

Intellectual disability (ID) is a lifelong, debilitating condition affecting 2% to 3% Canadians (1). It is defined by deficits in cognitive functioning (IQ <70) and adaptive skills, and termed global developmental disability (GDD) in children <5 years of age (defined as deficits in >2 developmental domains, eg, fine/gross motor skills, speech and interaction) (2,3). In Canada, approximately 7600 to 11,500 children are born with GDD annually (1). In the present commentary, the term ID will be used to refer to ID/GDD.
ID is associated with the highest health care costs of any disease, nearly equalling the combined economic impact of stroke, heart disease and cancer (4). Associated comorbidity is significant and includes epilepsy, autism, psychiatric/behavioural disturbances, sensory deficiencies and systemic organ involvement (eg, congenital heart defects, liver disease).
While the etiology of ID is diverse, including infectious, traumatic and toxic origins, genetic etiologies constitute the most frequent cause, demonstrable in >50% of patients; this number is increasing with the advent of novel genomic technologies (5). Genetic causes range from numerical and structural chromosomal abnormalities and submicroscopic copy number variants, to methylation abnormalities and single-gene defects (6).
Given the etiological heterogeneity, diagnostic evaluation of children with ID is a challenge for neurologists, geneticists and paediatricians. Current guidelines for the clinical evaluation of genetic causes of ID are based on frequencies of single conditions and yield of diagnostic methods and procedures (7–10). However, causal therapy (ie, targeting the pathophysiology) is not available for many of the conditions identified by this approach, thereby limiting any potential therapeutic benefit.
By contrast, inborn errors of metabolism (IEM) represent the largest category of genetic conditions amenable to causal therapy. However, the literature suggests low diagnostic yields associated with metabolic testing (0.8% to 2.5%). The need for multiple tests to exclude a few rare to ultra-rare conditions and the limited availability of laboratories offering comprehensive diagnostic testing explain why, outside of highly specialized centres, metabolic work-up of patients with ID is time-consuming, expensive and often remains incomplete. Due to these limitations, diagnostic yield is not representative, varies according to publication and is reportedly low in patients presenting with ID. However, several studies indicate a higher yield for metabolic testing performed in the tertiary care setting (11,12). As of 2013, a comprehensive metabolic evaluation, as we suggest here, has not been reported for larger groups of ID patients (13).
In 2012, our group was the first to publish a systematic literature review identifying 81 treatable IEM with ID as a distinguishing feature (14). We sought to investigate the number of treatable IEM presenting with ID, and to characterize the types of treatments and evidence for their effect. Furthermore, our literature search identified 91 causal therapies including medical diets, vitamin supplements, substrate inhibitors, stem cell transplant and gene therapy. Only one expensive drug is included; the remaining therapies are relatively affordable, accessible and safe. Effects range from improvement of cognition and development, to stabilization or prevention of brain damage. The evidence level is limited, a factor inherent to rare diseases such as these. While an overview of all therapies for each IEM is beyond the scope of the present article, a complete list of identified treatments together with corresponding evidence levels and therapeutic effects is available at www.treatable-id.org.
Motivated by the cost savings generated by early identification of these diseases, the potential for improved health outcomes and prevention of brain damage through timely therapeutic initiation, we used evidence generated by our literature review to develop a two-tiered diagnostic protocol prioritizing genetic, treatable conditions presenting with ID (5).The protocol is currently implemented as the Treatable Intellectual Disability Endeavor (TIDE) study (www.tidebc.org) in our institution, the BC Children’s Hospital (BCCH) (Vancouver, British Columbia) and throughout British Columbia, in a joint 18-month pilot initiative with Child Health BC.
Diagnostic protocol for treatable IDs
The two-tiered TIDE protocol is designed to identify treatable IDs at the outset of diagnostic work-up in at-risk patients. The first tier involves biochemical group tests, which potentially indicate 60% of the currently known treatable IDs (Figure 1). This first tier can and should be applied for all patients with unexplained ID by community paediatricians and specialists, to effectively exclude a majority of treatable IEM without referral to a specialized centre. Generally, these tests are provided by most biochemical genetics laboratories across Canada, with reasonable turnaround times and affordable prices. In British Columbia, these tests are funded by the medical services plan for a total cost of approximately $527.97 per patient (15).
Figure 1).
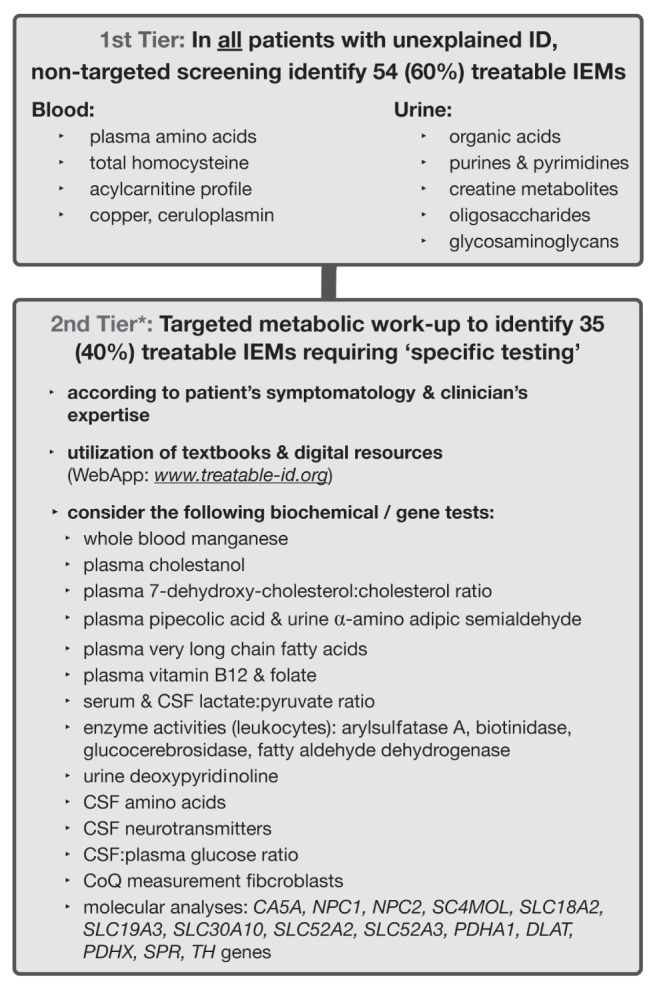
Treatable Intellectual Disability Endeavor (TIDE) protocol. The asterisk at the 2nd tier denotes the step at which the current practice parameters for the comprehensive evaluation of intellectual disability should be implemented (American Academy of Pediatrics, 2014 [8]). IEM Inborn errors of metabolism; CSF Cerebrospinal fluid; CoQ Coenzyme Q
The next step is to apply the current diagnostic practice parameters for ID, published by the American Academy of Pediatrics in 2014 (8), which include chromosome microarray as a first-line test and, in selected cases, Fragile X testing, neuroimaging and other tests, in combination with the second tier of the TIDE algorithm for the identification of the remaining 35 treatable IDs. These conditions require a more targeted approach, including single metabolite or primary molecular analysis, which is designated as the second tier. Because these tests can be difficult to obtain, and/or require invasive sampling procedures (eg, spinal tap for cerebrospinal fluid collection, skin biopsy to cultivate fibroblasts) and/or extensive funding, a clinical differential diagnosis is necessary to facilitate an efficient diagnostic work-up.
Together with a corresponding interactive website (www.treatable-id.org) and a freely available Treatable ID App (via www.treatable-id.org and the Apple App store as the TIDE-BC App), an adapted version of the TIDE protocol has been implemented in our tertiary-care institution as of 2011, for the evaluation of ID patients seen in the divisions of medical genetics, paediatric neurology and biochemical diseases. This local version comprises urine glycosaminoglycans and oligosaccharides as second-tier rather than first-tier tests due to insufficient laboratory capacity at present.
The WebApp can mitigate the complexity and time-consuming nature of this task by enabling recognition of treatable IDs and maximizing the efficiency of diagnostic work-up. Usable on all types of handheld devices (eg, BlackBerry [BlackBerry Limited, Canada], iPad [Apple Inc, USA]), these digital tools enable review of all treatable IDs according to biochemical defects and categories, diagnostic tests, clinical features and treatment modalities with the levels of evidence and effect. They have been designed for clinicians and scientists active in the diagnostic evaluation of ID (paediatricians, neurologists, biochemical/clinical geneticists, metabolic specialists) as well as laboratory scientists, ranging from student to expert level.
Notably, during the period since the literature search was performed in 2011, new ‘treatable IDs’ have emerged through gene discovery and/or generation of evidence for new treatments including but not limited to: dihydrofolate reductase deficiency (oral folinic acid supplements), SC4MOL deficiency (oral cholesterol supplements); Lesch-Nyhan syndrome (hematopoietic stem cell transplantation); pyridoxine-dependent epilepsy (lysine-restricted diet); carbonic anhydrase II deficiency (carglumic acid, sick day formula) (16–20). Consequently, the guidelines have been and will continue to be modified to incorporate these and other exciting diagnostic and therapeutic advances as necessary; the latest publication comprises 89 treatable IEMs (5).
To enable immediate use of the results of the evidence-based systematic review, see www.sciencedirect.com/science/article/pii/S1096719211006081.
INITIAL RESULTS
Evaluation of the TIDE protocol is currently underway via a study funded by the BCCH Foundation as the first Collaborative Area of Innovation. The 2.5-year pilot study, performed in our tertiary care centre, is gathering quantitative data (ie, screening, referral and testing volumes) and qualitative data (usefulness and uptake of the protocol) to determine the utility for both diagnostic frequency and screening costs associated with the protocol. Furthermore, with informed consent, phenotypic data and DNA are collected and stored for future research. In parallel, studies using genomics and metabolomics technologies are underway to identify new diseases.
Results from the initial 24-month implementation period show that clinicians at BCCH identified treatable IEMs in >5% of 410 patients with ID. Further analysis comparing these patients with those diagnosed at BCCH between 2000 and 2009 showed that the screening protocol reduced ‘time to diagnosis’ by as much as six months (range one to 50 months), and costs of unnecessary testing (>$1,500 per patient) (21). The final results from data on >450 patients will be published by the end of 2014.
Given the positive preliminary data from our pilot study and the fact that early identification of individuals with ID/GDD enables the initiation of treatment before irreversible brain damage occurs, thereby improving patient outcomes and reducing unnecessary costs, implementing the TIDE protocol throughout BC would enable further, systematic evaluation of the protocol while also providing more equitable access to screening.
Consequently, we undertook a second, parallel pilot study, involving 18 community-based paediatricians, to evaluate implementation of the protocol’s first-tier metabolic screening outside BCCH in every patient presenting with unexplained ID. Additionally, we refined the consensus criteria for referral of patients for further evaluation at our tertiary care centre. This effort is meant to improve access to screening for the 2% to 3% of our provincial youth who exhibit ID, to facilitate early diagnosis, to streamline referrals, to improve patient outcomes, and to provide health care economies by mitigating diagnostic delay and unnecessary testing. The data generated through this provincial initiative (which, to date, show good uptake/yield and positive experiences) will be used to expand the implementation process province-wide, with the support of Child Health BC. This model could serve as an example for other provinces and centres in Canada to motivate clinicians to screen for treatable conditions in patients with unexplained ID, thereby leading the way globally for improvement in clinical practice.
Acknowledgments
Drs Graham Sinclair and Hilary Vallance (Biochemical Genetics Laboratory) for data on cost of first-tier testing; Mr Brian Sayson, TIDE Research Coordinator; Suud Nahdi BSc MPH, TIDE Research Evaluator; Ms Claire Sowerbutt (medical writer) for text editing; Mr Roderick Houben and Mr Jeff Joa (Health2Media) for App design and updates.
Footnotes
CONFLICT OF INTEREST: Drs van Karnebeek and Stockler-Ipsiroglu developed the content of, and contributed to the design of the freely available App described (www.treatable-id.org).
FUNDING: This work was supported by funding from the BC Children’s Hospital Foundation as the first Collaborative Area of Innovation. Dr van Karnebeek was supported by the Michael Smith Foundation for Health Research, via the Scholar Award.
REFERENCES
- 1.Statistics Canada . Ottawa: Government of Canada.; Date modified: September 26, 2013. < www.statcan.gc.ca/tables-tableaux/sum-som/l01/cst01/demo04a-eng.htm> (Accessed May 29, 2014). [Google Scholar]
- 2.Luckasson R, Reeve A. Naming, defining, and classifying in mental retardation. Ment Retard. 2001:47–52. doi: 10.1352/0047-6765(2001)039<0047:NDACIM>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 3.Shevell M. Present conceptualization of early childhood neurodevelopmental disabilities. J Child Neurol. 2010:120–6. doi: 10.1177/0883073809336122. [DOI] [PubMed] [Google Scholar]
- 4.Meerding WJ, Bonneux L, Polder JJ, Koopmanschap MA, van der Maas PJ. Demographic and epidemiological determinants of healthcare costs in Netherlands: Cost of illness study. BMJ. 1998:111–7. doi: 10.1136/bmj.317.7151.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Karnebeek CDM, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: Diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab. 2014;111:428–38. doi: 10.1016/j.ymgme.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Moeschler J. Genetic evaluation of intellectual disabilities. Semin Ped Neurol. 2008:2–9. doi: 10.1016/j.spen.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 7.van Karnebeek CDM, Jansweijer MCE, Leenders AGE, Offringa MA, Hennekam RCM. Diagnostic investigations in individuals with mental retardation: A systematic literature review. Eur J Hum Genet. 2005:6–25. doi: 10.1038/sj.ejhg.5201279. [DOI] [PubMed] [Google Scholar]
- 8.Moeschler J, Shevell M, Committee on Genetics Comprehensive evaluation of the child with intellectual disability or global developmental delay. Pediatrics. 2014;134:e903–18. doi: 10.1542/peds.2014-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AJ, Ashwal S. Evidence report: Genetic and metabolic testing on children with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011:1629–35. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 10.Curry CJ, Stevenson RE, Aughton D, et al. Evaluation of mental retardation: Recommendations of a Consensus Conference: American College of Medical Genetics. Am J Med Genet. 1997:468–77. doi: 10.1002/(sici)1096-8628(19971112)72:4<468::aid-ajmg18>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 11.Papavasiliou AS, Bazigou H, Paraskevoulakos E, Kotsalis C. Neurometabolic testing in developmental delay. J Child Neurol. 2000:620–2. doi: 10.1177/088307380001500909. [DOI] [PubMed] [Google Scholar]
- 12.Engbers HM, Berger R, van Hasselt P, et al. Yield of additional metabolic studies in neurodevelopmental disorders. Ann Neurol. 2008:212–7. doi: 10.1002/ana.21435. [DOI] [PubMed] [Google Scholar]
- 13.van Karnebeek CD, Jansweijer MC, Leenders AG, Offringa M, Hennekam RC. Diagnostic investigations in individuals with mental retardation: A systematic literature review. Eur J Hum Genet. 2005:6–25. doi: 10.1038/sj.ejhg.5201279. [DOI] [PubMed] [Google Scholar]
- 14.van Karnebeek CDM, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: A systematic literature review. Mol Genet Metab. 2012:368–81. doi: 10.1016/j.ymgme.2011.11.191. [DOI] [PubMed] [Google Scholar]
- 15.van Karnebeek CD, Houben RF, Lafek M, Giannasi W, Stockler S. The treatable intellectual disability APP www.treatable-id.org: A digital tool to enhance diagnosis & care for rare diseases. Orphanet J Rare Dis. 2012;7:47. doi: 10.1186/1750-1172-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banka S, Blom HJ, Walter J, et al. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am J Hum Genet. 2011:216–25. doi: 10.1016/j.ajhg.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He M, Kratz LE, Michel JJ, et al. Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. J Clin Invest. 2011:976–84. doi: 10.1172/JCI42650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kállay Z K, Liptai G, Benyó C, et al. Successful unrelated umbilical cord blood transplantation in Lesch-Nyhan syndrome. Metab Brain Dis. 2012:193–6. doi: 10.1007/s11011-012-9279-9. [DOI] [PubMed] [Google Scholar]
- 19.van Karnebeek CDM, Hartmann H, Jaggumantri S, et al. Lysine restricted diet for pyridoxine-dependent epilepsy: First evidence and future trials. Mol Genet Metab. 2012:335–44. doi: 10.1016/j.ymgme.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 20.van Karnebeek CDM, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet. 2014;94:453–61. doi: 10.1016/j.ajhg.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Karnebeek CD. Diagnosis and discovery of treatable inborn errors of metabolism causing intellectual disability. Mol Genet Metab. 2014:111–227. doi: 10.1016/j.ymgme.2011.11.191. [DOI] [PubMed] [Google Scholar]