

Abstract
Improving the therapeutic efficacy and reducing systemic side effects of drugs is an important aspect in chemotherapy. The strategy presented here is the use of cisplatin loaded, temperature-sensitive, hydrogel nanoparticles (CisPt-NPs) and their ability to deliver and release chemodrugs selectively, based on thermal stimuli. The specially synthesized CisPt-NPs show a temperature-dependent increase of cisplatin release, at neutral pH (as in blood and normal tissue), in both the presence and absence of common metallic ions, as well as at the low pH found in lysosomes, where endocytosed NPs often localize. These CisPt-NPs were uptaken by breast cancer MDA-MB-435 cells, via endocytosis, and then mostly localized in the lysosomes. The in vitro cytotoxicity tests show that these CisPt-NPs have a significantly better efficacy at the slightly elevated temperatures. Potential applications are discussed.
The development of hydrogel nanoparticles (NPs) that transport and deliver chemodrugs selectively to the tumor area is a recent strategy for improving therapeutic efficacy and avoiding systemic side effects, such as renal toxicity, phlebitis, bone marrow suppression, and nausea.1,2 Such selective delivery may be achieved by active and/or passive targeting of such NPs.3
Two important aspects for consideration in the development of drug delivering NPs are the chemodrug loading capacity and the control over its release. An optimal design of the NPs would facilitate incorporation of a large amount of the drug, with efficient release to the tumor region, preferably in a controlled manner, while limiting release elsewhere.4 The latter is one of the most challenging aspects. Designing the NP matrix to enhance and enable controlled release of the drug is often based on environmental stimuli,3 e.g., temperature, pH,1 light,5,6 glucose,7 antigen,8 and reducing agents, such as glutathione.9
Using temperature as a stimulus to control the drug release from NPs is particularly attractive because it can exploit the variation in the local temperature, typical for tumor tissues.10 Tumor tissues have been shown to have slightly elevated temperatures, compared to the host basal temperature, due to an increased metabolic rate.11 Also, additional temperature differentials can be induced by external heating of the tumor region, e.g., by ultrasonic, magnetic field, or light-mediated heating, targeted to the nanoparticles.12−14
The integration of temperature-sensitive properties into the design of hydrogel nanoparticles has shown promise for enhancement of the drug release to the somewhat hotter tumor tissues, while limiting the release elsewhere.10 Due to the flexible structures of these NPs, the polymer interactions inside the NPs can be made temperature sensitive, thereby altering the NP size and its polymer density; consequently, the release efficiency of the loaded drugs changes.
Many different kinds of temperature-sensitive nanoparticle matrices have been formulated, such as poly(vinyl alcohol),15,16 cellulose derivatives,17 and complex core–shell polymer designs.18 Among them, one of the most common matrix systems for hydrogel NPs is poly(N-isopropylacrylamide) (PNIPAM), which shows good biocompatibility as well as temperature sensitivity, a so-called lower critical solution temperature (LCST) behavior in aqueous solution, across a biologically relevant temperature and pressure range.10,19,20 Hydrogen bonding is formed between the amide of PNIPAM and water, and a cage-like structure is formed around the isopropyl group below the LCST; this solvates PNIPAM and expands the nanoparticles.10,21,22 On the other hand, these structures are broken above the LCST; the latter shrinks the NPs, thereby “squeezing out” chemodrugs from the NPs or, alternatively, tightening the pores of the hydrogel, to reduce the chemodrug’s release.1,20−22 This matrix is often combined with other components, such as SiO2-coated, Fe3O4 nanoparticles or butyl methacrylate, to enhance functionality and shift the LCST.23,24
We emphasize that an upper critical solution temperature (UCST)-like behavior, characterized by swelling rather than deswelling at elevated temperature, potentially lends itself to being very valuable as well because swollen hydrogels do enhance the release of chemodrugs due to their lower polymer density.25,26 Some examples of UCST-based hydrogels are given in the review by Seuring and Agarwal.27
The combination matrix of acrylamide and acrylic acid is an example of an UCST-like hydrogel in the presence of a salt such as NaCl.27 This combination has been studied for both bulk hydrogel and NPs, since acrylic acid forms hydrogen bonds with acrylamide, bonds that may break at elevated temperatures, causing the hydrogel matrix to swell.25,28−30 Echeverria and Mijangos have further demonstrated the ability to control the specific swelling properties of acrylamide-co-acrylic acid particles by altering the cross-linker content and incorporating gold nanoparticles into the matrix.31 Also, acrylic acid binds a certain type of drugs tightly, due to its carboxyl group.1 Although these acrylamide-acrylic acid hydrogels show promising properties, as of yet no application of this matrix for temperature-responsive drug delivery systems has been reported, to the best of our knowledge, especially for cancer treatment, where the tumor is at a temperature slightly higher than that of the normal body temperature (37 °C).
Here we present NPs based on the combination of acrylic acid and acrylamide; they are specifically designed for temperature-sensitive release of chemodrugs. We incorporated cisplatin into these copolymer NPs (CisPt-NPs). Cisplatin, which is a chemodrug commonly used for carcinoma and melanoma,2,32 was chosen as a model drug. We present the temperature-sensitive behavior of the cisplatin release, under physiological conditions, with and without some of the ions that are abundantly present in vivo. We also monitored the intracellular localization of the NPs in cancer cells and observed them to accumulate primarily in the lysosomes. We thus also investigated the temperature-sensitive release of cisplatin in a lysosome-mimicking environment. CisPt-NPs showed increased cytotoxicity to tumor cells with increasing temperature. The results presented here show that acrylamide-co-acrylic acid hydrogel NPs may be a promising candidate for a stable and reliable temperature-sensitive drug delivery system.
The CisPt-NPs were synthesized in two steps (Figure 1A and B and Supporting Information). Similar NPs have shown temperature-dependent swelling property near the physiological temperature.25 We have also confirmed that our CisPt-NPs have a temperature sensitivity by using dynamic light scattering (Figure S1, Supporting Information), with emphasis on the lysosomal pH of 4.33
Figure 1.

(A) CisPt-NP synthesis, (B) postloading procedure, and (C) release mechanism.
The feeding ratio of acrylamide to acrylic acid was chosen to have equal moles of the monomers after the polymerization.25 When the NPs are synthesized only from acrylamide or only from acrylic acid, the temperature sensitivity decreases.25 The emulsifier concentration and solvent were chosen based on Owens and our standard protocol.25,34,35 The combination produces NPs whose size distribution is narrow and with a size that is ideal for drug delivery application (i.e., avoiding rapid clearance from the body and targeting the tumor passively in vivo).3
Before cisplatin loading, the size of the p(AA-co-AAm) NPs, at room temperature, was found to be 90 (±2) nm with a polydispersity index (PDI) of 0.30 (±0.01), while after the loading the size was 132 (±3) nm with a PDI of 0.30 (±0.01), using dynamic light scattering (Figure S2A, Supporting Information). The images from transmission electron microscopy showed a uniform NP size distribution (Figure S2B, Supporting Information). The zeta-potential of the p(AA-co-AAm) NPs was −57 (±5) mV before the cisplatin loading, while it was −56 (±5) mV after the loading. Thus, there was significant size expansion with cisplatin postloading. However, there was little change in surface potential, indicating that the cisplatin was not just adsorbing to the surface. The loading of cisplatin was 11 (±3) (cisplatin/CisPt-NPs) wt % of the unloaded particles.
Acrylic acid enhances the incorporation of cisplatin by the substitution of its chlorides with carboxyl groups of the NPs (Figure 1B).1,36 What is more, the bonding of the carboxyl group and the platinum of cisplatin is reversible, under physiological conditions.1,36,37 In the presence of a high concentration of Cl–, hydronium ions, or metallic cations, the carboxyl group interaction with the platinum center of cisplatin gets looser (Figure 1C).1,29 This may also explain the pH-enhanced release of cisplatin from the CisPt-NPs, discussed below.
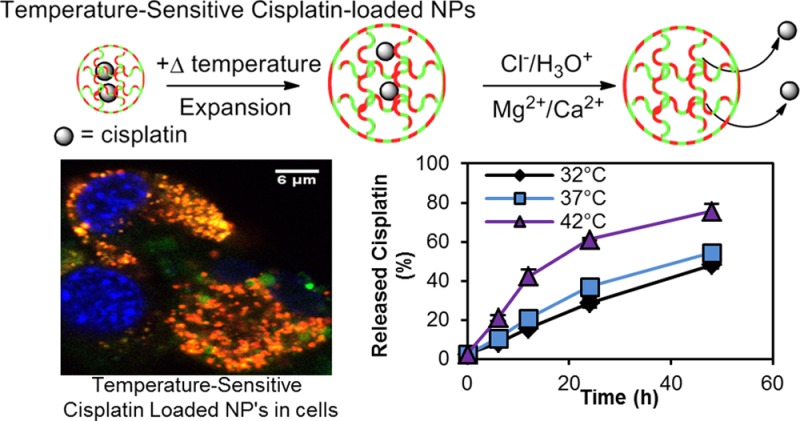
The temperature-dependent release of cisplatin was assessed in phosphate-buffered saline (PBS) (pH 7.4), which mimics serum conditions, at three different temperatures: 32 °C, 37 °C, and 42 °C (Figure 2). The percent release of the cisplatin (η) was calculated according to the following equation (eq 1).
| 1 |
where t is the time point of the drug release study; T is the temperature of the drug release study; C(t,T) is the concentration of cisplatin released at that time point, and C0(T) is the initial (t = 0) concentration of cisplatin in the CisPt-NPs. Also, the percent enhancement of the cisplatin release in PBS (Figure 2, inset) was calculated by the following equation (eq 2).
| 2 |
where η0(t) is the percent release of the cisplatin at 32 °C, at a given time point, t.
Figure 2.
Cisplatin release in PBS at three different temperatures: 32 °C, 37 °C, and 42 °C. Error bars show the standard deviation. Inset: % Enhancement of cisplatin release at 37 and 42 °C, compared to 32 °C.
We monitored the cisplatin release over a period of 48 h, starting at 6 h. Over the time scale we monitored, cisplatin always showed the highest release percentage at 42 °C, compared to 37 and 32 °C. After 48 h, 25% of cisplatin got released from the CisPt-NPs at 42 °C, while only 19% and 17% of the cisplatin got released at 37 and 32 °C, respectively. Although the average cisplatin release is slightly higher at 37 °C, compared to 32 °C, this is not statistically significant.
The above temperature sensitivity of the drug release is attributable to three different factors. First, the swelling property of the CisPt-NPs decreases the density of the matrix at higher temperature, which could facilitate the escape of cisplatin from the matrix. Second, the reverse substitution reaction rate of the carboxyl group, attached to the platinum center of the cisplatin with chloride, is enhanced at higher temperature, because of the higher accessibility of the Cl– ions.1,26 Third, the rate of diffusion of the cisplatin molecules increases with temperature.
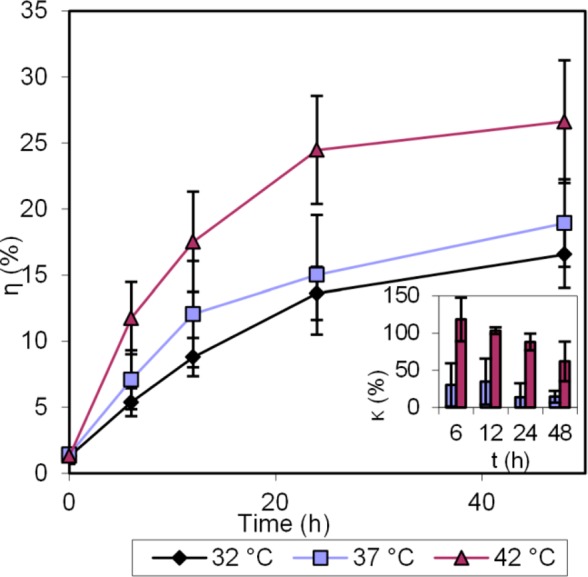
As described above, another major factor that governs the release of the drug is the presence of divalent ions.29 Therefore, we evaluated the effects of such ions on the drug release profile, particularly those of Mg2+ and Ca2+, which are abundantly present in the bloodstream, as shown in Figure 3. The concentration of each ion used is 1 mM, similar to their blood levels.38
Figure 3.
Cisplatin release in PBS, containing 1 mM each of Ca2+ and Mg2+, at three different temperatures: 32 °C, 37 °C, and 42 °C. Error bars show the standard deviation. Inset: % Enhancement of cisplatin release at 37 and 42 °C, compared to 32 °C.
The difference in the release of cisplatin becomes more evident after 6 h. We observe that, over 48 h, 32.0% of the cisplatin got released when the solution was at 42 °C, while only 22.0% and 17.8% of cisplatin got released at 37 and 32 °C, respectively.
The increase in drug release observed in the presence of divalent ions over longer incubation times may be attributed to the disruption by the divalent cations of the loose interaction between the carboxyl groups of the NP matrix and the cisplatin molecule’s center.29
We evaluated the release profile of CisPt-NPs in PBS with 10% fetal bovine serum and confirmed the temperature sensitivity (Figure S3, Supporting Information).
Determining the intracellular fate of the p(AA-co-AAm) NPs is essential to designing a drug release process, due to the importance of the local chemical environment inside the cells. The p(AA-co-AAm) NPs can be taken up by the cells via various intracellular pathways that dictate the intracellular fate of the p(AA-co-AAm) NPs. The intracellular colocalization of the p(AA-co-AAm) NPs was monitored using fluorescence confocal microscopy. We observe that these p(AA-co-AAm) NPs mostly colocalize with lysosomes, as shown in Figure 4. The overlapping of the fluorescence (seen as yellow/orange) from the 5-FTSC-labeled NPs (green) with the lysotracker labeled acidic vesicles (red) shows the colocalization. We have previously observed similar phenomena for the amine-functionalized hydrogel nanoparticles.35 Late endosomes and lysosomes are acidic in nature and have a pH value in the range of 4–5.33 Since we observe that most of the p(AA-co-AAm) NPs are trapped into low pH environments, we next studied the temperature-sensitive release of drugs at acidic conditions (Figure 5).
Figure 4.
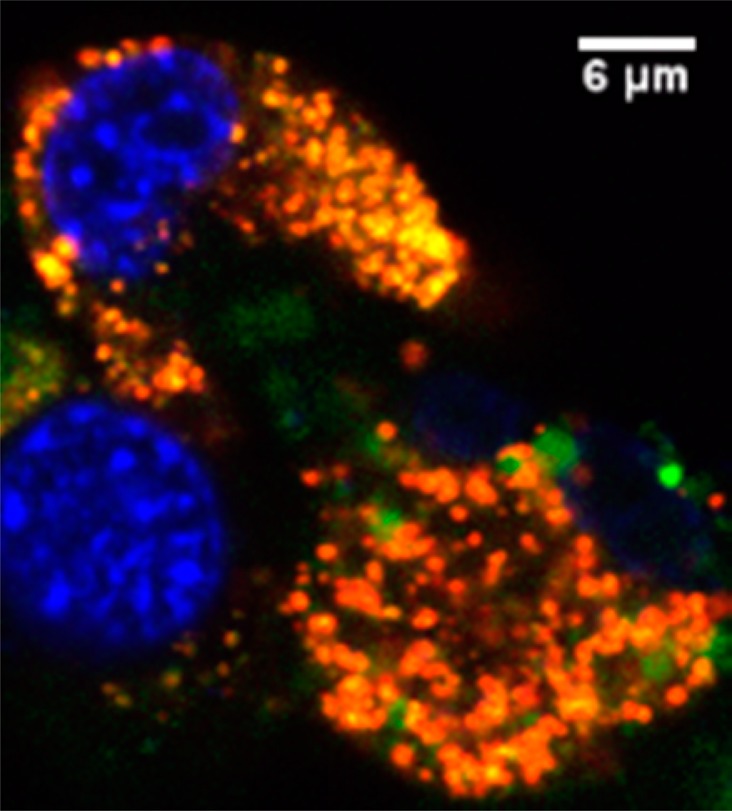
Cellular uptake study of 5-FTSC-loaded p(AA-co-AAm) NPs using MDA-MB-435. Blue indicates the nucleus, stained with Hoechst Blue. Green indicates the 5-FTSC-loaded p(AA-co-AAm) NPs. Red indicates the lysotracker. A significant amount of colocalization of red and green is observed (shown in yellow/orange).
Figure 5.
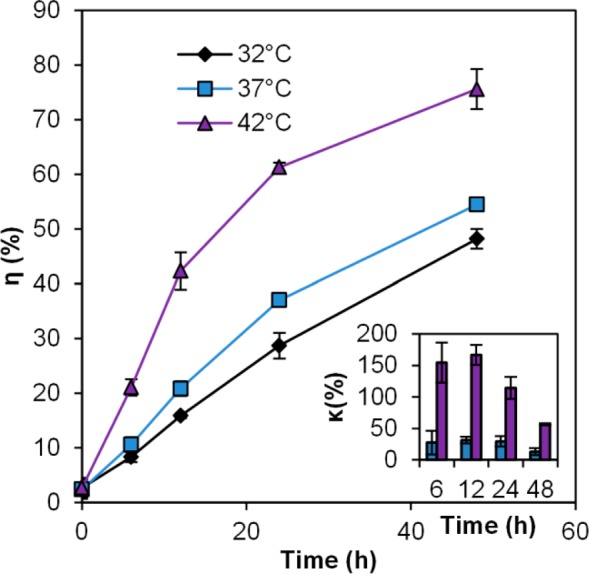
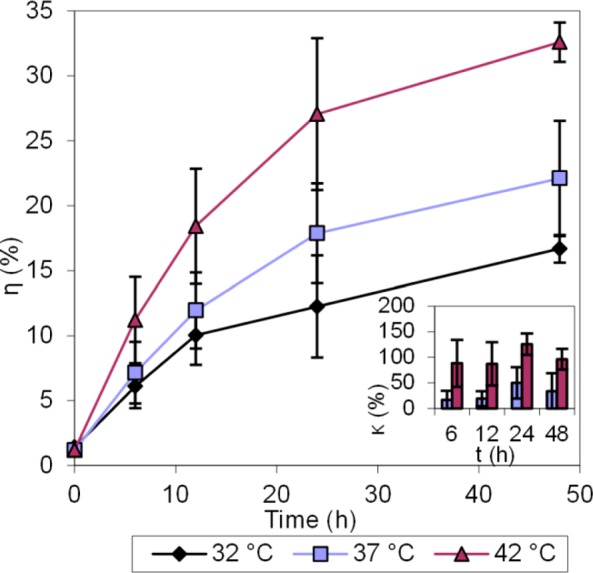
Cisplatin release study in pH 4 buffer at three different temperatures: 32 °C, 37 °C, and 42 °C. Error bars represent standard deviations. Inset: % Cisplatin release enhancements, relative to release at 32 °C.
The release studies were performed by suspending the CisPt-NPs in a pH 4 buffer (50 mM phthalate buffer containing 150 mM NaCl), which mimics the lowest pH level in the intact lysosome.33
The release of cisplatin in 48 h at 42 °C was 75.6%, while the release at 37 and 32 °C was 54.5% and 48.2%, respectively. The difference in the drug release became evident within the first 6 h and remained so over the rest of the observed time period. More cisplatin was released at this low pH than in PBS and in PBS with metallic ions, which is consistent with what was reported previously with rhodamine 6G.39 The higher release, under acidic conditions, such as inside the tumor tissue as well as the lysosomes, will further increase the tumor selectivity of the delivered cisplatin.33,40
Acrylamide-co-acrylic acid hydrogel shrinks at low pH because of the protonation of the carboxyl groups and formation of more hydrogen bonds.29,41 However, when the system was heated, these hydrogen bonds were broken, which resulted in the decrease of the matrix density (Figure S1, Supporting Information).25 In addition, the carboxyl groups on the conjugated cisplatin were substituted by the hydronium ions, and cisplatin was released from the CisPt-NPs.1
Also, we compared the total cisplatin release from CisPt-NPs to that from cisplatin-loaded nanoparticles made of poly(acrylic acid-co-methyl methacrylate), which reported about 80% of cisplatin released in PBS at 37 °C.42 This is a 4 times higher release of cisplatin than from our CisPt-NPs. However, as is shown above, increasing amount of cisplatin was released from the CisPt-NPs at the elevated temperature, at the pH of lysosomes, where we showed that CisPt-NPs were trapped. This selective and controlled release can further increase the therapeutic index.
As a proof of principle, we demonstrated the efficacy of this technique, with a view toward in vivo applications. The temperature-enhanced increase in NP drug release also resulted in increased cytotoxicity (Figure S4, Supporting Information), whereas free cisplatin did not show any statistically significant increase with temperature (Figure S5, Supporting Information).
In summary, temperature-sensitive NPs (CisPt-NPs), with a matrix of p(AA-co-AAm), were synthesized as a carrier for the chemotherapeutic drug, cisplatin, with the aim of reducing side effects; the latter is expected from the lowering of the release of free cisplatin in the blood stream and by selectively increasing its release in the tumor region, due to the temperature difference. We evaluated the temperature-dependent release profiles of the drug from the matrix, as well as its in vitro cytotoxicity. With increasing temperature, these NPs showed a very significant increase in the release of cisplatin, in PBS, even in the absence of divalent metallic ions. Furthermore, adding divalent ions, which are physiologically present in the body, further accelerated the drug release with increasing temperature. Intracellular fluorescence imaging showed that most of the nanoparticles colocalize with lysosomes. The release of cisplatin showed an even stronger correlation with the temperature at the low lysosomal pH. Furthermore, we have shown that the in vitro cytotoxicity of the CisPt-NPs also increases with higher temperature, correlating well with the temperature-enhanced drug release. We believe that the above results demonstrate both the feasibility and the potential utility of such temperature-sensitive NPs as drug carriers with a high therapeutic index.
Acknowledgments
Financial support was provided by a National Institutes of Health grant, R21 NS084275 (RK). The authors thank Yue Hou for help with the drug release study. They also thank the Microscopy and Image Analysis Laboratory of the University of Michigan and the Chemistry Instrument Shop for their support.
Supporting Information Available
Experimental procedure and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
‡ Chemistry and Biochemistry, Colorado College, 14 East Cache La Poudre St., Colorado Springs, CO 80903.
Author Present Address
§ NIST, 325 Broadway, Boulder, CO 80305.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Peng J.; Qi T.; Liao J.; Chu B.; Yang Q.; Li W.; Qu Y.; Luo F.; Qian Z. Biomaterials 2013, 34, 8726–8740. [DOI] [PubMed] [Google Scholar]
- Decatris M. P.; Sundar S.; O’Byrne K. J. Cancer Treat. Rev. 2004, 30, 53–81. [DOI] [PubMed] [Google Scholar]
- Koo Lee Y.; Kopelman R. In Multifunctional Nanoparticles for Drug Delivery Applications; Svenson S., Prud’homme R. K., Eds.; Nanostructure Science and Technology; Springer US: Boston, MA, 2012; pp 225–255. [Google Scholar]
- Koo Lee Y.-E.; Orringer D. A.; Kopelman R. In Polymer-based Nanostructures; Broz P., Ed.; RSC Publishing: Cambridge, UK, 2010; pp 333–353. [Google Scholar]
- Suzuki A.; Tanaka T. Nature 1990, 346, 345–347. [Google Scholar]
- Mamada A.; Tanaka T.; Kungwatchakun D.; Irie M. Macromolecules 1990, 23, 1517–1519. [Google Scholar]
- Kataoka K.; Miyazaki H.; Bunya M.; Okano T.; Sakurai Y. J. Am. Chem. Soc. 1998, 120, 12694–12695. [Google Scholar]
- Miyata T.; Asami N.; Uragami T. Nature 1999, 399, 766–769. [DOI] [PubMed] [Google Scholar]
- Lin C.; Zhong Z.; Lok M. C.; Jiang X.; Hennink W. E.; Feijen J.; Engbersen J. F. J. Bioconjugate Chem. 2007, 18, 138–145. [DOI] [PubMed] [Google Scholar]
- Schmaljohann D. Adv. Drug Delivery Rev. 2006, 58, 1655–1670. [DOI] [PubMed] [Google Scholar]
- Stefanadis C.; Chrysochoou C.; Markou D.; Petraki K.; Panagiotakos D. B.; Fasoulakis C.; Kyriakidis A.; Papadimitriou C.; Toutouzas P. K. J. Clin. Oncol. 2001, 19, 676–681. [DOI] [PubMed] [Google Scholar]
- Lammers T.; Kiessling F.; Hennink W. E.; Storm G. Mol. Pharmaceutics 2010, 7, 1899–1912. [DOI] [PubMed] [Google Scholar]
- Alvarez-Berríos M. P.; Castillo A.; Mendéz J.; Soto O.; Rinaldi C.; Torres-Lugo M. Int. J. Nanomed. 2013, 8, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry T.; Epstein T.; Smith R.; Kopelman R. Nanomedicine (London, U.K.) 2013, 8, 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. K.; Wang N.; Wu X. S. J. Controlled Release 1998, 56, 117–126. [DOI] [PubMed] [Google Scholar]
- Ghugare S. V.; Mozetic P.; Paradossi G. Biomacromolecules 2009, 10, 1589–1596. [DOI] [PubMed] [Google Scholar]
- Lu X.; Hu Z.; Gao J. Macromolecules 2000, 33, 8698–8702. [Google Scholar]
- Wu W.; Aiello M.; Zhou T.; Berliner A.; Banerjee P.; Zhou S. Biomaterials 2010, 31, 3023–3031. [DOI] [PubMed] [Google Scholar]
- Ebeling B.; Eggers S.; Hendrich M.; Nitschke A.; Vana P. Macromolecules 2014, 47, 1462–1469. [Google Scholar]
- Fan T.; Li M.; Wu X.; Li M.; Wu Y. Colloids Surf. B: Biointerfaces 2011, 88, 593–600. [DOI] [PubMed] [Google Scholar]
- Cho E. C.; Lee J.; Cho K. Macromolecules 2003, 36, 9929–9934. [Google Scholar]
- Deshmukh S. a; Sankaranarayanan S. K. R. S.; Suthar K.; Mancini D. C. J. Phys. Chem. B 2012, 116, 2651–2663. [DOI] [PubMed] [Google Scholar]
- Lien Y.-H.; Wu J.-H.; Liao J.-W.; Wu T.-M. Macromol. Res. 2013, 21, 511–518. [Google Scholar]
- Jeong B.; Gutowska A. Trends Biotechnol. 2002, 20, 305–311. [DOI] [PubMed] [Google Scholar]
- Owens D. E.; Jian Y.; Fang J. E.; Slaughter B. V.; Chen Y.-H.; Peppas N. A. Macromolecules 2007, 40, 7306–7310. [Google Scholar]
- Amsden B. Macromolecules 1998, 31, 8382–8395. [Google Scholar]
- Seuring J.; Agarwal S. Macromol. Rapid Commun. 2012, 33, 1898–1920. [DOI] [PubMed] [Google Scholar]
- Okano T. Adv. Polym. Sci. 1993, 110, 179–197. [Google Scholar]
- Tang Q.; Wu J.; Lin J.; Li Q.; Fan S. J. Mater. Sci. 2008, 43, 5884–5890. [Google Scholar]
- Echeverria C.; López D.; Mijangos C. Macromolecules 2009, 42, 9118–9123. [Google Scholar]
- Echeverria C.; Mijangos C. Macromol. Rapid Commun. 2010, 31, 54–58. [DOI] [PubMed] [Google Scholar]
- Rosenberg B.; Vancamp L.; Trosko J. E.; Mansour V. H. Nature 1969, 222, 385–386. [DOI] [PubMed] [Google Scholar]
- Poole B. J. Cell Biol. 1981, 90, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen A. K.; Arleth L.; Almdal K.; Scharff-Poulsen A. M. J. Colloid Interface Sci. 2007, 306, 143–153. [DOI] [PubMed] [Google Scholar]
- Ray A.; Lee Y. K.; Kim G.; Kopelman R. Small 2012, 8, 2213–2221. [DOI] [PubMed] [Google Scholar]
- Nishiyama N.; Yokoyama M.; Aoyagi T.; Okano T.; Sakurai Y.; Kataoka K. Langmuir 1999, 15, 377–383. [Google Scholar]
- Howe-Grant M. E.; Lippard S. J. In Metal Ions in Biological Systems; Sigel H., Sigel A., Eds.; Dekker: New York, 1980; pp 63–125. [Google Scholar]
- Walser M. J. Clin. Invest. 1961, 40, 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Chen Q.; Patel K.; Li L.; Li X.; Wang Q.; Zhang G.; Zheng J. Soft Matter 2012, 8, 7848. [Google Scholar]
- Vaupel P.; Kallinowski F.; Okunieff P. Cancer Res. 1989, 49, 6449–6465. [PubMed] [Google Scholar]
- Thakur A.; Wanchoo R. K.; Singh P. Chem. Biochem. Eng. Q. 2011, 25, 181–194. [Google Scholar]
- Lee K. D.; Jeong Y.; Kim D. H.; Lim G.; Choi K.-C. Int. J. Nanomed. 2013, 8, 2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.