1/cip1) Oncogene. 2003;22:117–130. doi: 10.1038/sj.onc.1206065. [DOI] [PubMed] [Google Scholar]
- Chin YE, Kitagawa M, Kuida K, et al. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol Cell Biol. 1997;17:5328–5337. doi: 10.1128/mcb.17.9.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister N, Shalaby T, von Bueren AO, et al. Interferon-gamma mediated up-regulation of caspase-8 sensitizes medulloblastoma cells to radio- and chemotherapy. Eur J Cancer. 2007;43:1833–1841. doi: 10.1016/j.ejca.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Sironi JJ, Ouchi T. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem. 2004;279:4066–4074. doi: 10.1074/jbc.M307774200. [DOI] [PubMed] [Google Scholar]
- Chin YE, Kitagawa M, Su WC, et al. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science. 1996;272:719–722. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- Townsend PA, Scarabelli TM, Davidson SM, et al. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem. 2004;279:5811–5820. doi: 10.1074/jbc.M302637200. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Brar BK, Knight RA, et al. Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-x promoters. Cell Death Differ. 2000;7:329–330. doi: 10.1038/sj.cdd.4400656. [DOI] [PubMed] [Google Scholar]
- Nayernia Z, Turchi L, Cosset E, et al. The relationship between brain tumor cell invasion of engineered neural tissues and in vivo features of glioblastoma. Biomaterials. 2013;34:8279–8290. doi: 10.1016/j.biomaterials.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Francis OE, Bendall M, Manimaran S, et al. Pathoscope: species identification and strain attribution with unassembled sequencing data. Genome Res. 2013;23(10):1721–1729. doi: 10.1101/gr.150151.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende DR, Waller AS, Sunagawa S, et al. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS One. 2012;7:e31386. doi: 10.1371/journal.pone.0031386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.