

Abstract
Testing for high-risk (hr) types of human papillomavirus (HPV) is highly sensitive as a screening test of high-grade cervical intraepithelial neoplastic (CIN2/3) disease, the precursor of cervical cancer. However, it has a relatively low specificity. Our objective was to develop a prediction rule with a higher specificity, using combinations of human and HPV DNA methylation. Exfoliated cervical specimens from colposcopy-referral cohorts in London were analyzed for DNA methylation levels by pyrosequencing in the L1 and L2 regions of HPV16, HPV18, HPV31 and human genes EPB41L3, DPYS and MAL. Samples from 1,493 hrHPV-positive women were assessed and of these 556 were found to have CIN2/3 at biopsy; 556 tested positive for HPV16 (323 CIN2/3), 201 for HPV18 (73 CIN2/3) and 202 for HPV31 (98 CIN2/3). The prediction rule included EPB41L3 and HPV and had area under curve 0.80 (95% CI 0.78–0.82). For 90% sensitivity, specificity was 36% (33–40) and positive predictive value (PPV) was 46% (43–48). By HPV type, 90% sensitivity corresponded to the following specificities and PPV, respectively: HPV16, 38% (32–45) and 67% (63–71); HPV18, 53% (45–62) and 52% (45–59); HPV31, 39% (31–49) and 58% (51–65); HPV16, 18 or 31, 44% (40–49) and 62% (59–65) and other hrHPV 17% (14–21) and 21% (18–24). We conclude that a methylation assay in hrHPV-positive women might improve PPV with minimal sensitivity loss.
Keywords: cervical intraepithelial neoplasia, DNA methylation, early detection of cancer, human papillomavirus 16, human papillomavirus 18, human papillomavirus 31, human papillomavirus DNA tests, uterine cervical neoplasms
Persistent infection with high-risk (hr) human papillomavirus (HPV) types is the main cause of cervical cancer. hrHPV testing is becoming the preferred primary screening test because it detects almost all women with high-grade cervical intraepithelial neoplasia (CIN2/3), for whom treatment may prevent cervical cancer. However, an important drawback is low specificity and positive predictive value (PPV) compared with cytology, which is the established cervical screening test. It is therefore of clinical interest to develop a molecular reflex test for triage that maintains the high sensitivity of HPV testing but has better specificity. Our objective was to develop such a prediction rule using combinations of human and HPV DNA methylation.
Quantitative measurement of DNA methylation at CpG sites of human or viral genes has shown promise for cervical and other cancers.1–4 We recently showed that DNA methylation in HPV16 infections was a good predictor of CIN2/3 in women with low-grade abnormalities detected by cervical cytology.5 In a similar vein, research by other groups has suggested using methylation of CpGs in the promoters or introns of human genes, including CADM, MAL, EPB41L3, TERT, PAX1, SOX1 and LMX1.6–9 Building on this research, the primary aim of our study was to develop a molecular test using HPV16, HPV18, HPV31 and human gene DNA methylation to predict whether women had CIN2/3 at the time of sampling. Around three in four cervical cancers are caused by these three HPV types,10 and the human genes might help to determine the others. The molecular test results may be used to predict risk by the development of a multivariate classification score, formed as a weighted average of the methylation results. Such a score may have clinical relevance by being used in triage to spare some women colposcopy and subsequent anxiety and overtreatment. The concern over missing some high-grade disease might be addressed by repeat HPV testing 1 year later those who are predicted to be at low risk.
What’s New? —
While high-risk (hr) human papillomavirus (HPV) testing is a highly sensitive technique for the early detection of high grade cervical intraepithelial neoplastic disease (CIN 2/3), its relatively low specificity and positive predictive value (PPV) limits its utility. To improve specificity and PPV, the authors of this study developed a prediction rule based on combinations of DNA methylation in the human gene EPB41L3 and three hrHPVs: HPV16, HPV18, and HPV31. Highly significant superior performance was obtained with the predictive rule, suggesting that methylation assays may be useful in cervical screening programs for the triage of hrHPV-positive women to colposcopy.
Our study used two large cohorts from colposcopy-referral populations in London, with biopsy-proven CIN2/3 status. Methylation was measured in 7 HPV16 sites in L1 and L2 regions as Ref.5; 29 HPV18 sites in regions L2, L1, URR and E6 and 29 HPV31 sites in regions L2, L1 and URR; and CpGs in human genes MAL, DPYS and EPB41L3. MAL and EPB41L3 were selected on the basis of previous data in cervical cancer; while DPYS has shown potential in other cancers.2,8,9 A multivariate classification score was developed and validated in independent data sets.
Material and Methods
Patients
The study included 1,493 women from the Predictors 1 (P1) and 2 (P2) studies at St. Mary’s and Hammersmith Hospitals in London, where they had been referred because of an abnormal screening smear (persistent borderline or mild, moderate or severe dyskaryosis). All women underwent colposcopic examination, with biopsy and treatment as appropriate. The studies were approved by the local research ethics committees and all women analyzed provided written consent; full details are available elsewhere.11,12
Specimen characteristics
Prior to colposcopy samples from a Cervex brush were placed into PreservCyt (Hologic, Danbury, USA) and stored at −70°C until the methylation assays were run. PreservCyt of 300 µl was centrifuged at 13,200 rpm for 2 min and the pellet was resuspended in 200 µl of phosphate buffered saline. DNA isolation and bisulfite conversion was according to manufacturer’s instructions. The genomic DNA was extracted with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), with an exception that DNA was eluted in 60 µl AE buffer. DNA of 250 ng was used in the bisulfite conversion reactions where unmethylated cytosines were converted to uracil with the EZ DNA methylation kit (Zymo research, Irvine).
Assay methods
Samples for HPV16, HPV18 and HPV31 methylation assays were selected by the Linear Array (Roche Molecular Systems, Pleasanton) and a quantitative polymerase chain reaction (PCR) test in P1, and the BD HPV test (BD Diagnostics, Burlington) in P2. The tests provided separate typing results, and are functionally equivalent to other hrHPV tests.12
Methylation assays were based on PCR and quantitative pyrosequencing as previously described.2,3,5 For HPV16, two sets of primers in L1 and L2 from Ref.5 were used. For HPV18 primer sets for 8 amplicons in L2, L1, URR and E6 covering 29 CpG positions, and for HPV31 7 primer sets in L2, L1 and URR covering 29 CpG positions were obtained using PyroMark Assay Design software version 2.0.1.15 (Qiagen). The number of CpGs selected for HPV16 was less than HPV18 and HPV31 because they were preselected from earlier analysis on HPV16.5 Primers covered dense CpG areas in a single amplicon less than 300 bp and did not overlap any CpG dyads. The internal control for total bisulfite conversion was a non-CpG cytosine in the region for pyrosequencing. Human genes CpG sites were tested within the vicinity of the promoters.
HPV16 DNA methylation assays were as described earlier.5 The concentration of each primer for the HPV31 assays and human genes was 0.2 µM; see Supporting Information for HPV18 assays.
Thermal cycling conditions for HPV18 were 95°C for 15 min, an optimized number of cycles: 30 sec at 94°C; 30 sec at the optimized annealing temperature; 30 sec at 72°C and a final extension between 5 and 10 min at 72°C. For HPV31, they were 95°C for 15 min and then 45 cycles: 30 sec at 94°C; 30 sec at the optimized annealing temperature; 30 sec at 72°C and a final extension between 5 and 10 min at 72°C. Finally, for the human genes, they were 95°C for 15 min then 45 cycles: 30 sec at 94°C; 30 sec at the optimized annealing temperature; 30 sec at 72°C, and a final extension of 10 min at 72°C.
PCRs were performed using a converted DNA equivalent of 1,600 cells using the PyroMark PCR kit (Qiagen). The cell genome equivalents of DNA calculations assumed 6.6 pg DNA per diploid cell. Master mix of 12.5 µl, 2.5 µl Coral red, 2 µl of DNA, 1 µl primer and an optimized concentration of MgCl2 adjusted with water to give a 25-µl reaction volume were combined. The size of the amplified DNA was confirmed by the QiaExel capillary electrophoresis instrument (Qiagen). PCR product of 10 µl was pyrosequenced using a PyroMarkQ96 ID (Qiagen) as Ref.2.
In each run, a nontemplate negative control was run in addition to a standard curve consisting of 1 pg/ml 0, 50 and 100% methylated HPV plasmid in a background of 10 ng/ml human DNA for HPV assays. For human gene assay, the standard curves of 0, 50 and 100% methylated human DNA were run as positive controls. The annealing temperature and MgCl2 concentrations were optimized by running four-gradient PCRs with ranging temperatures 49–56° with 0.5 mM increase in MgCl2. The controls showed a clear trend with inputs of increasing percentages of methylated versus unmethylated DNA showing linearly higher quantitative measurements. Observed versus expected ratios of the controls were close to 1 (data not shown).
Further details of primers and PCR conditions are in Supporting Information Tables 1–3.
Study design
A subset of the referral samples from the P1 (between 2005 and 2007) and P2 (2007 to 2009) studies were assayed; exclusions were solely due to sample availability, negative hrHPV test and cancer diagnosis (Fig. 1). The primary endpoint was histologically confirmed CIN2/3, taking the highest grade of abnormality seen in the punch or treatment biopsy specimen. Histopathology was first reported locally and then centrally reviewed.
Figure 1.
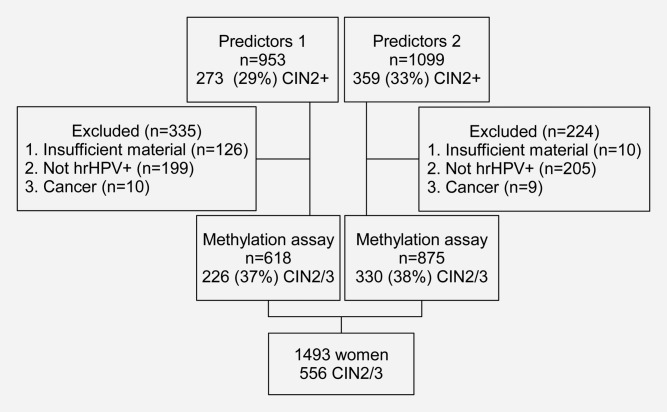
Study flow chart.
The CpGs listed in Supporting Information Tables 1–3 were assessed for inclusion in a classifier. HPV16, HPV18 and HPV31 genotype positivity was also considered where results were defined positive if valid DNA methylation, including a zero value, was detected at any of the CpGs assayed.
The rationale for sample size was based on the number of CIN2/3 cases in the HPV18 and 31 subtypes: in earlier work 25 CIN2/3 cases gave enough power for CpG sites in HPV16.5
Statistical analysis methods
Three stages were used. First, the assay results from P1 were analyzed, and a multivariate model was developed when blinded to the results from P2. Second, this model was validated on the P2 samples. Third, the model was refined based on the P2 samples and practical assay considerations, and a more simple classification score was evaluated using all of the data. Logistic regression was applied with a forward-stepwise algorithm for the development phase, so that variables were added based on their likelihood-ratio contributions. CpG combinations considered were based on exploratory analysis from P1, using the mean methylation or proportion of CpGs methylated. These were chosen because earlier analysis with HPV16 indicated that for some sites the most information might be whether a site is methylated or not (cf. HPV16 L2), and for others it is in the absolute value (cf. HPV16 L1).5 Failed methylation of a CpG site was imputed as zero methylation if methylation was detected in other CpG sites for the gene or HPV type. Model fit was assessed using likelihood-ratio χ2 statistics, and discrimination by receiver–operating characteristics (ROCs) and the area under the curve (AUC). Log10 p values were presented based on the χ2 statistics with one degree of freedom. HPV types were classified based on detectable methylation, where a sample was deemed positive if any of the CpGs was measured successfully (including zero methylation). These methylation-positive HPV types were tabulated by CIN categories. Plots of AUC by CpG site were used to summarize their discrimination ability; the distributions of methylation by CIN status at CpGs in the final model were also plotted. Attention was focused on 90% sensitivity cut points because this was considered appropriate for triage, where it might be unacceptable to miss more than 10% CIN2/3. The classification score distribution was compared by cytology categories to assess overlap. DeLong confidence intervals were used for AUC statistics,13 and Wilson confidence intervals for binary outcomes.14 Analysis was undertaken by using the statistical software GNU R 2.15.1.15
Results
Table 1 tabulates the HPV16, HPV18 and HPV31 types detected by the methylation assays by CIN status. It shows that the CIN2/3 proportion in the P1 and P2 studies was comparable by HPV type.
Table 1.
Summary of sample HPV results in total and in the Predictors 1 and 2 (P1 and P2) studies
| Group | n | <CIN1 | CIN1 | CIN2 | CIN3 | CIN2/31 |
|---|---|---|---|---|---|---|
| Total | 1,493 | 543 (36%) | 394 (26%) | 183 (12%) | 373 (25%) | 556 (37%) |
| P1 | 618 | 325 (53%) | 67 (11%) | 61 (10%) | 165 (27%) | 226 (37%) |
| P1 HPV16 | 238 | 83 (35%) | 21 (9%) | 24 (10%) | 110 (46%) | 134 (56%) |
| P1 HPV31 | 69 | 30 (43%) | 8 (12%) | 12 (17%) | 19 (28%) | 31 (45%) |
| P1 HPV18 | 100 | 54 (54%) | 13 (13%) | 5 (5%) | 28 (28%) | 33 (33%) |
| P1 other | 240 | 167 (70%) | 30 (12%) | 21 (9%) | 22 (9%) | 43 (18%) |
| P2 | 875 | 218 (25%) | 327 (37%) | 122 (14%) | 208 (24%) | 330 (38%) |
| P2 HPV16 | 318 | 53 (17%) | 76 (24%) | 54 (17%) | 135 (42%) | 189 (59%) |
| P2 HPV31 | 133 | 23 (17%) | 43 (32%) | 22 (17%) | 45 (34%) | 67 (50%) |
| P2 HPV18 | 101 | 26 (26%) | 35 (35%) | 17 (17%) | 23 (23%) | 40 (40%) |
| P2 other | 397 | 125 (31%) | 188 (47%) | 46 (12%) | 38 (10%) | 84 (21%) |
The number of women (n) in each group is split by CIN status and HPV result based on detectable methylation, i.e., a woman is classified to be HPV type positive if any of the CpGs tested were successful.
Sum of previous CIN2 and CIN3 columns.
Abbreviations: CIN: cervical intraepithelial neoplastic; HPV: human papillomavirus.
Stage 1: Model development using P1
Six methylation variables were selected from the forward-stepwise logistic regression algorithm applied in P1: (1) mean EPB41L3, (2) mean DPYS1, (3) HPV16 score from Ref.5 (based on CpGs in L1 and L2), (4) HPV16 methylation-assay positivity (also includes 0% methylation), (5) mean HPV18 L2 sites 4256, 4261, 4265, 4269, 4575 and 4281 and (6) mean HPV31 L1 sites 6352 and 6364. The CpG groupings were partly chosen to simplify the design of an assay, because the same primer could be used for each group. Other regions and groupings were excluded because they did not add to the multivariable model fit in P1. Figure 2 shows that the individual CpG sites in these regions were more univariately important than those not included such as MAL, or URR regions of HPV. Supporting Information Table 5 provides the model fit, which shows that all components were also multivariately significant. The ROC from the fitted model in P1 is shown in Figure 4. As a large number of possible predictor variables were assessed for inclusion in the model, the next stage was used to verify the model performance in an independent data set.
Figure 2.

AUC and p values for individual CpGs, excluding failed assays (no detectable methylation). The p value is represented by the shade of the bar, with key shown in the legend; the sample size differs between the four plots. Plot (a) is for samples that were not positive for HPV16, HPV18 or HPV31; (b) is based on specimens positive for HPV16, and so adds this particular set of HPV CpGs to the human CpGs; (c) is for HPV18 samples and (d) is for HPV31.
Figure 4.
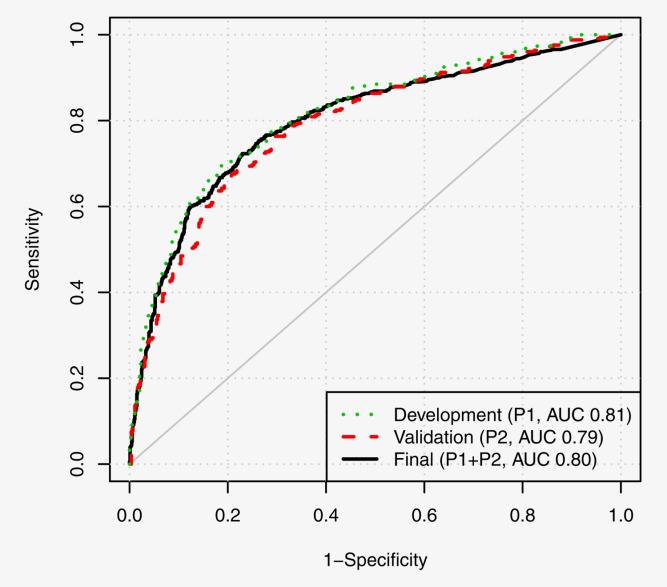
ROCs from the three stages of model development [final AUC 0.80 (95% CI 0.78–0.82)]. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Stage 2: Validation with P2
When applied to P2 the model derived from P1 had AUC 0.79 (95% CI 0.76–0.82), χ2 = 220.54, p < 0.00001. As expected the performance was slightly inferior to the fitted score in P1, but it was still promising and highly significant. Supporting Information Table 5 shows that all components of the model were significant (p values <0.05) in both training and validation datasets. The replication of the model components provides further reassurance that the classification score provides a real signal to predict CIN status in hrHPV positive women with abnormal cytology.
Stage 3: Final classifier using P1 and P2
Although statistically significant, model fit suggested that DPYS and HPV16 positivity were not practically important, so to keep the assay and score simple they were dropped. The decision to drop the variables was based on coefficient weights in the score, p values and sample size. For example, HPV18 was not dropped even though it had a larger p value than DPYS when dropped from the model in P2, because it had a smaller sample size of 101 samples, compared with 875 for the human gene DPYS. The proposed score fitted from P1 and P2 was
where the genomic regions of HPV types are indicated following a hyphen. The terms are the mean methylation of the CpGs within a region, except for the proportion of CpGs methylated in the HPV16 L2 region as in Ref.5. Each component ranges between 0 and 1, so that R ranges between 0 and 100.
Table 2 shows a summary of this final model, where all terms had (i) significant univariate power, (ii) added to the model in a stepwise manner and (iii) information was lost when they were dropped from the full model.
Table 2.
Final model summary using all the samples from P1 and P2
| Predictor | Score weight (95% CI) | 1. Univariate LR-χ2 (−log10 p) | 2. Drop from full model LR-χ2 (−log10 p) | 3. Stepwise LR-χ2 (−log10 p) | EPB41L3 Spearman |
|---|---|---|---|---|---|
| EPB41L3 | 38.8 (29.7–48.6) | 152.5 (34.3) | 87.4 (20.1) | 152.5 (34.3) | |
| HPV16 L1 | 17.2 (12.5–22.2) | 214.2 (47.8) | 59.4 (13.9) | 174.7 (39.2) | 0.18 |
| HPV16 L2 | 5.4 (3.4–7.3) | 190.6 (42.6) | 30.1 (7.4) | 26.2 (6.5) | 0.24 |
| HPV31 L1 | 28.1 (18.3–38.8) | 31.4 (7.7) | 38.5 (9.3) | 35.6 (8.6) | 0.10 |
| HPV18 L2 | 10.5 (5.5–16.1) | 14.1 (3.8) | 17.8 (4.6) | 17.8 (4.6) | 0.28 |
The overall likelihood ratio χ2 was 406.65. The score weights (model coefficients) and 95% CIs are scaled so that the score runs between 0 and 100. The performance of individual predictors is shown in three ways: (i) the univariate log-likelihood ratio (LR) χ2 (1 df) and associated p values on a log10 scale so that—log10 p = 1, 2 and 3, respectively, when p = 0.1, 0.01, 0.001, etc.; (ii) the drop in LR-χ2 when removing the variable from the full model and (iii) the stepwise increase in LR-χ2 when starting with the human gene EPB41L3, that is common to all samples, and then adding the HPV methylation predictors to a model that includes all the variables above it in the table. The Spearman correlation coefficient between EPB41L3 and the HPV methylation predictors is also given.
Abbreviations: CIN: cervical intraepithelial neoplastic; CI: confidence interval; df: degree of freedom; HPV: human papillomavirus.
The methylation distributions of CpGs used in the model are shown in Figure 3, and with failure rates in Supporting Information Table 4. The failure rates were between 0.6 and 2.0% for those not in the L2 HPV region, but they were higher for HPV L2 region CpGs. In particular 33/128 (26%) of controls failed in the HPV18-L2 CpGs but had detectable methylation elsewhere; for cases this was 9/73 (12%). However, because the failure rate in controls was twice that of cases, imputing zero methylation on such occasions actually helped prediction performance.
Figure 3.

Distribution of methylation in the CpGs selected for the final model. The boxes in (a) show the interquartile range and median when methylated in <CIN2 (white) and CIN2/3 (black) when methylated. The bars in (b) show the proportion of individuals methylated (values greater than 0%) for each CpG for <CIN2 (white) and CIN2/3 (black). Failed assays (methylation negative) are excluded from these plots.
The final model ROC is shown in Figure 4, where for a cutoff of 0.5 with 90% sensitivity, specificity was 36% (33–40) and PPV was 46% (43–48). Thus, in triage around one in three <CIN2 women would not need to be referred to colposcopy if 10% of CIN2/3 women were allowed to be missed. By HPV type, 90% sensitivity corresponded to the following specificities and PPV, respectively: HPV16, 38% (32–45) and 67% (63–71); HPV18, 53% (45–62) and 52% (45–59); HPV31, 39% (31–49) and 58% (51–65); HPV16, 18 or 31, 44% (40–49) and 62% (59–65) and other types than HPV16, 18 or 31, 17% (14–21) and 21% (18–24). Supporting Information Tables 6–9 provide further details for 60, 70, 80 and 90% sensitivity operating points.
An investigation of the score by cytology grade showed that the score distribution (and thereby methylation distribution) by CIN status and cytology were quite similar. For example, normal smears that were <CIN2 (n = 201) had a mean R score of 2.0 (interquartile range (IQR) 0.2–2.2); for mild dyskaryosis (n = 411), the mean R score was 2.1 (IQR 0.4–2.3). A complete summary is given in Supporting Information Table 10.
Discussion
We have developed and evaluated a new classifier to predict CIN2/3 histology from a sample taken prior to colposcopy in an abnormal cytology referral population in London, UK. The final model used methylation measurements of selected CpG sites in the human gene EPB41L3, and HPV late regions of HPV16-L1, HPV16-L2, HPV18-L2 and HPV31-L1. The results confirmed earlier findings for HPV16 in a different population and with a much larger sample set,5 and extended them to HPV18 and HPV31,16 and to other types than HPV16, HPV18 or HPV 31 by incorporating the human gene EPB41L3.9
A three-stage design of model development, validation and updating was used to mitigate issues associated with overfitting and to retain simplicity.17 The validation ROC from the initial model was similar at the upper specificity points to the final amended model, which lends credibility to the results for this population at high sensitivity.
Our findings are directly applicable to cytology-abnormal hrHPV+ women, where missing approximately 10% of CIN2/3 by recalling those with R > 0.5 had specificity 36% (33–40). This improvement in specificity was obtained because the women with low or negligible DNA methylation were more likely to be < CIN2. Although only approximately 17% specificity at 90% sensitivity was obtained when EPB41L3 was methylated for women not infected with HPV16, HPV18 or HPV31, the human gene helped to improve classification performance by enabling the stratification of risk at the overall 90% sensitivity cutpoint. Further, as the sensitivity operating points decreased, EPB41L3 became even more useful (Supporting Information Tables 6–9).
A limitation of cutpoint analysis is that women with normal cytology were not represented in our cohorts, but they will be detected through primary hrHPV screening. Thus, the specificity and PPV might be different for 90% sensitivity than was observed. To help address this issue future work is planned to examine the distribution of the developed score in hrHPV+ women with normal cytology. However, if the improvements seen in this cytology-abnormal hrHPV+ population transfer to the wider population, then they might be practically useful if cost-effective methylation assays can be implemented in the routine clinical setting. The methylation tests could be performed on the original samples in a reflex manner and may reduce subsequent anxiety and overtreatment in some women spared colposcopy. Concern over missing approximately 10% of CIN2/3 might be addressed by referring women predicted to be low risk by the DNA methylation classifier to repeat HPV testing in 1 year. Only a very small proportion of CIN2/3 with low methylation would be expected to progress to cancer within a year, and our earlier results in Ref.5 suggested that HPV16 L1 methylation might also predict persistence.
The proportion of CIN2/3 was greater for multiple infections than for single infections (data not shown). This provided some support to the model assumption that multiple infections act independently, so that, for example, a woman with high methylation in HPV16 and HPV18 is more likely to be CIN2/3 than one with only high methylation in HPV16. However, a larger data set would be needed to verify whether the classifier might be improved by a more sophisticated model for type-specific interactions.
An issue raised by the results is that L2 PCRs in HPV16 and HPV18 had a poorer success rate than L1 (Supporting Information Table 4). A possible reason for L2 failing is that the PCR primers had a lower amplification efficiency than the L1 primers. Another possibility is that L2 may be more prone to deletions than L1 as the virus integrates into the host genome. Integration of HPV16 and HPV18 occur mostly in cancers but also in some CIN3, while integration of HPV31 and other hrHPVs have been documented less frequently.18 Also, L2 commonly failed in the assays where L1 also failed, and the low viral load in low-grade lesions in combination with the lower amplification efficiency of L2 primers might explain why imputation of 0% methylation was useful for classification.
The final model may be implemented to clinical tests in practice by the design of a suitable assay. Although multiple PCRs are required for the assay, CpGs selected for each component in the final model are in the same amplicon, which facilitates the design of an assay for clinical testing.
A strength of the current study is that the HPV16 DNA methylation classifier from Ref.5 has been extended to other hrHPV+ women, by incorporating a human gene and by examining a wide range of CpG sites for HPV18 and HPV31. Further, the number of CIN2/3 cases for HPV subtypes appears to be greater than previous studies in the field. For example, the sample size for HPV18 and HPV31 provided more power to detect a methylation marker than in our previous studies5,16; the number of HPV16 CIN2 and CIN3 was also much greater than previous analysis of this type, including.3 A limitation is that the analysis was based on referral cohorts rather than a primary hrHPV screening group. More work is needed to help address the issues this raises for triage, and to further validate the model.
Acknowledgments
The authors gratefully recognize the patients who participated in this study. We thank four anonymous reviewers who helped to improve the article.
Glossary
- AUC
area under the ROC curve
- CIN
cervical intraepithelial neoplasia
- hrHPV
high-risk human papillomavirus
- HPV
human papillomavirus
- HPV16
human papillomavirus type 16
- HPV18
human papillomavirus type 18
- HPV31
human papillomavirus type 31
- IQR
interquartile range
- P1
Predictors 1
- P2
Predictors 2
- PCR
polymerase chain reaction
- PPV
positive predictive value
- qPCR
quantitative polymerase chain reaction
- ROC
receiver–operating characteristic
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Vasiljević N, Wu K, Brentnall AR, et al. Absolute quantitation of DNA methylation of 28 candidate genes in prostate cancer using pyrosequencing. Dis Markers. 2011;30:151–61. doi: 10.3233/DMA-2011-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Schiffman M, Ghosh A, et al. Elevated methylation of HPV16 DNA indicates increased risk for the development of high grade cervical intraepithelial neoplasia. Int J Cancer. 2013;132:1412–22. doi: 10.1002/ijc.27750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wojdacz TK, Su YHH. Challenges for the application of DNA methylation biomarkers in molecular diagnostic testing for cancer. Expert Rev Mol Diag. 2013;13:283–94. doi: 10.1586/erm.13.9. [DOI] [PubMed] [Google Scholar]
- Lorincz AT, Brentnall AR, Vasiljević N, et al. HPV16 L1 and L2 DNA methylation predicts high-grade cervical intraepithelial neoplasia in women with mildly abnormal cervical cytology. Int J Cancer. 2013;133:637–44. doi: 10.1002/ijc.28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HCC, Lin YWW, Huang RLL, et al. Quantitative DNA methylation analysis detects cervical intraepithelial neoplasms type 3 and worse. Cancer. 2010;116:4266–74. doi: 10.1002/cncr.25252. [DOI] [PubMed] [Google Scholar]
- Overmeer RM, Louwers JA, Meijer CJ, et al. Combined CADM1 and MAL promoter methylation analysis to detect (pre-)malignant cervical lesions in high-risk HPVpositive women. Int J Cancer. 2011;129:2218–25. doi: 10.1002/ijc.25890. [DOI] [PubMed] [Google Scholar]
- Hesselink AT, Heideman DA, Steenbergen RD, et al. Combined promoter methylation analysis of CADM1 and MAL: an objective triage tool for high-risk human papillomavirus DNA-positive women. Clin Cancer Res. 2011;17:2459–65. doi: 10.1158/1078-0432.CCR-10-2548. [DOI] [PubMed] [Google Scholar]
- Eijsink JJ, Lendvai A, Deregowski V, et al. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J Cancer. 2012;130:1861–9. doi: 10.1002/ijc.26326. [DOI] [PubMed] [Google Scholar]
- Muñoz N, Bosch XX, de Sanjosé S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Szarewski A, Ambroisine L, Cadman L, et al. Comparison of predictors for High-Grade cervical intraepithelial neoplasia in women with abnormal smears. Cancer Epidemiol Biomarkers Prev. 2008;17:3033–42. doi: 10.1158/1055-9965.EPI-08-0508. [DOI] [PubMed] [Google Scholar]
- Szarewski A, Mesher D, Cadman L, et al. A comparison of seven tests for high grade cervical intraepithelial neoplasia in women with abnormal smears: the predictors 2 study. J Clin Microbiol. 2012;50:1867–73. doi: 10.1128/JCM.00181-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, et al. Proc: an open-source package for R and S+ to analyze and compare roc curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta A, Cai TT, Brown LD. Confidence intervals for a binomial proportion and asymptotic expansions. Ann Stat. 2002;30:160–201. [Google Scholar]
- R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- Wentzensen N, Sun C, Ghosh A, et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J Natl Cancer Inst. 2012;104:1738–49. doi: 10.1093/jnci/djs425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moons KGM, Kengne AP, Grobbee DE, et al. Risk prediction models: II. External validation, model updating, and impact assessment. Heart. 2012;98:691–8. doi: 10.1136/heartjnl-2011-301247. [DOI] [PubMed] [Google Scholar]
- Schmitz M, Driesch C, Jansen L, et al. Non-random integration of the HPV genome in cervical cancer. PLoS ONE. 2012;7:e39632+. doi: 10.1371/journal.pone.0039632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.