

Abstract
Background and Aims
Hypothermia provides neuroprotection after cardiac arrest, hypoxic-ischemic encephalopathy, and in animal models of ischemic stroke. However, as drug development for stroke has been beset by translational failure, we sought additional evidence that hypothermia protects human neurons against ischemic injury.
Methods
Human embryonic stem cells were cultured and differentiated to provide a source of neurons expressing β III tubulin, microtubule-associated protein 2, and the Neuronal Nuclei antigen. Oxygen deprivation, oxygen-glucose deprivation, and H2O2-induced oxidative stress were used to induce relevant injury.
Results
Hypothermia to 33°C protected these human neurons against H2O2-induced oxidative stress reducing lactate dehydrogenase release and Terminal deoxynucleotidyl transferase dUTP nick end labeling-staining by 53% (P ≤ 0·0001; 95% confidence interval 34·8–71·04) and 42% (P ≤ 0·0001; 95% confidence interval 27·5–56·6), respectively, after 24 h in culture. Hypothermia provided similar protection against oxygen-glucose deprivation (42%, P ≤ 0·001, 95% confidence interval 18·3–71·3 and 26%, P ≤ 0·001; 95% confidence interval 12·4–52·2, respectively) but provided no protection against oxygen deprivation alone. Protection (21%) persisted against H2O2-induced oxidative stress even when hypothermia was initiated six-hours after onset of injury (P ≤ 0·05; 95% confidence interval 0·57–43·1).
Conclusion
We conclude that hypothermia protects stem cell-derived human neurons against insults relevant to stroke over a clinically relevant time frame. Protection against H2O2-induced injury and combined oxygen and glucose deprivation but not against oxygen deprivation alone suggests an interaction in which protection benefits from reduction in available glucose under some but not all circumstances.
Keywords: brain, hypothermia, ischemic stroke, neuroprotection, stem cells, treatment
Introduction
Obstruction of an artery supplying the brain initiates a cascade of events leading ultimately to necrotic and apoptotic cell death. With the most extreme perfusion deficits, survival is unlikely and cell death is rapid. However, in areas adjacent to the ischemic core (the ischemic penumbra), residual blood flow can preserve tissue vitality for a limited time until the obstruction resolves spontaneously or is removed by thrombolysis 1,2. After these acute events delayed neuronal death may progress for up to three-days 3. Both these phenomena (the penumbra during ischemia and delayed neural injury after ischemia) provide targets for treatment and in animal models of ischemic stroke over 500 ‘neuroprotective’ treatment strategies reported improved outcome. However, none have proved robustly effective in randomized controlled clinical trials 4. Importantly with the exception of therapeutic hypothermia 5, clinical trials of neuroprotection for stroke have largely been abandoned (http://www.clinicaltrials.gov).
The reasons for this translational failure have been hotly debated. Contamination of the preclinical data set by falsely positive results influenced by bias and lack of statistical power offer some explanation 6–8. The molecular targets of therapy may be present in rodents but not in man 9, either because of differences in genetic background 10 or because evolution of the ischemic cascade might proceed at a different pace in different species 11–13. Additionally, laboratory and clinical thrombolysis experiments show similar, and unfortunately short, windows of opportunity 2,14, which are frequently unachievable in most trials of neuroprotection 15,16. Targeting single components of the ischemic cascade might also limit our chances of success 17,18, but combinatorial pharmacotherapy therapy brings its own challenges 19.
The preclinical data set for therapeutic hypothermia is large (3353 animals) and demonstrates substantial and consistent efficacy, with only modest effects of publication bias and failure to randomize or blind 20. Hypothermia influences multiple molecular targets 21,22 and provides benefit over a wide range of times to treatment in animals 20. Clinically, evidence of a relationship between body temperature and outcome after human stroke 23–25, together with the proven neuroprotective benefit of hypothermia in adults with global ischemia after cardiac arrest 26,27 and neonates with hypoxic-ischemic encephalopathy 28,29, suggest that the targets that provide benefit in animal models of stroke will also be present in human stroke. However, the absence of clear therapeutic benefit in traumatic brain injury 30, which accrues damage similarly to stroke 31, suggests caution should be taken. A Cochrane systematic review of stroke patients treated with physical cooling devices suggests there may be a trend toward improvement. However, in most cases, treatment was initiated (8–12 h) when penumbral tissue, the main putative target, may no longer be present. Overall, the analysis supports the view that we still have insufficient data 32 with trials too small to allow firm conclusions to be made about the efficacy of hypothermia in stroke patients 33–35.
In this report, we show that hypothermia (33°C) protects human stem-cell derived neurons from oxidative stress induced by hydrogen peroxide and from oxygen-glucose deprivation but not hypoxia alone. Importantly, protection against oxidative stress is observed at later times (six-hours) than obtained under oxygen-glucose deprivation. The results were replicated in three independent series of experiments employing randomization and blinding of operator-dependent, outcome analyses.
Methods
Human embryonic stem cell (hESC) Culture
Experiments with these cells were carried out in accordance with the guidelines and regulations of the National Health and Medical Research Council and with the approval of the Austin Health Human Research Ethics Committee (Approval number H2008/03194) and University of Melbourne Human Research Ethics Committee (Approval number 0605017).
H9 (WA-09, WiCell, Madison, Wisconsin, USA), human embryonic stem cells (hESC), were cultured on mitomycin-C treated mouse embryonic fibroblasts (MEFs) in hESC medium consisting of high-glucose Dulbecco's modified Eagle's minimal essential medium (DMEM) without sodium pyruvate, supplemented with insulin/transferrin/selenium 1%, β-mercaptoethanol 0·1 mM, nonessential amino acids (NEAAs) 1%, glutamine 2 mM, penicillin 25 U/ml, streptomycin 25 μg/ml (all from Invitrogen, Victoria, Australia) and fetal calf serum (FCS) 20% (HyClone, Australia) or on mitomycin-C treated human foreskin fibroblasts (HFF; ATCC, CRL-2097) in KSR media consisting of DMEM/nutrient mixture F-12, supplemented with β-mercaptoethanol 0·1 mM, NEAAs 1%, glutamine 2 mM, penicillin 25 U/ml, streptomycin 25 μg/ml, and knockout serum replacement 20% (all from Invitrogen). All cells were cultured at 37°C in 5% CO2. Colonies were mechanically dissected every seven-days and transferred to freshly prepared MEFs or HFFs. Media was changed every second day.
Neuronal differentiation and growth
Neuronal differentiation was achieved using the noggin induction method described for mouse neurospheres 36 as adapted by Dottori for human neurospheres 37.The colonies were maintained (37°C, 5% CO2) in hESC medium supplemented with 500 ng/ml of Noggin (6057-NG, R&D systems, Australia) for 14 days with the media and Noggin replaced every second day.
At this point, the colonies were again mechanically dissociated, but at this time the central (differentiated) part of the colony was also cut into smaller pieces using a 26-gauge needle. The pieces were transferred to individual wells in a low adherent 96-well plate containing Neural Basal Media (NBM) that contains Neurobasal A (10888-022, Invitrogen) supplemented with 2% B27 (17504-044, Invitrogen), 1% Insulin Transferrin Selenium-A (51500-056, Invitrogen), 1% N2 (17502-048, Invitrogen), 2 mM glutamine, 0·5% Penicillin-Streptomycin Solution (15070-063, Invitrogen), 20 ng/ml human recombinant epidermal growth factor (EGF) (PHG0314, Invitrogen) and 20 ng/ml human recombinant basic fibroblast growth factor (bFGF) (13256-029, Invitrogen). Media was changed every two- to three-days to allow neurosphere formation over two-weeks.
In order to facilitate neuronal differentiation, neurospheres were again separated into smaller pieces under a dissection microscope with three to four pieces transferred to each well of a 96-well plate [precoated with poly-D-lysine (10 μg/ml) and mouse laminin (5 μg/ml)]. The cells were then grown for 11 days (with media changed every two-days) in NBM lacking growth factors prior to induction of injury and assessment of hypothermia.
Injury and hypothermia induction
On the day of experiments, the medium was changed to NBM+N2 containing a B27 preparation lacking the usual antioxidants (10889-038, Invitrogen) (NBM-AO) to eliminate their confounding effects.
Three different injury models were used to assess the effect of hypothermia on these cells. Oxygen deprivation was induced by placing the cells into a hypoxic chamber flushed with nitrogen for 20 min and maintained at the appropriate temperature (33°C or 37°C) for four-hours inside a standard 5% CO2 incubator. Culture supernatant was removed after this four-hour period and stored at 4°C until analysed for lactate dehydrogenase activity (LDH). The media was replaced with fresh NBM-AO, and the cells were incubated for a further 20 h at the appropriate temperature (33°C or 37°C) before again measuring LDH. For combined oxygen and glucose deprivation (ODG), 25 mM 2-deoxy-D-glucose was added to NBM-AO medium and equilibrated for 30 min at room temperature before the initial media change as described above. The third injury model used was oxidative stress induced by adding 50 μM of fresh H2O2 (H1009, Sigma-Aldrich, Australia) to the NBM-AO for four-hours (33°C or 37°C, 5% CO2) when LDH was measured, and NBM-AO without H2O2 was returned to the culture, which was maintained for a further 20 h before again measuring LDH. To examine whether the effects of injury continued beyond four-hours, the media containing the above stressors was removed and replaced with fresh growth factor negative NBM before resampling for LDH activity and assessment of apoptotic cell death by terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining at 24 h.
To evaluate the effects of hypothermia over time from the induction of injury, incubation at 33°C was started immediately, or one-, or three-, or six-hours after induction of injury and maintained until a total of 24 h in each of the models.
LDH, a marker of total cell death and TUNEL staining and a marker of DNA damage typical of apoptosis, were performed according to the kit manufacturer's instructions (1164479001 and 11684795910, respectively, Roche, Australia). For LDH assays, three control measurements were made in addition to the test sample measurements. The cell culture medium without cells was assayed to assess background activity. Uninjured cells were assayed to detect activity due to basal cell death, and maximum possible cell death (100%) was detected by measuring LDH activity of lysed cells.
Statistical analysis
Each experiment was repeated three times. To avoid systematic effects on individual cultures because of the arrangement of wells in the tissue culture plates, control and injury cultures were distributed across the plates at random. Complete experiments contained within each tissue culture plate were then placed alternatively at either 37OC or 33OC. To minimize the potential impact of systematic bias, before counting of TUNEL positive cells, wells were imaged and the images recoded independently before quantitation. No additional blinding was performed for the machine-read LDH assay process. Within experiments, each comparison was performed at least in triplicate and the mean of these values taken forward into group comparisons. Two-way analysis of variance was performed, followed by post hoc Dunnett's multiple comparison test with significance set at P < 0·05 using spss (Statistics 20, IBM, Armonk, NY, USA). All values are presented as mean ± standard error of the mean.
Results
The method of stem cell culture and neuronal differentiation employed in this study gives rise to neurons with a mature projecting phenotype (Fig. 1) that express the markers β III tubulin, microtubule-associated protein 2 (MAP2), and neural nuclei (NeuN). β III tubulin positive cells comprise 70·4% ± 1·8 of the cells present before injury (obtained by calculating the ratio of 4',6-diamidino-2-phenylindole dihydrochloride (DAPI) and β III tubulin cells from 10 fields per well in three independent experiments). Dose response assays were performed to establish injury assay conditions (hypoxia, hypoxia plus glucose deprivation, and oxidative stress) that reproducibly induced 30–40% of maximal signal at four-hours to ensure that values lay within the working range of the assays used. In control cultures without injury, hypothermia had no effect on the basal level of cell death measured by LDH release at either 4 or 24 h (Figs 5).
Figure 1.
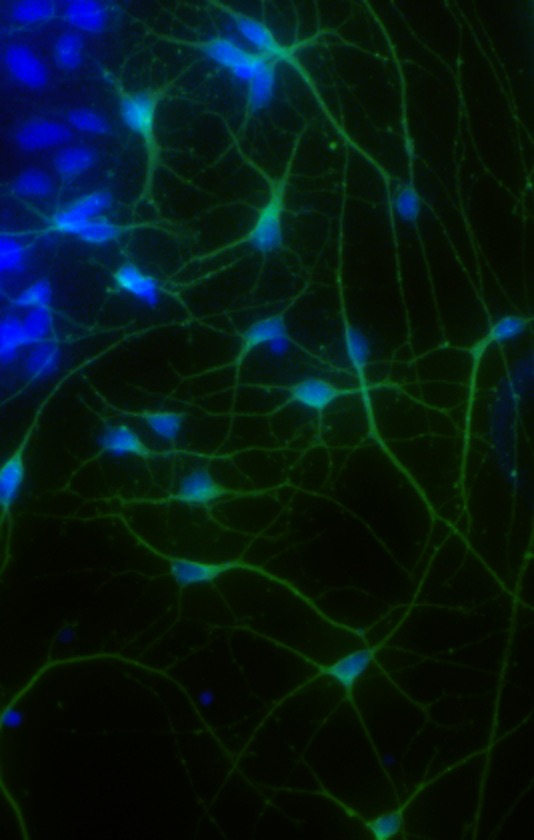
β III tubulin positive human neurons after 11 days in culture with nuclei counterstained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI).
Figure 5.
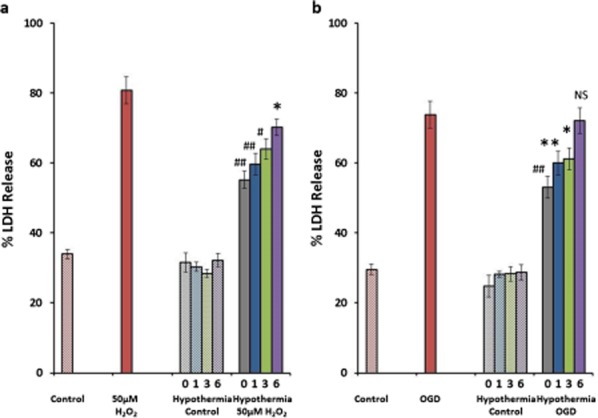
Time-dependent effects of hypothermia on H2O2 and OGD-induced cell death. Hypothermia reduces LDH-detected cell death induced with H2O2 by 52%, 43%, 34%, and 21% (a) and OGD-induced injury by 45%, 30%, 27%, and 4% when started at 0, one-, three-, and six-hours (b), respectively. *P ≤ 0·05, **P ≤ 0·01, #P ≤ 0·0005, and ##P ≤ 0·0001. Data presented as mean ± SEM.
H2O2-induced oxidative stress increased LDH-detected cell death 3·8-fold after four-hours. Removal of the H2O2 by replacing the culture media at four-hours dramatically slowed but did not halt the H2O2-induced injury (Fig. 2a,b). Hypothermia to 33°C reduced H2O2-induced cell death at four-hours by 45% [P ≤ 0·0001; 95% confidence interval (CI) 27·5–62·6] (after correcting for basal injury in controls) (Fig. 2a). Hypothermia effectively abolished (92% reduction) (P ≤ 0·001; 95% CI 45·3–131·3) the delayed injury that continued to accrue between removal of H2O2 at four-hours and completion of the experiment at 24 h (Fig. 2b). The net effect at 24 h was a reduction of LDH release of 53% (P ≤ 0·0001; 95% CI 34·8–71·04) (Fig. 2c). H2O2-induced oxidative stress increased TUNEL-detected apoptotic cell death 2·9-fold after 24 h, with 22% of cells present in culture killed by this mechanism upon introduction of H2O2. Hypothermia prevented 42% (P ≤ 0·0001; 95% CI 27·5–56·6) of this death (after correcting for basal injury in the control) (Fig. 2d).
Figure 2.
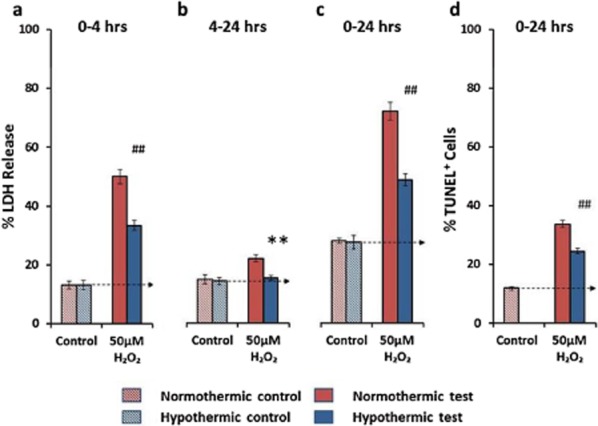
The effects of hypothermia on H2O2-induced cell death. Hypothermia reduces LDH detected cell death by 45% at four-hours (a) and effectively abolished (92% reduction) the delayed injury (b). The net effect at 24 h was reduction of 53% (c). H2O2-induced oxidative stress increased TUNEL-detected apoptotic cell death 2.9-fold after 24 h and hypothermia prevents 42% of this death (d). Statistical significance detected at *P ≤ 0·05, **P ≤ 0·01, #P ≤ 0·0005, and ##P ≤ 0·0001. Data presented as mean ± SEM.
Oxygen deprivation alone increased LDH-detected cell death approximately twofold after four-hours. Restoring the culture to a normal air/5% CO2 incubator and replacing the culture media at four-hours again slowed but did not completely halt the oxygen depletion induced injury (Fig. 3a,b). Hypothermia to 33°C had no discernible effect on oxygen depletion-induced cell death at either 4 or 24 h by either LDH or TUNEL assays (Fig. 3a–d).
Figure 3.
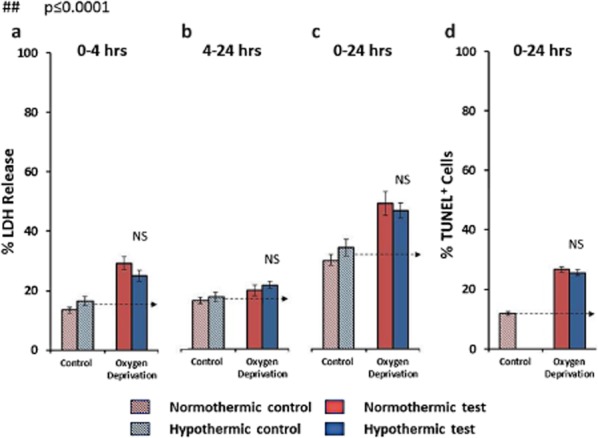
The effects of hypothermia on oxygen deprivation induced cell death. Hypothermia has no beneficial effect at four hours (a), 4–20 h (b), or in the combined 24 h LDH measurement of cell death (c) as well as on apoptotic cell death detected by TUNEL staining at 24 h (d). NS = not significant.
Combined OGD caused greater injury, increasing LDH-detected cell death 3·9-fold after four-hours. This death continued at a slower rate on restoration of normal culture conditions (Fig. 4a,b). Hypothermia to 33°C reduced OGD induced cell death at four-hours by 37% (P ≤ 0·006; 95% CI 6·8–44·8) (after correcting for basal injury in controls) (Fig. 4a). Hypothermia reduced the delayed injury that occurred between removal of OGD at four-hours and completion of the experiment at 24 h by 80% (P ≤ 0·015; 95% CI 13·8–146·8) (Fig. 4b). The net effect at 24 h was a reduction of LDH release of 42% (P ≤ 0·001; 95% CI 18·3–71·3) (Fig. 4c). TUNEL staining for DNA damage typical of apoptosis at 24 h suggested that 22% of cell death occurred by this mechanism and that hypothermia prevented 26% (P ≤ 0·001; 95% CI 12·4–52·2) of this death (after correcting for basal injury in the control) (Fig. 4d).
Figure 4.
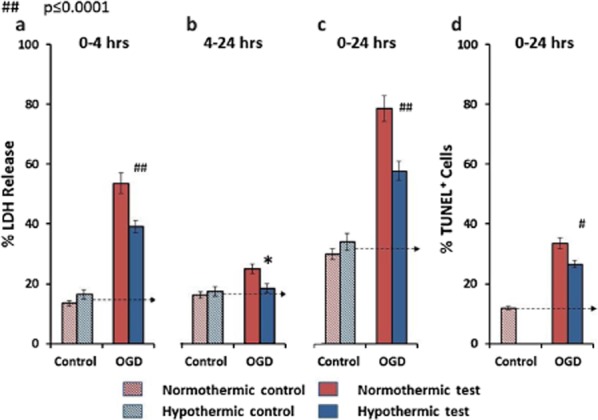
The effects of hypothermia on oxygen glucose deprivation (OGD)-induced cell death. Hypothermia reduced OGD-induced cell death at four hours by 37% (a). Hypothermia reduced the delayed injury that occurred between removal of OGD at four hours and completion of the experiment at 24 h by 80% (b). The net effect at 24 h was a reduction of LDH release of 42% (c). TUNEL staining for apoptosis at 24 h suggested that 22% of cell death occurred by this mechanism and that hypothermia prevented 26% of this death (d). *P ≤ 0·05, #P ≤ 0·0005, and ##P ≤ 0·0001. Data presented as mean ± SEM.
To confirm these observations, the H2O2 and OGD experiments were repeated as before with hypothermia throughout the period of exposure to injury (from 0 h) and with initiation of hypothermia delayed for one-, three-, or six-hours, with outcome recorded as the total % of LDH release at 24 h. As before hypothermia had no impact on cell death in control cultures but when initiated at 0, one-, three-, or six-hours protected against H2O2 toxicity by 52% (P ≤ 0·0001; 95% CI 29·8–73·5), 43% (P ≤ 0·0001; 95% CI 20·9–64·5), 34% (P ≤ 0·001; 95% CI 12·1–55·8), and 21% (P ≤ 0·05; 95% CI 0·57–43·1) (Fig. 5a), respectively, and against OGD-induced injury by 45% (P ≤ 0·0004; 95% CI 18·5–71·9), 30% (P ≤ 0·023; 95% CI 3·4–56·9), 27% (P ≤ 0·041; 95% CI 0·84–54·3), and 4% (P = 0·99; 95% CI −23·02 to 30·4) (Fig. 5b), respectively.
Discussion
This study provides the first description of protection of embryonic stem cell-derived human neurons by hypothermia. This is broadly consistent with data from ∼20 publications (Table 1) that have examined the effects of hypothermia in tissues cultured from rats, mice, gerbils, guinea pigs, and three papers utilizing human teratoma lines 40–50,52,54–56. In this literature, glutamate release 40, calcium accumulation 38, and glucose utilization 45 are reduced while membrane potential 41,51 and cellular morphology are restored 49. These are consistent with reduced injury and cell death 39,44–46,48 measured by LDH release 40,45, propidium iodide uptake 46,48, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) staining 54, caspase activity 44, and other immunohistochemical techniques 42,50,52,53,57.
Table 1.
Summary of hypothermia use to protect cells in vitro
| Year | Hypothermia temperature | Type of culture | Injury | Duration of injury | Outcome measure | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| 1991 | 35°C, 33°C, and 31°C | Gerbil hippocampal slices | OGD | Unknown | Intracellular Ca2+ accumulation | Ca2+ accumulation was delayed following hypothermia treatment | 38 |
| 1992 | 32°C | Rat cerebellar granule cells and GABAergic neurons | OGD | 3–6 h | LDH | Neuroprotective, reduces LDH caused cell death | 39 |
| 1998 | 33°C | Guinea pig E40, E60, and adult slices | OGD | 2 h and 12 h | HPLC | Reduced glutamate release was only observed in the immature cultures | 40 |
| 1998 | 35°C, 33°C, and 27°C | Rat hippocampal slices | OGD | Unknown | Extracellular membrane potential recordings | 27°C and 33°C the membrane potential was restored to the control conditions | 41 |
| 2000 | 34°C | Rat hippocampal slices | OGD | 10–20 min | Immunohistochemistry | Hypothermia reduced CA1 cell loss | 42 |
| 2001 | 31°C | Guinea pig hippocampal slices | OGD | 20, 30, or 40 min | Recovery of energy metabolism | When hypothermia added at 0 or 2 h postinjury induction it reduced energy metabolic rate | 43 |
| 2002 | 33°C | Rat cortical cultures | OGD | Unknown | Caspase 3–8 and −9 activity | Hypothermia neuronal apoptotic cell death | 44 |
| 2003 | 30°C and 33°C | Rat retina | OGD | 1–2 h | LDH | Hypothermia reduced glucose utilization and lactate production | 45 |
| 2004 | 31°C, 33°C, and 35°C | Rat hippocampal slices | OGD | 60 min | Propidium iodide (PI) | Reduced PI detected cell death only at 31°C and 33°C | 46 |
| 2004 | 33°C preconditioning | Rat cerebellar slices | OGD | 20 min | Immunohistochemistry | Hypothermic preconditioning induces acute neuroprotection | 47 |
| 2005 | 35°C | Rat hippocampal slices | OGD | 1 h | Propidium iodide | Protects all regions of hippocampus from cell death | 48 |
| 2005 | 27°C or 33°C | Dissociated mouse hippocampal neurons | OGD | 10–25 min | Spine shape and motility assessed by microscopy | Decrease in temperature reduced the spine modality, and the length of spines was reduced | 49 |
| 2005 | 35°C and 31°C | Rat hippocampal slices | OGD | 40 min | Immunohistochemistry | Decrease in temperature – increased protection in all the hippocampal regions | 50 |
| 2006 | 34°C pre treatment | Rat hippocampal slices | OGD | 20 min | Electrophysiology | Temperature lowering reduced OGD-induced cell death | 51 |
| 2006 | 34°C, 30°C, or 22°C | Rat hippocampal neurons | OGD | Unknown | Expression of Bcl-2, fluorescence magnitude of intracellular Ca2+ | Expression of Bcl-2 was increased with decrease in temperature | 52 |
| 2006 | 33°C preconditioning for 20 min, 1 h before OGD | Rat cerebellar cells | OGD | 20 min | Immunohistochemistry | Hypothermic preconditioning increased survival of Purkinje neurons in rat cerebellar slices after OGD | 53 |
| 2009 | 33°C | Human NT2-N neurons | OGD | 20 min | MTT | Posthypoxic hypothermia is protective in human NT2-N neurons | 54 |
| 2010 | 33°C and 30°C | Rat cortical neurons | Hypoxia | 24 h | Cell viability | Increased cell survival following hypothermia | 55 |
Hypothermia significantly protected embryonic stem cell-derived human neurons against oxidative stress and from hypoxia when glucose concentration was reduced but was ineffective against hypoxia alone. Neuroprotective effects were seen with delays in initiation of hypothermia of up to six-hours, with the magnitude of benefit progressively decreasing as the time to hypothermia initiation increased. In our preparations protection against apoptosis was also detected, consistent with data from mouse neuronal cultures 44 and focal cerebral ischemia experiments in animals 58 showing that mild hypothermia attenuates DNA damage typical of apoptotic neuronal cell death. However, the difference between degree of injury detected at 24 h by LDH and TUNEL assays suggests cell death is also occurring in the cultures by nonapoptotic mechanisms such as necrosis.
There are a number of studies using NT2-N neurons in similar experiments with conflicting data evident. Although one study reports essentially the same result as here 54, another reports that hypothermia protects against the effects of oxygen restriction alone 59. The reasons for these differences are unclear but suggest differential expression of the metabolic or signaling machinery involved in ischemic injury. NT2-N cells are derived from the human NTera2 embryonal carcinoma stem cell line 60 and are known to differ substantially in their broad pattern of gene expression when compared with different hESC lines 61 and therefore may not be the most appropriate models to study human disease 62.
Intriguingly, hypothermia had no effect on the basal level of cell death in the uninjured controls. The contrasting response to different injury models, lack of effect on culture dependent cell death, and different behaviors to experiments reported in other cell lines suggests interaction specifically in the processes of ischemic injury rather than cell death itself and requires further study. These differences also do not appear to be consistent with hypothermia-inducing protection simply by slowing overall metabolic activity 63 but could still be consistent with decreasing the cerebral metabolic rates of glucose and oxygen consumption 64 and slowing adenosine triphosphate (ATP) breakdown 65.
It is not clear how the time frame of protection of human neurons in vitro will be predictive of time frame in stroke. However, detection of protection against oxidative stress even after cessation of injury offers hope for the clinical setting. This may be particularly relevant if reperfusion injury 66–68 acts to lessen the potential benefit afforded by thrombolysis or thrombectomy and suggests this may be an important target for clinical trials of hypothermia. The data are certainly consistent with the recent observation that mild hypothermia reduces the deleterious side effects of tissue plasminogen activator treatment after thromboembolic stroke in rats 69. Different time frames of benefit via different mechanisms for a single therapeutic approach suggest we still have much to learn about the details of the ischemic cascade and the therapeutic opportunities it might offer. The window of opportunity detected here in human neurons in vitro is certainly consistent with a wide window in rodent models of stroke 20 and with the planned six-hour window for EuroHYP-1 70.
As in most experiments, our data and its interpretation have limits. The current experiments have been performed in cells that exhibit a branching phenotype expressing β III tubulin. However, these cells have spent only 11 days in culture and thus might be better viewed as immature. It is not yet clear whether these cells can stand in for a 60-year-old neuron from a typical stroke victim. However, it seems reasonable to believe that we can devise strategies to rapidly and repeatedly stress them much as a car manufacture would stress test a car. Nevertheless, we might see different effects when cultures have matured neurochemically. Moreover, clinically relevant protection might be dependent on effects on neurotransmission itself 71 or regulation of edema 72. Furthermore, beneficial effects might be restricted to specific neurochemical profiles present at high abundance in our culture but not in areas of the brain affected by most common forms of human stroke. Another limitation of the current experiments was ethical approval to study only one hESC cell line. It will be important to confirm these observations in other hESC lines, and when the technology is more mature 73,74 to examine whether the same effects are seen in induced pluripotent stem cells (iPSCs) from patients with stroke or relevant defects of oxidative metabolism such as seen in inborn errors of mitochondrial function.
Conclusion
This study provides the first description of protection of embryonic stem cell derived human neurons by hypothermia. If hypothermia does have a predilection for ischemic processes, this offers hope for use of hypothermia to treat ischemic stroke and spinal cord injuries where cord compression results in secondary ischemia 75. This study also provides proof of principle that human neurons derived from hESCs can be used to screen new drugs for therapeutic effect. For screening large chemical libraries, this will be more specific than screening in cell lines from other species, and more cost-effective than initial screening in animal models of stroke.
Acknowledgments
We thank Clare Parish for her contributions toward this study.
The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the NHMRC and the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
References
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695–1703. doi: 10.1016/S0140-6736(10)60491-6. [Meta-Analysis] [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Kollmar R, Gebhardt B, Schwab S. [EuroHYP-1 trial: EU-funded therapy study on the effectiveness of mild therapeutic hypothermia for acute ischemic stroke.] Nervenarzt. 2012;83:1252–1259. doi: 10.1007/s00115-012-3533-6. [DOI] [PubMed] [Google Scholar]
- Sena ES, van der Worp HB, Bath PM, Howells DW, Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. 2010;8:e1000344. doi: 10.1371/journal.pbio.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod MR, van der Worp HB, Sena ES, Howells DW, Dirnagl U, Donnan GA. Evidence for the efficacy of NXY-059 in experimental focal cerebral ischaemia is confounded by study quality. Stroke. 2008;39:2824–2829. doi: 10.1161/STROKEAHA.108.515957. [DOI] [PubMed] [Google Scholar]
- Macleod MR, O'Collins T, Horky LL, Howells DW, Donnan GA. Systematic review and metaanalysis of the efficacy of FK506 in experimental stroke. J Cereb Blood Flow Metab. 2005;25:713–721. doi: 10.1038/sj.jcbfm.9600064. [DOI] [PubMed] [Google Scholar]
- Antonic A, Howells DW. Human in vitro models of ischaemic stroke: a test bed for translation. Translational Stroke Research. 2012;3:306–309. doi: 10.1007/s12975-012-0201-x. [DOI] [PubMed] [Google Scholar]
- Zhao S, Shetty J, Hou L, et al. Human, mouse, and rat genome large-scale rearrangements: stability versus speciation. Genome Res. 2004;14(10A):1851–1860. doi: 10.1101/gr.2663304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos A, Castillo J, Serena J, Noya M. Duration of glutamate release after acute ischemic stroke. Stroke. 1997;28:708–710. doi: 10.1161/01.str.28.4.708. [Research Support, Non-U.S. Gov't] [DOI] [PubMed] [Google Scholar]
- Takagi K, Ginsberg MD, Globus MY, et al. Changes in amino acid neurotransmitters and cerebral blood flow in the ischemic penumbral region following middle cerebral artery occlusion in the rat: correlation with histopathology. J Cereb Blood Flow Metab. 1993;13:575–585. doi: 10.1038/jcbfm.1993.75. [DOI] [PubMed] [Google Scholar]
- Nilupul Perera M, Ma HK, Arakawa S, et al. Inflammation following stroke. J Clin Neurosci. 2006;13:1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Sena ES, Briscoe CL, Howells DW, Donnan GA, Sandercock PA, Macleod MR. Factors affecting the apparent efficacy and safety of tissue plasminogen activator in thrombotic occlusion models of stroke: systematic review and meta-analysis. J Cereb Blood Flow Metab. 2010;30:1905–1913. doi: 10.1038/jcbfm.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell CS, Liebeskind DS, Starkman S, Saver JL. Trends in acute ischemic stroke trials through the 20th century. Stroke. 2001;32:1349–1359. doi: 10.1161/01.str.32.6.1349. [DOI] [PubMed] [Google Scholar]
- Ferguson KN, Kidwell CS, Starkman S, Saver JL. Hyperacute treatment initiation in neuroprotective agent stroke trials. J Stroke Cerebrovasc Dis. 2004;13:109–112. doi: 10.1016/j.jstrokecerebrovasdis.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- Gladstone DJ, Black SE, Hakim AM, Heart, Stroke Foundation of Ontario Centre of Excellence in Stroke R. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Howells DW. Evaluation of combination therapy in animal models of cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:585–597. doi: 10.1038/jcbfm.2011.203. [Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain. 2007;130:3063–3074. doi: 10.1093/brain/awm083. [DOI] [PubMed] [Google Scholar]
- Zhao H, Steinberg GK, Sapolsky RM. General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab. 2007;27:1879–1894. doi: 10.1038/sj.jcbfm.9600540. [DOI] [PubMed] [Google Scholar]
- Yenari M, Kitagawa K, Lyden P, Perez-Pinzon M. Metabolic downregulation: a key to successful neuroprotection? Stroke. 2008;39:2910–2917. doi: 10.1161/STROKEAHA.108.514471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith J, Jorgensen HS, Pedersen PM, et al. Body temperature in acute stroke: relation to stroke severity, infarct size, mortality, and outcome. Lancet. 1996;347:422–425. doi: 10.1016/s0140-6736(96)90008-2. [DOI] [PubMed] [Google Scholar]
- Castillo J, Davalos A, Marrugat J, Noya M. Timing for fever-related brain damage in acute ischemic stroke. Stroke. 1998;29:2455–2460. doi: 10.1161/01.str.29.12.2455. [Research Support, Non-U.S. Gov't] [DOI] [PubMed] [Google Scholar]
- den Hertog HM, van der Worp HB, van Gemert HM, et al. The Paracetamol (Acetaminophen) In Stroke (PAIS) trial: a multicentre, randomised, placebo-controlled, phase III trial. Lancet Neurol. 2009;8:434–440. doi: 10.1016/S1474-4422(09)70051-1. [DOI] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Arrich J, Holzer M, Herkner H, Mullner M. Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst Rev. 2009;(4) doi: 10.1002/14651858.CD004128.pub2. CD004128. [DOI] [PubMed] [Google Scholar]
- Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574–1584. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Hunt R, Tarnow-Mordi W, Inder T, Davis P. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2007;(4) doi: 10.1002/14651858.CD003311.pub2. CD003311. [DOI] [PubMed] [Google Scholar]
- Sydenham E, Roberts I, Alderson P. Hypothermia for traumatic head injury. Cochrane Database Syst Rev. 2009;(2) doi: 10.1002/14651858.CD001048.pub3. CD001048. [DOI] [PubMed] [Google Scholar]
- Leker RR, Shohami E. Cerebral ischemia and trauma-different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Brain Res Rev. 2002;39:55–73. doi: 10.1016/s0165-0173(02)00157-1. [DOI] [PubMed] [Google Scholar]
- Den Hertog HM, van der Worp HB, Tseng MC, Dippel DW. Cooling therapy for acute stroke. Cochrane Database Syst Rev. 2009;(1) doi: 10.1002/14651858.CD001247.pub2. CD001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Georgia MA, Krieger DW, Abou-Chebl A, et al. Cooling for Acute Ischemic Brain Damage (COOL AID): a feasibility trial of endovascular cooling. Neurology. 2004;63:312–317. doi: 10.1212/01.wnl.0000129840.66938.75. [DOI] [PubMed] [Google Scholar]
- Guluma KZ, Hemmen TM, Olsen SE, Rapp KS, Lyden PD. A trial of therapeutic hypothermia via endovascular approach in awake patients with acute ischemic stroke: methodology. Acad Emerg Med. 2006;13:820–827. doi: 10.1197/j.aem.2006.03.559. [Multicenter Study Randomized Controlled Trial] [DOI] [PubMed] [Google Scholar]
- Hemmen TM, Raman R, Guluma KZ, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS-L): final results. Stroke. 2010;41:2265–2270. doi: 10.1161/STROKEAHA.110.592295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dottori M, Pera MF. Neuronal differenitation of human embryonic stem cells. Methods Mol Biol. 2007;438:19–30. doi: 10.1007/978-1-59745-133-8_3. [DOI] [PubMed] [Google Scholar]
- Mitani A, Kataoka K. Critical levels of extracellular glutamate mediating gerbil hippocampal delayed neuronal death during hypothermia: brain microdialysis study. Neuroscience. 1991;42:661–670. doi: 10.1016/0306-4522(91)90035-m. [DOI] [PubMed] [Google Scholar]
- Yue TL, Gu JL, Lysko PG, Cheng HY, Barone FC, Feuerstein G. Neuroprotective effects of phenyl-t-butyl-nitrone in gerbil global brain ischemia and in cultured rat cerebellar neurons. Brain Res. 1992;574:193–197. doi: 10.1016/0006-8993(92)90816-r. [DOI] [PubMed] [Google Scholar]
- Berger R, Jensen A, Hossmann KA, Paschen W. Effect of mild hypothermia during and after transient in vitro ischemia on metabolic disturbances in hippocampal slices at different stages of development. Brain Res Dev Brain Res. 1998;105:67–77. [PubMed] [Google Scholar]
- Onitsuka M, Mihara S, Inokuchi H, Shigemori M, Higashi H. Mild hypothermia protects rat hippocampal CA1 neurons from irreversible membrane dysfunction induced by experimental ischemia. Neurosci Res. 1998;30:1–6. doi: 10.1016/s0168-0102(97)00110-7. [DOI] [PubMed] [Google Scholar]
- Popovic R, Liniger R, Bickler PE. Anesthetics and mild hypothermia similarly prevent hippocampal neuron death in an in vitro model of cerebral ischemia. Anesthesiology. 2000;92:1343–1349. doi: 10.1097/00000542-200005000-00024. [DOI] [PubMed] [Google Scholar]
- Garnier Y, Pfeiffer D, Jensen A, Berger R. Effects of mild hypothermia on metabolic disturbances in fetal hippocampal slices after oxygen/glucose deprivation depend on depth and time delay of cooling. J Soc Gynecol Investig. 2001;8:198–205. doi: 10.1016/s1071-5576(01)00119-8. [DOI] [PubMed] [Google Scholar]
- Xu L, Yenari MA, Steinberg GK, Giffard RG. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab. 2002;22:21–28. doi: 10.1097/00004647-200201000-00003. [DOI] [PubMed] [Google Scholar]
- Quinones-Hinojosa A, Malek JY, Ames A, 3rd, Ogilvy CS, Maynard KI. Metabolic effects of hypothermia and its neuroprotective effects on the recovery of metabolic and electrophysiological function in the ischemic retina in vitro. Neurosurgery. 2003;52:1178–1186. discussion 86–7. [PubMed] [Google Scholar]
- McManus T, Sadgrove M, Pringle AK, Chad JE, Sundstrom LE. Intraischaemic hypothermia reduces free radical production and protects against ischaemic insults in cultured hippocampal slices. J Neurochem. 2004;91:327–336. doi: 10.1111/j.1471-4159.2004.02711.x. [DOI] [PubMed] [Google Scholar]
- Yuan HB, Huang Y, Zheng S, Zuo Z. Hypothermic preconditioning increases survival of purkinje neurons in rat cerebellar slices after an in vitro simulated ischemia. Anesthesiology. 2004;100:331–337. doi: 10.1097/00000542-200402000-00023. [DOI] [PubMed] [Google Scholar]
- Feiner JR, Bickler PE, Estrada S, Donohoe PH, Fahlman CS, Schuyler JA. Mild hypothermia, but not propofol, is neuroprotective in organotypic hippocampal cultures. Anesth Analg. 2005;100:215–225. doi: 10.1213/01.ANE.0000142129.17005.73. [DOI] [PubMed] [Google Scholar]
- Gisselsson LL, Matus A, Wieloch T. Actin redistribution underlies the sparing effect of mild hypothermia on dendritic spine morphology after in vitro ischemia. J Cereb Blood Flow Metab. 2005;25:1346–1355. doi: 10.1038/sj.jcbfm.9600131. [DOI] [PubMed] [Google Scholar]
- Rytter A, Cardoso CM, Johansson P, et al. The temperature dependence and involvement of mitochondria permeability transition and caspase activation in damage to organotypic hippocampal slices following in vitro ischemia. J Neurochem. 2005;95:1108–1117. doi: 10.1111/j.1471-4159.2005.03420.x. [DOI] [PubMed] [Google Scholar]
- Lipski J, Park TI, Li D, et al. Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res. 2006;1077:187–199. doi: 10.1016/j.brainres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Liu RG, Wang WJ, Song N, Chen YQ, Li LH. Diazoxide preconditioning plus subsequent hypothermia increased resistance of rat cultured hippocampal neurons against hypoxia-reoxygenation injury. Chin Med J (Engl) 2006;119:887–893. [PubMed] [Google Scholar]
- Yuan HB, Huang Y, Zheng S, Zuo Z. Hypothermic preconditioning reduces Purkinje cell death possibly by preventing the over-expression of inducibl nitric oxide synthase in rat cerebellar slices after an in vitro simulated ischemia. Neuroscience. 2006;142:381–389. doi: 10.1016/j.neuroscience.2006.06.053. [DOI] [PubMed] [Google Scholar]
- Dalen ML, Froyland E, Saugstad OD, Mollnes TE, Rootwelt T. Post-hypoxic hypothermia is protective in human NT2-N neurons regardless of oxygen concentration during reoxygenation. Brain Res. 2009;1259:80–89. doi: 10.1016/j.brainres.2008.12.055. [DOI] [PubMed] [Google Scholar]
- Hua Y, Hisano K, Morimoto Y. Effect of mild and moderate hypothermia on hypoxic injury in nearly pure neuronal culture. J Anesth. 2010;26:67–76. doi: 10.1007/s00540-010-0999-x. [DOI] [PubMed] [Google Scholar]
- Mitani A, Kadoya F, Kataoka K. Temperature dependence of hypoxia-induced calcium accumulation in gerbil hippocampal slices. Brain Res. 1991;562:159–163. doi: 10.1016/0006-8993(91)91201-b. [DOI] [PubMed] [Google Scholar]
- Yuan HB, Huang YM, Zheng SQ, Zuo ZY. Hypothermic preconditioning increases survival of Purkinje neurons in rat cerebellar slices after an in vitro simulated ischemia. Anesthesiology. 2004;100:331–337. doi: 10.1097/00000542-200402000-00023. [DOI] [PubMed] [Google Scholar]
- Eberspacher E, Werner C, Engelhard K, et al. Long-term effects of hypothermia on neuronal cell death and the concentration of apoptotic proteins after incomplete cerebral ischemia and reperfusion in rats. Acta Anaesthesiol Scand. 2005;49:477–487. doi: 10.1111/j.1399-6576.2005.00649.x. [DOI] [PubMed] [Google Scholar]
- Rootwelt T, Dunn M, Yudkoff M, Itoh T, Almaas R, Pleasure D. Hypoxic cell death in human NT2-N neurons: involvement of NMDA and non-NMDA glutamate receptors. J Neurochem. 1998;71:1544–1553. doi: 10.1046/j.1471-4159.1998.71041544.x. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Page C, Lee VM. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12:1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz CM, Spivak CE, Baker SC, et al. NTera2: a model system to study dopaminergic differentiation of human embryonic stem cells. Stem Cells and Development. 2005;14:517–534. doi: 10.1089/scd.2005.14.517. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 6:11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod YK, Diringer MN. Temperature management in acute neurologic disorders. Neurol Clin. 2011;26:585–603. doi: 10.1016/j.ncl.2008.02.005. [Review], xi 2008. [DOI] [PubMed] [Google Scholar]
- Hagerdal M, Harp J, Nilsson L, Siesjo BK. The effect of induced hypothermia upon oxygen consumption in the rat brain. J Neurochem. 1975;24:311–316. doi: 10.1111/j.1471-4159.1975.tb11881.x. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. J Cereb Blood Flow Metab. 2003;23:513–530. doi: 10.1097/01.WCB.0000066287.21705.21. [DOI] [PubMed] [Google Scholar]
- Mowlavi A, Neumeister MW, Wilhelmi BJ, Song Y-H, Suchy H, Russell RC. Local hypothermia during early reperfusion protects skeletal muscle from ischemia-reperfusion injury. Plastic Reconstr Surg. 2003;111:242–250. doi: 10.1097/01.PRS.0000034936.25458.98. [DOI] [PubMed] [Google Scholar]
- Strbian D, Karjalainen-Lindsberg M-L, Kovanen PT, Tatlisumak T, Lindsberg PJ. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation. 2007;116:411–418. doi: 10.1161/CIRCULATIONAHA.106.655423. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cerebral Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- Kallmunzer B, Schwab S, Kollmar R. Mild hypothermia of 34 degrees C reduces side effects of rt-PA treatment after thromboembolic stroke in rats. Exp Transl Stroke Med. 2012;4:3. doi: 10.1186/2040-7378-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Worp HB, Macleod MR, Kollmar R. Therapeutic hypothermia for acute ischemic stroke: ready to start large randomized trials[quest] J Cereb Blood Flow Metab. 2010;30:1079–1093. doi: 10.1038/jcbfm.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Ouyang Y, Kennedy BR, Rothman SM. Cooling blocks rat hippocampal neurotransmission by a presynaptic mechanism: observations using 2-photon microscopy. J Physiol. 2005;567(Pt 1):215–224. doi: 10.1113/jphysiol.2005.088948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreckinger M, Marion DW. Contemporary management of traumatic intracranial hypertension: is there a role for therapeutic hypothermia? Neurocrit Care. 2009;11:427–436. doi: 10.1007/s12028-009-9256-2. [Review] [DOI] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of Fragile X syndrome by human embryonic stem cells and induced-pluripotent stem cells. Cell Stem Cell. 2010;6:407. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Lu SJ, Klimanskaya I, et al. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells. 2010;28:704–712. doi: 10.1002/stem.321. [DOI] [PubMed] [Google Scholar]
- Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15–26. doi: 10.3171/jns.1991.75.1.0015. [DOI] [PubMed] [Google Scholar]