

Abstract
This article provides practical guidance on the use of passive sampling methods (PSMs) that target the freely dissolved concentration (Cfree) for improved exposure assessment of hydrophobic organic chemicals in sediments. Primary considerations for selecting a PSM for a specific application include clear delineation of measurement goals for Cfree, whether laboratory-based “ex situ” and/or field-based “in situ” application is desired, and ultimately which PSM is best-suited to fulfill the measurement objectives. Guidelines for proper calibration and validation of PSMs, including use of provisional values for polymer–water partition coefficients, determination of equilibrium status, and confirmation of nondepletive measurement conditions are defined. A hypothetical example is described to illustrate how the measurement of Cfree afforded by PSMs reduces uncertainty in assessing narcotic toxicity for sediments contaminated with polycyclic aromatic hydrocarbons. The article concludes with a discussion of future research that will improve the quality and robustness of Cfree measurements using PSMs, providing a sound scientific basis to support risk assessment and contaminated sediment management decisions. Integr Environ Assess Manag 2014;10:210–223. © 2014 The Authors. Integrated Environmental Assessment and Management published by Wiley Periodicals, Inc. on behalf of SETAC.
Keywords: Bioavailability, Contaminated sediment, Equilibrium partitioning, Passive sampling methods, Porewater
Editor's note
This paper represents 1 of 6 papers in the special series “Passive Sampling Methods for Contaminated Sediments,” which was generated from the SETAC Technical Workshop “Guidance on Passive Sampling Methods to Improve Management of Contaminated Sediments,” held November 2012 in Costa Mesa, California, USA. Recent advances in passive sampling methods (PSMs) offer an improvement in risk based decision making, since bioavailability of sediment contaminants can be directly quantified. Forty four experts, representing PSM developers, users, and decision makers from academia, government, and industry, convened to review the state of science to gain consensus on PSM applications in assessing and supporting management actions on contaminated sediments.
Introduction
Sediments are repositories of past and ongoing discharges of organic and metal contaminants that are potentially available to the aquatic food chain. Assessments of pollutant fate, transport, bioaccumulation, and toxicity of impacted sediments have historically been based on total pollutant concentrations and geochemical properties (e.g., total organic carbon) of bulk sediment (USEPA 2012a). Such assessments have been challenged by a gradually evolving understanding of the complexity of chemical sequestration in sediments and the involvement of various geochemical phases such as black carbons that are often difficult to quantify and accurately characterize (NRC 2003; Ghosh and Hawthorne 2010). Researchers have, therefore, focused on developing measures that account for bioavailability and better reflect the potential of chemicals to cause impact, be mobilized from, or be degraded in a given sediment. These measures, as illustrated in Figure 1 for hydrophobic organic chemicals (HOCs), typically fall into 2 broad categories: 1) chemical activity-based bioavailability methods focused on freely dissolved concentrations in porewater (Cfree), and 2) desorption-based methods aimed at determining the bioaccessible fraction of HOCs (Reichenberg and Mayer 2006). Although desorption based methods can also be used for assessing potential for bioaccumulation and biodegradation, the focus of this article is on chemical activity-based passive sampling methods (PSMs) that target Cfree of HOCs in sediment.
Figure 1.
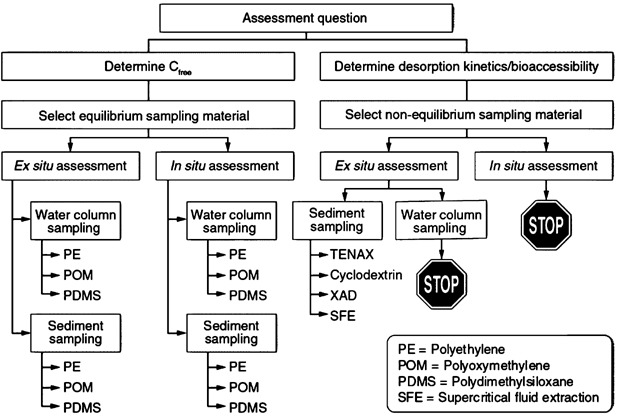
General flow chart for selecting passive sampling devices for applications involving organic contaminants present in sediments and the overlying water column.
In the last few decades, a wide range of methods has been used to measure or estimate Cfree in sediment porewater, and these measures have been reported to correlate well with contaminant uptake in organisms and toxicity (Lydy et al. this issue). Direct measurement methods for porewater have focused on the challenge of removing interference from particulates, especially colloids, and losses to glassware. Methods using centrifugation and alum flocculation have shown some success in directly measuring freely dissolved concentrations in porewater (Ghosh et al. 2000; Hong et al. 2003). Even with these separation techniques, however, accurate measurement of low concentrations of organic chemicals with high octanol–water partition coefficients (Kows) remains challenging. Recent work, therefore, has focused on the development of passive sampling with organic polymers such as polyethylene (PE), polyoxymethylene (POM), or polydimethylsiloxane (PDMS) to accurately measure freely dissolved concentrations of HOCs in porewater or surface water (Figure 1). For porewaters with low interference from dissolved and/or colloidal organic matter, and especially for less hydrophobic organic chemicals (log Kow < 4), direct measurement using liquid–liquid solvent extraction may also be feasible.
A method that uses commercially available PDMS-coated solid phase micro extraction (SPME) fibers to extract porewater and estimate Cfree (ASTM 2007) has been shown to be a good predictor of toxicity (and lack of toxicity) of polycyclic aromatic hydrocarbons (PAHs) to benthic infauna (Kreitinger et al. 2007; Kane Driscoll et al. 2009). In this method, sediment is centrifuged to obtain porewater, which is flocculated with alum to remove colloids, resulting in porewater with PAHs that are partitioned between the water (Cfree) and dissolved organic carbon. Depending on the use of internal standards, the data can be reported for total dissolved concentrations or Cfree. Although this method has been used in site assessments to eliminate PAHs as contaminants of concern and to develop clean-up strategies (McArdle et al. 2010), a key barrier to wider implementation of this method is that only a few commercial laboratories can perform the measurement currently.
The reader is referred to Lydy et al. (this issue) for a detailed literature review of passive sampler use in sediment assessments and Mayer et al. (this issue) for a theoretical description of the rationale behind passive sampling. In the present article, we provide guidance on the use of PSMs including both laboratory-based ex situ assessments and field-based in situ assessments, along with key research questions and other considerations that influence the selection of particular PSMs. This article presents guidance on methods for porewater measurement with PSMs, calibration approaches, selection of polymer partitioning parameters, and quality assurance measures. The article also describes how Cfree data can be used in site decisions and concludes with an illustrative hypothetical example.
Considerations for using PSMs to measure Cfree
The primary research and/or investigative question for a site drives many of the choices to be made in the selection of a PSM (and other measures), as indicated in Figure 1. Once established, the 2 key considerations for selecting a PSM are the choice of polymer type and/or configuration and whether the technology will be applied ex situ or in situ. These considerations are discussed below.
Polymer type
For HOCs, the polymers most common used to determine Cfree are PE, POM, and PDMS. Both PE and POM are most often deployed as thin sheets with large surface areas. In contrast, PDMS has been most often deployed as a coating on a thin glass fiber (i.e., SPME) or as coatings inside vials. However, other types of silicone polymers or rubbers are available for passive sampling in fiber and thin sheet or film configurations (see Lydy et al. [this issue] for a review). Chemical structural differences between PDMS and these other silicones have to do primarily with the dimethyl group being replaced by another hydrocarbon group (e.g., ethyl, phenyl). Furthermore, SPME fibers can be coated with polymers other than PDMS (e.g., polyacrylate). All of these polymers are moderately sorbing organic phases that exhibit similar sorption capacities and have demonstrated the ability to reliably determine Cfree for a wide variety of HOCs (e.g., polychlorinated biphenyls [PCBs], PAHs, and chlorinated pesticides) in both laboratory and field applications (Lydy et al. this issue). Although these commonly used polymers are commercially available, certain polymer configurations, such as custom thicknesses or vial coatings, may not be commercially available and will need to be synthesized in the laboratory or specially ordered.
Ex situ and in situ deployments
Passive sampling methods can be deployed in the field (in situ) or used in the laboratory (ex situ) for the assessment of Cfree. Table1 lists factors to consider when selecting between the ex situ and in situ approaches. The ex situ approach will often be simpler to perform and more acceptable for many sediment research and management applications. The objective of the ex situ approach is to determine Cfree that is representative of equilibrium conditions for the sediment sample under consideration, while maintaining the practicality, relevance, control, and interpretation possibilities (e.g., hypothesis testing) that are afforded by laboratory experiments. In the ex situ PSM approach, sediment samples collected from the field site are transported to the laboratory, where the sampler is added to the sediment and mixed well for a duration typically sufficient for the contaminants to achieve equilibrium between porewater, environmental phases (e.g., colloids), and the polymer. Laboratory-spiked sediments can also be analyzed in this manner. Common ex situ applications include partitioning investigations, sediment toxicity testing, and bioaccumulation assessments (Vinturella et al. 2004; Friedman et al. 2009; Fagervold et al. 2010; Gschwend et al. 2011).
Table 1.
Factors to consider when selecting between ex situ or in situ application of PSMs
| Approach |
||
|---|---|---|
| Factor | Ex situ | In situ |
| Ability to estimate equilibrium Cfree | Laboratory conditions can be controlled to better attain equilibrium. | Uncertainty can occur; need to use PRCs, multiple polymer thicknesses, or time series sampling to confirm equilibrium. Time series interpretation can be impacted by temporal changes in the field. |
| Comparison to independent confirmatory methods (e.g., air bridge) can be applied. | ||
| Spatial scale (e.g., to differentiate between biologically active zones and underlying sediments or contaminant migration through a cap) | Sediments are frequently composited and/or homogenized to avoid concentration variability caused by vertical and horizontal spatial heterogeneity. | Fine-scale spatial (vertical and horizontal) patchiness in concentrations can be measured (e.g., identify gradients). |
| Coring followed by passive sampling in intact cores can maintain spatial characteristics if not influenced dramatically by site dynamics. | Best approach to capture field conditions. | |
| Contaminant depletion | Mixing (e.g., tumbling of sample) during equilibration period is used to limit localized depletion. | Contaminant depletion may occur in the zone around samplers; use of multiple polymer thicknesses or time series analysis may be used to evaluate depletion. |
| Statistical design | Multiple treatments and replication are possible; hypothesis testing can be performed. | Multiple treatments, replication, and hypothesis testing are possible, but logistically challenging and expensive. |
| Ease of experimentation | Experiments are simpler to perform under laboratory conditions. | Expense, achieving experimental and statistical design goals, safety concerns, weather, adverse site conditions, and vandalism. |
| Ability to capture field conditions (e.g., currents, tidal cycles, groundwater intrusion, sediment-water column fluxes, bioturbation, temperature and salinity change) | Laboratory conditions are frequently standardized, but can be altered to attempt to replicate some field conditions. | Best approach for capturing field conditions. |
PRCs = performance reference compounds; PSMs = passive sampling methods.
In contrast, the in situ approach is used when it is critical to capture conditions in the field. In this approach, the polymer is inserted directly into sediments or suspended in the water column above the sediment in the field and left in place for sufficient duration to allow the derivation of Cfree (Fernandez et al. 2009a, 2009b, 2012; Oen et al. 2011; Beckingham and Ghosh 2013; Lampert et al. 2013). However, the ability to attain equilibrium and demonstrate that equilibrium has been achieved is often more difficult for the in situ approach as compared to the ex situ approach (see Table1). In situ approaches may be preferable where it is important to understand groundwater intrusion, currents, bioturbation, depth-varying contaminant porewater concentration profiles, and sediment–water column gradients and fluxes. In general, these conditions are difficult to recreate or model in the laboratory. Groundwater movement can transport contaminants from deeper contaminated sediments to surface sediments (Gidley et al. 2012). Uncontaminated groundwater intrusion through sediment and tidal pumping in coastal regions can reduce porewater concentrations of HOCs (McCoy and Corbett 2009), although the degree of dilution will depend on the time scales of the fresh groundwater movement and kinetics of the release of contaminants from the sediment phase. If the groundwater flow is very slow, the impact on porewater concentrations may not be appreciable.
Calibration of passive sampling materials
Mathematical basis for passive sampler calibration
Calibration of passive samplers involves deriving the concentrations of target analytes in the medium of interest (i.e., sediment porewater) from their concentrations in the passive sampling material. For substances that reversibly sorb to the passive sampling polymer, the mass transfer into or out of the polymer is governed by diffusion within the polymer matrix, diffusion through the external aqueous boundary layer, and desorption and diffusion of contaminant from the sediment solids. For simplicity, this exchange is usually described by a first order kinetic model as described in Lydy et al. (2014). This assumes that the passive sampling application does not produce a significant reduction of the analyte concentration in the medium sampled (i.e., depleted) and that Cfree is constant over the sampling time.
Figure 2 illustrates data for PCB 153 (hexachlorobiphenyl) fractional uptake in 77 μm POM sheet (Hawthorne et al. 2009) from a sediment slurry and a first order model fit to the kinetics data. During the initial period of deployment, the analyte concentration increases in a linear fashion such that Cfree can be determined from the slope of the linear increase of the analyte concentration in the passive sampler (CP) over time. For first order uptake, a near-linear response between CP and t is maintained during the initial time period that is less than approximately 0.5/ke representing 40% uptake of the analyte's equilibrium concentration, where ke is the analyte's exchange rate constant between the sampled media and the sampler. The initial linear uptake period is followed by a period of time where the analyte concentration increases in a nonlinear fashion with time. Finally, the concentration in the passive sampling polymer reaches a steady state (i.e., dCP/dt = 0) and the analyte concentration in the sampler is constant over time. If the analyte does not degrade in the sampling polymer or in the sampled medium, this situation represents a thermodynamic equilibrium (i.e., an equilibrium controlled by the relative affinities of the analyte for the sampling material and the sampled media). This chemical equilibrium is represented by the polymer–water partition coefficient (Kpw). The time required to reach equilibrium is controlled by ke. The time to reach 95% of the analyte's equilibrium concentration is approximately 3/ke.
Figure 2.
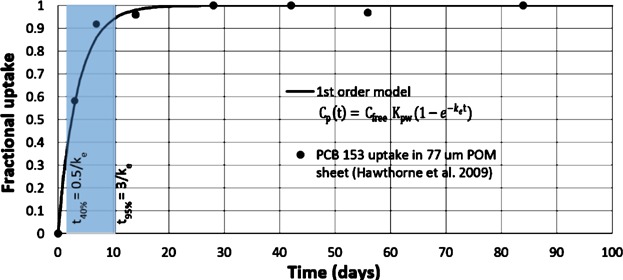
Time course of fractional uptake of PCB 153 (hexachlorobiphenyl) in a 77 μm POM sheet (Hawthorne et al., 2009) from a sediment slurry. A first order model fit illustrates the conditions required for equilibrium sampling (i.e., t ≥ 3/ke) and for linear non-equilibrium sampling (i.e., t ≤ 0.5/ke). The intermediate, shaded region illustrates the period when uptake into the polymer is nonlinear.
Three basic passive sampler calibration strategies
Based on the above-mentioned 3 time phases, 3 possible passive sampling strategies are possible.
Equilibrium sampling
The first and most commonly used strategy is suitable for substances that can reach equilibrium between the polymer and the sampled media within a realistically short exposure time. For these substances, the deployment time, td, should be equal to or greater than t95 or 3/ke. The relationship between Cp and Cfree is then given by Kpw, and Cfree can be found from Cp as Cp/Kpw. Equilibrium samplers require relatively fast exchange kinetics and/or sufficient equilibration time. Fast exchange rates can be achieved by keeping the polymer thickness small, thus reducing diffusion distances by mixing the sampler during equilibration. The equilibration times are related to the magnitude of Kpw, with larger Kpw producing longer equilibration times. Equilibrium conditions can be readily, and in most cases, more rapidly attained in well-mixed laboratory systems (i.e., ex situ) than under primarily static field conditions (i.e., in situ). The equilibrium uptake approach is the most convenient approach for determining Cfree and has been used with PE, POM, and PDMS (Lydy et al. this issue; Mayer et al. this issue).
Nonlinear uptake rate
A second sampling strategy is based on the nonlinear relationship between Cp and t, and may be appropriate if deployment times are between 0.5/ke and 3/ke. This sampling strategy is used less frequently than the equilibrium approach as it requires more data to relate Cp to Cfree and such data are not always available. Also, more sophisticated data analysis tools such as nonlinear regression, curve fitting, and diffusion modeling tools are required to establish the relationship between Cp and Cfree. The use of performance reference compounds (PRCs) to characterize nonlinear uptake falls under this category of sampling strategy and is described in “Calibration of polymer exchange kinetics (Ke)” (see below).
Linear uptake rate
A third strategy (that is rarely implemented for Cfree assessments in sediment) can be used for organic contaminants for which the passive sampler deployment time is much shorter than 0.5/ke, representing sampler analyte concentrations that are small compared to their equilibrium values (Huckins et al. 1999). For these substances, the concentration in the sampled medium is determined from the measured uptake rate divided by the analyte's uptake rate constant ke. This sampling and calibration strategy is therefore a function of td and the ke of the analyte in the passive sampling device.
Calibration of the polymer–water partition coefficient
The most important and commonly used parameter necessary for calibration of PSMs is Kpw. Given the challenges involved in accurately measuring Kpw values, there is practical value in using reliably determined published partition coefficients for commonly used polymers (i.e., PE, POM, PDMS). However, it is important to use the same source of polymer, or to verify that Kpw values for target HOCs are sufficiently close to those described below if using materials from different batches and/or suppliers. A list of provisional Kpw values that are judged to be reliable for the 3 most commonly used polymers are included in the Supplemental Data for PAHs and PCBs (Tables S1 and S2, respectively) as discussed below.
KPDMS-w
Values for PDMS-coated glass fibers are based on Smedes et al. (2009) for SR-TF (J-Flex Industrial Rubber Produces) and are consistent with a variety of other published and unpublished measurements using Supelco, Polymicro (Difilippo and Eganhouse 2010), and Prime Optical Fibers (Taiwan) (Hsieh et al. 2011), as well as PDMS sheets from Dow Corning (US) and Vizo (Zeewolde, the Netherlands) (Smedes et al. 2009). The KPDMS variation among these sources is approximately 0.2 log units with up to 0.4 log units for specific compounds listed.
For PDMS sheets (AlteSil™) manufactured by ALtecWeb (UK), the values are based on Smedes et al. (2009). The expected variation in Kpw for a given chemical for AlteSil PDMS sheets is ± 0.2 log units for PAHs and low molecular weight PCBs (Smedes et al. 2009). Based on other literature sources, the expected variation for high molecular weight (HMW) PCBs is ± 0.4 log units.
KPE-w
Values for PE sheets (obtained from Brentwood Plastics) presented are also from Smedes et al. (2009). Variation of ± 0.2 log units is expected for PCBs and 2–3 ring PAHs (Booij et al. 2003; Adams et al. 2007; Cornelissen et al. 2008; Fernandez et al. 2009a; Perron et al. 2009; Choi et al. 2013). The few reported KPE-w measurements for >4 ring PAHs show variability up to ± 1 log unit.
KPOM-w
Values for commercially available POM sheets (77 μm; from CS Hyde Company) are presented based on Hawthorne et al. (2009, 2011). Variation of ± 0.1 (with a maximum of 0.3) log units were observed between the 2 laboratories participating in a comparison (Gschwend et al. 2011). Independent measurements presented in Hale et al. (2010) for the same POM agree mostly within a factor of 3 or ± 0.5 log units to those determined by Hawthorne et al. (2009).
Correlation of Kpw with Kow
Correlations with Kow can be used to extrapolate predicted Kpw values for contaminants in the same chemical class (e.g., specific PCB congeners), but with the caveat that Kow values should be from a reliable and consistent source as described here. The following correlations relate log Kpw for PDMS (Smedes et al. 2009), log Kow values for PAHs (SPARC estimates) (Hilal et al. 2004), and log Kow values for PCBs (Hawker and Connell 1988):
The following correlations relate log Kpw for PE (log KPE-w) (Smedes et al. 2009), log Kow values for PAHs (Hilal et al. 2004), and log Kow values for PCBs (Hawker and Connell 1988):
The following correlation relates the log Kpw (Hawthorne et al. 2009, 2011) for POM, the log Kow for PAHs (Hilal et al. 2004), and the log Kow for PCBs (Hawker and Connell 1988):
Polymers from different suppliers may differ in properties such as the degree of cross-linking, which can impact uptake kinetics and Kpw. Thus, until the selection of polymers is universally standardized, it is advisable for laboratories performing PSM studies to obtain a large quantity (i.e., a multiple-year supply) of 1 type of polymer, for which partitioning characteristics are available or will be determined. When a new batch is purchased or a new supplier is used, partitioning characteristics should be reconfirmed. This approach is suggested because at this time little information exists on differences in the same polymer obtained from different sources. However, as described previously in “Calibration of the polymer–water partition coefficient,” the differences in Kpw between different sources of PDMS and POM are small, and this may not pose a significant source of error in assessments when compared to other assessments of bioavailability (i.e., use of default equilibrium partitioning assumptions).
Calibration of polymer exchange kinetics (ke)
Calibration that considers the kinetics of deployment is often challenging but is needed when equilibrium sampling is not practical. For HMW HOCs, it may take months or more for polymers to reach equilibrium with the sediment porewater when deployed in the field (Tomaszewski and Luthy 2008). Therefore, it is often more practical to employ nonequilibrium sampling techniques in the field rather than wait until equilibrium is established. In this situation, calibration of the passive sampler for mass transfer kinetics is required. Generally, the uptake of HOCs into passive samplers has been shown to be dependent not only on the mass transfer kinetics between the passive sampler and the sediment porewater but also on the compound release rate from the sediment (Fernandez et al. 2009a). In addition, kinetics are temperature-dependent and may be affected by biofouling of the membrane surface (Huckins et al. 1999). This suggests that the linear uptake assumption may not be sufficiently accurate in some cases and a new calibration study is required whenever the exposure conditions for passive sampling are changed.
To overcome these challenges, Huckins et al. (1993, 2002) suggested the use of PRCs to calculate Cfree from nonequilibrium PSM measurements (CP). PRCs are analytically noninterfering chemicals that are embedded in the passive sampler before environmental exposure (Huckins et al. 2002). Examples of surrogate chemicals are stable isotope-labeled or deuterated forms of the analytes of interest, substances with a log Kow that is similar to that of the target analytes (Huckins et al. 2002; Fernandez et al. 2009a), or rare PCB congeners (Tomaszewski and Luthy 2008). A good PRC should 1) allow precise measurement of its loss, 2) follow the same kinetics as the target analyte, and 3) not exist in the target environment (Huckins et al. 2002; Fernandez et al. 2009a). The depletion rate of a PRC during sampler deployment reflects the uptake rates of a target analyte, assuming isotropic exchange kinetics occur (Figure 3). Because of the differences in the compound properties for the PRC and the target analyte, correction is needed to calculate the fractional approach to equilibrium for the target analyte (C(t)/C(ss)) from the fractional PRC dissipation (1–CPRC(t)/CPRC0) at time t. In addition, PRC correction becomes difficult if sorption in the surrounding media is concentration dependent. Several approaches for the calibration using PRC data have been suggested (Huckins et al. 2006; Tomaszewski and Luthy 2008; Fernandez et al. 2009b; Reible and Lotufo 2012).
Figure 3.
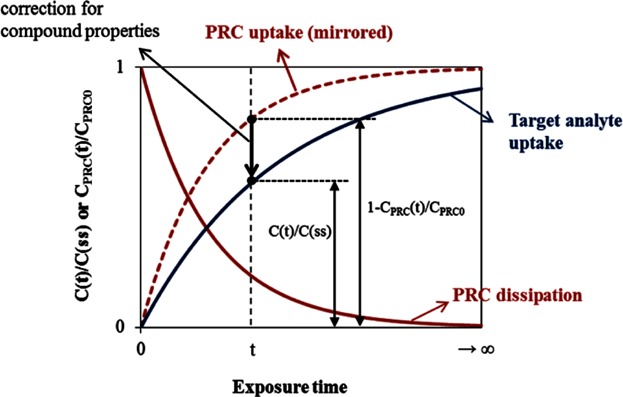
PRC dissipation and compound uptake kinetics generally assumed for the performance reference compound (PRC) approach. C(t) and C(ss) refer to target analyte concentrations in the passive sampler at time t and steady state, respectively; CPRC(t) and CPRC(0) refer to PRC concentrations in the passive sampler at time t and 0, respectively.
Use of multiple polymer thicknesses to assess equilibrium
An alternative method for confirming equilibrium involves the application of multiple polymer thicknesses of the sampling material (Mäenpää et al. 2011). In this approach, attainment of the same Cp for different thicknesses confirms that equilibrium has been obtained. This approach is particularly suitable if reaching equilibrium within the deployment time is achievable.
Temperature and salinity corrections
Both temperature and salinity can influence Kpw and mathematical approaches for corrections are available (Schwarzenbach et al. 2003) and described in more detail in the Supplemental Data. Adjustments for temperature and salinity can be performed when it is necessary and viable to validate the data and assess the accuracy of the adjustments. To ensure transparency whenever Cfree results are presented, it should be clearly stated if and how corrections for temperature and salinity have been performed. As long as extreme conditions are not expected at the field site, the error introduced by performing ex situ exposures at room temperature is expected to be relatively small compared to other causes of uncertainty and may be within limits acceptable for regulatory purposes (e.g., a factor of 2).
Application of PSMs in laboratory and field situations
Pre-exposure considerations and preparations
For assessments performed ex situ, preparations include cleaning and/or pre-extracting the passive sampler and glassware, preparing aqueous medium containing at least a biocide, and homogenization of the sediment sample. For in situ exposures, preparations include precleaning the samplers and their carrier devices, loading the samplers with PRCs (if required and possible), and the production of procedural field blanks (i.e., samplers taken into and from the field without exposing them). The different preparation steps are discussed in detail below.
Selection of sampling materials
Figure 4 illustrates various considerations related to the selection of a specific passive sampler for a given application. Considerations include, but are not restricted to:
Required detection limits. PE and POM sheets generally have lower detection limits than PDMS-coated SPME fibers due to their larger absorptive capacities. Although direct injection of an SPME fiber and thermal desorption can reduce detection limits by transferring all sorbed analytes into the analytical instrument, direct injection without analyte cleanup may be impacted by high background noise that increases detection limits. Thus, a relatively large polymer absorptive mass is generally preferable when low detection limits are desired. The mass of polymer needed depends on the detection limit of the chosen analytical method (e.g., regular GC-ECD or GC-MS vs HR-GC/HR-MS), anticipated porewater concentrations (more polymer needed for low concentrations), and Kpw. A quick calculation method for the required polymer mass is provided in the Supplemental Data.
Equilibration kinetics. Thinner and less sorbing polymers have faster kinetics and PDMS-coated SPME fibers are expected to equilibrate faster than PE and POM sheets, making them preferable for static in situ measurements. PDMS-coated jars with thin layers of polymer have been shown to equilibrate in 14 days (Jahnke et al. 2012).
Sampler fouling. It has been suggested that POM is less prone to fouling by black C particles and nonaqueous-phase liquids (NAPLs) than SPME and PE (Jonker and Koelmans 2001; Van der Heijden and Jonker 2009). Housings constructed of biocidal metals (e.g., Cu) have been designed to provide structural rigidity and reduce the potential for accumulation of biofilms on the surfaces of deployed polymers (Maruya et al. 2009). However, there is a lack of clear understanding on polymer susceptibility to fouling and additional studies are needed to address this issue. Furthermore, the use of PRCs may aid in addressing potential artifacts of fouling.
Figure 4.

Selection considerations for passive sampling devices.
Assuming fouling and background contamination do not confound measurements, Cfree values determined with any of the polymers described should be comparable if equilibrium conditions are attained and appropriate Kpw and sediment–polymer ratios are used.
The use of commercially available polymers is advantageous because they are expected to be more uniform and homogeneous and are also available to the general public, which increases the possibilities for standardization. For example, SPME fibers with various thicknesses of PDMS (e.g., 7, 30, and 100 μm) are commercially available, can be cut to desired lengths, and have been calibrated and used in both laboratory and field exposures for several classes of HOCs (Maruya et al. 2009).
Pre-extraction of sampling materials
Before exposure, sampling polymers need to be extracted. This pre-extraction and cleaning process ensures that background levels of target contaminants and interfering compounds in the sampling material are minimized. Because of the high absorptive capacity of the materials, they can accumulate ambient chemicals from the atmosphere during transport and storage such that background contamination is introduced easily. Additionally, interfering contaminants might be introduced during the polymer production process. For instance, oligomers are present in many polymers and can interfere with the accurate quantification of target contaminants during the analysis of the final extracts after exposure. Pre-extraction should be performed using appropriate solvents, the choice of which depends on the sampling material and the target compounds. Generally, polar solvents are applied, either alone or in combination with a nonpolar solvent. The selection of a solvent depends on the resistance of the polymer to the solvent, the polarity of the polymer, and the ability of the solvent to remove oligomers and other background (target) contaminants. For instance, methanol is well-suited for pre-extracting POM, but methanol extraction is insufficient for removing oligomers from PDMS sheets. The pre-extraction processes thus differ too much to generalize among passive sampling devices. Several different approaches have been used, some of which have been listed in Table2. The pre-extraction solvent, as well as the extraction duration, should be selected carefully after consulting the literature or experts.
Table 2.
Extraction solvents and times commonly applied for pre-extracting passive sampling polymers
| Polymer | Target compounds | Pre-extraction solvent | Extraction time (h) | Reference |
|---|---|---|---|---|
| POM | PAHs (HPLC, GC-MS), PCBs (GC-ECD, GC-MS) | Hexane, methanol, acetonitrile Hexane, methanol | 2 2 | Jonker and Koelmans (2001); Hawthorne et al. (2009, 2011) |
| Oil (GC-FID) | Hexane/acetone | 6 | Muijs and Jonker (2011, 2012) | |
| PE | PCBs, PAHs, DDTs, PBDEs, triclosan | Dichloromethane, hexane, acetone | 24 | Fernandez et al. (2009a, 2009b, 2012); Perron et al. (2009, 2013a, 2013b) |
| PDMS-SPME | PAHs (HPLC) | Methanol, water, acetonitrile | 3 | Muijs and Jonker (2009, 2012) |
| Oil (GC-FID) | Heptane | 3 | Muijs and Jonker (2011, 2012) | |
| PAHs, PCBs, other semivolatiles | Thermal desorption | 0.5 | ASTM (2007); Reible and Lotufo (2012) | |
| Silicone rubber | PAHs, PCBs | Ethyl acetate | 100 | Smedes et al. (2009) |
PDMS-SPME = polydimethylsiloxane-solid phase microextraction; PE = polyethylene; POM = polyoxymethylene.
Ex situ (“laboratory”) exposures
Exposure medium
Although wet field sediment with high water content can often be used directly for passive sampling, in many cases, additional clean water may need to be added to allow slurry formation and good mixing. For most HOCs (log KOW >3), the fraction transferred to the additional water is very small compared to the fraction sorbed to sediments. Adding a biocide is particularly necessary when targeting the Cfree of contaminants that are biodegradable (e.g., PAHs). Commonly used biocides include sodium azide or mercuric chloride, applied at concentrations of 25 to 200 mg/L (Van der Heijden and Jonker 2009; Fagervold et al. 2010). Furthermore, when adding aqueous medium to freshwater sediments, a salt-like calcium chloride (e.g., 0.01 M) is often added to maintain a natural ionic strength (Van der Heijden and Jonker 2009). In estuarine and marine studies, the exposure system should be designed to mimic natural salinities using natural or artificial seawater.
Sediment homogenization
The sediment sample under investigation is usually either mechanically or manually homogenized before the introduction of the sampler to reduce data variability. In addition, samples may be sieved to remove coarse particles that might potentially damage the sampler (SPME fibers are more fragile than other materials). This should be limited to coarse sieving (e.g., 500 μm) for removal of nonsorbing constituents like stones, because potentially any manipulation may cause changes in the sediment composition, leading to a matrix that does not fully reflect the in situ conditions to which the ultimate risk assessment should apply. To ensure homogeneity, it is also recommended that sufficiently large subsamples of sediment material be used in each exposure system, as this may help to limit variation caused by small-scale heterogeneity.
Negligible depletion
Accurate measurement of existing porewater Cfree requires the use of a sampler volume to matrix ratio that ensures negligible depletion of the matrix or porewater concentration (described as <1% depletion) when equilibrium is reached. For hydrophobic chemicals, the introduction of a passive sampler will inevitably start depleting the porewater, but desorption from the sediment will replenish the aqueous pool. If the mass that must be transferred to replenish the pool is too large, the standard exposure time may be insufficient to reach new equilibrium conditions or chemical transport may take place from domains where the chemicals are bound more strongly. This may result in a measurement performed under conditions that do not reflect the actual in situ conditions. To avoid depletive extractions, the sediment organic C-to-sampler ratio should be sufficiently large, as these are the 2 primary absorptive pools that compete for sorption of the hydrophobic contaminants. As a general rule (assuming sediment organic carbon and polymer matrices have similar partitioning characteristics), a ratio of 1:100 polymer mass to sediment organic carbon mass should reduce any depletion to an acceptable value of <1%. As described below, this ratio can be refined for sediment- and polymer-specific conditions if more accurate estimate of chemical-specific Koc and Kpw values are available:
where Mp is the mass of polymer, and Moc is the mass of sediment organic carbon.
If detection limit issues and other logistical considerations, such as lack of prior accurate estimates of Koc or Moc, do not allow maintenance of depletion at <1%, it may be possible to correct for the potential depletion as described in Fagervold et al. (2010). Such corrections are feasible when the depletion is still small (<10%) and within the range for which a linear relationship for partitioning characteristics of the sediment organic matter can be assumed. Also, when the goal of the Cfree measurements is to assess site-specific native partition constants (e.g., Koc), the decreased matrix concentrations can be measured and accounted for in the partitioning calculation.
Equilibration conditions
During equilibration in ex situ sampling, mixing is required to enhance exchange of chemicals among the sediment, water, and sampler phases. Equilibration in static systems is slow, especially for hydrophobic chemicals, and thorough mixing using a shaker, orbital mixing table, or other device is recommended. For PE and POM, shaking regimes of 100 to 150 rpm have commonly been applied, whereas for SPME a more gentle agitation (e.g., on a rock and roller apparatus) is needed to not damage the fragile fibers. In well-mixed systems, the thicknesses of aqueous boundary layers surrounding the sampler and the sediment particles are reduced, which enhances the equilibration kinetics.
As the methods discussed here are targeting the equilibrium Cfree of chemicals, it follows that care should be taken to measure the concentrations at (or near) thermodynamic equilibrium. As a general rule, for dynamic ex situ exposures, 4-week equilibrations are generally applied when assessing Cfree with SPME, PE, or POM (Jonker and Koelmans 2001; Gschwend et al. 2011; Hawthorne et al. 2011). Even for the most hydrophobic contaminants such as PCBs and HMW PAHs, this exposure time yields (near) equilibrium concentrations (Maruya et al. 2009; Hawthorne et al. 2011). Using vials and SPME fibers containing a thin PDMS coating (e.g., 10 μm PDMS), full equilibration can be achieved within a shorter period of time (e.g., 2 weeks) (Jahnke et al. 2012). In case of uncertainty about the equilibration status or if shorter exposure times are warranted, equilibrium conditions should be demonstrated by including a time series of exposures (i.e., exposures of various lengths of time) or by using samplers of different thicknesses. At equilibrium, samplers of different thicknesses should yield the same Cfree values. Alternatively, PRCs can be used to demonstrate equilibrium conditions (discussed below). Although this approach has been applied in situ, ex situ applications are still rare (Gschwend et al. 2011; Oen et al. 2011).
In situ (“Field”) deployment
Spatial coverage to address the extent and heterogeneity of contamination
Because of the relatively small size of passive samplers, and because they sample microscale environments (at most several mm surrounding the static sampler), samplers deployed in situ (e.g., SPME fibers) can be prone to very small-scale heterogeneity although the porewater can be an integrative medium in a dynamic sediment environment. As with bulk solid sampling, compositing can more accurately represent average values within a zone. If small-scale heterogeneity is suspected or known to be large, sampling designs may need to include spatial pooling of samplers or results, to obtain a spatially representative measurement. Conversely, measuring the spatial heterogeneity of Cfrees on a very small scale may be of interest, depending on the question being asked (e.g., high resolution of the depth distribution of contaminant, addressed in the next section).
Characterizing depth profiles
Passive samplers can be deployed in situ to characterize the depth profiles of contaminants in porewater (Fernandez et al. 2009b; Beckingham and Ghosh 2013; Lampert et al. 2013). Although sediment core profiles can also be studied in the laboratory, laboratory evaluation may not adequately characterize site-specific processes such as the effect of groundwater upwelling or hyporheic exchange. However, adequate interpretation and understanding of depth profile data may require additional extensive measurements of contaminant concentrations and total organic C and black C profiles with depth. If correctly measured and interpreted, depth profiles of Cfree can provide useful information on chemical gradients and potential directions of the diffusive fluxes of contaminants (Mayer et al. this issue). Several recent articles have reported in situ depth profiles using POM, SPME, and PE (Fernandez et al. 2009b; Oen et al. 2011; Beckingham and Ghosh 2013; Lampert et al. 2013). In these studies, passive sampler strips (POM or PE) or SPME fibers were either exposed directly, encased in a mesh, supported in Al frames, or placed in hollow perforated Cu tubes. Copper tubes or mesh provide the additional benefit of reducing biological growth on the sampler. Under these static conditions, kinetics are expected to be retarded, and care must be taken to ensure the equilibrium status of the samplers is understood (e.g., by adding PRCs).
Temporal variability
Short-term temporal variations cannot be determined effectively using porewater passive samplers, due to the requirement of a relatively long period of exposure time to reach equilibrium conditions. As discussed above, this is typically in the range of a few weeks (or months in the field). Passive samplers are more effective at assessing time-averaged conditions in the field over the period of deployment. However, annual changes, seasonal changes, or changes brought about due to episodic events can be measured. To ensure proper interpretation of results, it is critical to consider additional parameters, such as the net sediment deposition rate, loss of sediment from the area, episodic disturbances (e.g., resuspension events), and changes in bulk sediment concentrations over time. Also, it is important to be able to distinguish sampling errors and site heterogeneity from temporal effects. In such cases, the use of temperature-specific Kpw values may be important when assessing and comparing Cfree measured during different seasons (i.e., at different temperatures).
Validation of equilibrium conditions
In situ exposures of passive samplers in field sediments generally should be considered static or semistatic (due to some tidal-, current-, or wind-induced porewater and sediment movement). In particular for such exposures, it may take relatively long periods of time to reach full equilibrium conditions. To enhance the kinetics, the use of thin polymers or polymer coatings (on SPME) is recommended (see above) although the latter may result in reduced detection limits. Still, it is important to demonstrate the existence of equilibrium conditions. Investigation of the equilibrium status can be performed as described above for ex situ exposures, although including a time series of exposures may be laborious and costly under field conditions. Although PRCs have proven successful in water-only exposures (Huckins et al. 2002, 2006; Fernandez et al. 2012; Perron et al. 2013a, 2013b), this approach in static sediment-water systems is still being developed (Tomaszewski and Luthy 2008; Fernandez et al. 2009a, 2009b, 2012; Gschwend et al. 2011; USEPA 2012a, 2012b). One study with POM was recently performed successfully by Oen et al. (2011), but studies on the utility of PRCs for use with PDMS and POM have been limited so far (Reible and Lotufo 2012). Alternatively, equilibrium in the field can be established by performing 2 different thickness samplers (Reible and Lotufo 2012) or by time series deployment (Tomaszewski and Luthy 2008), staggering the removal schedule of individual samplers over a period within which the establishment of equilibrium can be expected. Assessment of equilibrium for in situ exposures can be time-consuming and costly and should be considered at the beginning of the planning process for passive sampler deployment.
Quality assurance and quality control guidelines
As with every analytical method, analysis, or experiment, quality assurance and quality control (QA/QC) precautions should be taken when performing measurements of Cfree. Because there are many guidance documents available that describe analytical QA/QC, the following sections focus on guidelines that apply specifically to PSMs. These typically include thorough cleaning procedures, processing of (field) blanks, the development and application of reference materials, and the use of dark glassware when targeting photodegradable compounds such as PAHs. As with other analytical methods, replication (e.g., at least triplicate measurements) is also highly recommended.
Blanks
Various types of blanks (i.e., solvent blanks for ex situ experiments and passive samplers transported to and from the field, without deploying them for in situ exposures) should be used to correct for the presence of any residual, analytical background concentrations, or contaminants introduced during any step in the deployment and recovery procedure. Reliable corrections are possible if blanks are replicated, and, at least, triplicate blank measurements with every exposure are recommended. It is desirable for blanks to be as low in contaminants (target and otherwise) as possible, as correcting for blank concentrations that constitute a substantial fraction of concentrations in samples will lead to unreliable results. Thorough cleaning of samplers and glassware with solvents, and reducing sampler exposure to air, will help to accomplish this goal. Field blanks should, however, be exposed to the air at the site for a period similar to that of total air exposure for deployed samplers. The rationale for this is that media such as PDMS can absorb contaminants from vapors and dusts during deployment and retrieval, and simply taking field blank passive samplers along on the trip without actually exposing them is not a true representation of incidental field exposure. General rules for blank corrections are not widely available, and corrections typically rely on expert judgment and experience.
Analysis of reference materials
To evaluate the accuracy of any results, the inclusion of a reference sediment sample is recommended. The results of the analysis of this sample should be compared to the existing database for this sample (e.g., Cfree as determined with passive samplers), as obtained by several laboratories or developed by a reliable source (e.g., US National Institute of Science and Technology [NIST] standard reference materials [SRMs]). Based on repeated analyses, quality criteria should be set for acceptable deviations from the mean. If the results do not meet these criteria, the analyses should be assessed for problem sources and then repeated. SRMs for Cfree measurements do not yet exist, but initiatives are being undertaken.
Interlaboratory comparison exercises
Distribution of reference materials among laboratories performing Cfree measurements would allow for the determination of interlaboratory variability in PSM measurements, which benefits the quality of the results and the robustness of the measurements. So far, very few interlaboratory studies have been reported; only recently have 3 different laboratories assessed the bioaccumulation of PCBs in a polychaete by 3 different PSMs (i.e., SPME, POM, and PE) and found general agreement but with considerable variability among PSMs (Gschwend et al. 2011). Interlaboratory comparability and data quality would also benefit from standardization of sampling materials (e.g., the type and supplier of a passive sampling polymer) and establishment of a gold standard data set of passive sampler Kpw values for each of the specific polymers (see previous section). The calculated values of Cfree from the application of passive samplers are dependent on the value of the partition coefficient applied, as mentioned above. A first step toward a standard data set was recently made for POM, for which Kpw values for PAHs and PCBs were determined by different laboratories (Hawthorne et al. 2011). The use of standard polymers and partition coefficients will undoubtedly minimize the differences in Cfree assessments obtained by multiple laboratories.
Instrumental analysis
The remaining variation in results will most likely originate from analytical issues, caused by the calibration of analytical equipment and handling (i.e., cleaning, extraction) of the passive sampling materials. The quality of chemical analyses (e.g., GC/MS, GC/ECD, HPLC/MS) should be established by using internal calibration standards. Internal standards should be compounds not existing in the samples or being used as PRCs, whereas calibrations standards should span the full concentration range of the target compounds in the sample extracts while preferably falling in the linear calibration range of the instrument used. Furthermore, the solvent in which the standards are prepared should be the same as that of the extracts. This implies that a solvent exchange may be required after the extraction of the sampling materials, as extraction solvents are not always suitable as injection solvents. The completeness of the solvent exchange should be confirmed, to ensure that remaining fractions of unwanted solvents do not adversely affect the analytical equipment or the quality of the results.
NAPL-containing sediments
Some contaminated sediments contain NAPLs such as oil or coal tar that may foul passive samplers and affect the final results. NAPLs will typically have high concentrations of organic contaminants (e.g., PAHs). Most importantly, NAPL fouling on passive samplers will lead to overestimations of Cfree if the NAPL is not properly removed from the sampler (Van der Heijden and Jonker 2009). In addition to visual observations, indications of NAPL fouling may include increased variability in Cp measurements or resulting Cfree estimates that are well above the aqueous solubility of the target compound or compounds. If NAPL appears to be present in a sediment sample or on a passive sampler, it should be recorded so that the resulting Cfree will be recognized as potentially affected by artifacts. For sites where NAPL is an issue, a useful QA/QC step would be to test the effectiveness of pre-analysis removal of NAPL fouling.
Dealing with measurement uncertainty
Sources and extent of uncertainty in the measurement of Cfree are key consideration when selecting a PSM for a given application. For equilibrium sampling, the primary uncertainties lie in the measurement errors associated with Kpw and Cp. In most cases, the error associated with Kpw is approximately 0.2 log units compared to ± 20% for analytical determination of Cp (Lydy et al. this issue). It is also well documented that the uncertainty for analytical measurements increases as one approaches the analyte-specific instrument detection limit. Practitioners should also be cognizant that a higher maximum uncertainty (factor of up to 10) currently exists for some combinations of HOCs and PSDs (see “Calibration of the polymer–water partition coefficient”). Addition of a third parameter (ke) for nonequilibrium sampling increases the magnitude of uncertainty in Cfree determination by PSMs. Moreover, this added uncertainty has the potential to increase the further one operates from equilibrium (Lydy et al. this issue; Mayer et al. this issue).
As practical guidance, PSMs offer the best reduction in uncertainty in estimating Cfree (a factor of 2 compared to a factor of 10 or more for currently available alternative approaches) when the number of estimated parameters and analytical measurements is minimized, and the sediments under investigation are homogeneous. For laboratory-based exposures, this corresponds to equilibrium sampling of a well-mixed sediment sample that produces a signal well above instrumental detection limits. In contrast, the uncertainty associated with in situ PSM measurements can be much higher, due to small- and large-scale sediment heterogeneity and suboptimal mass transfer conditions, resulting in nonattainment of equilibrium and the need for correction using PRCs or models.
Guidance on incorporating results from PSMS into decision-making frameworks
As part of the risk assessment of contaminated sediment sites, sediment samples are typically analyzed to determine the likelihood that contamination will result in adverse effects to benthic invertebrates and higher trophic level organisms. The results of these analyses can be used to develop site-specific measures of exposure as well as measures of biological effects. Resultant exposure-effect relationships can ultimately serve as the basis for risk estimates and remediation plans. However, considerable uncertainty in these relationships can result from an incomplete understanding of bioavailability, which can be highly variable among sites. The use of PSMs to determine and compare Cfree in porewater to observed effects can reduce uncertainty in sediment assessments and management decisions. Examples of the various ways in which PSMs can be used in site assessments are presented in companion articles in this series (Greenberg et al. this issue; Lydy et al. this issue; Mayer et al. this issue).
Use of PSMs to estimate equilibrium exposure of sediment-associated organisms
The use of PSMs assumes that the chemical activity of an HOC in sediment is directly proportional to its Cfree in porewater (i.e., not associated with particulates or dissolved and/or colloidal organic C), and it has been shown that the Cfree is a good surrogate for bioavailability assessment (Di Toro et al. 1991; Lu et al. 2011; Gschwend et al. 2011; Burgess et al. 2013; citations in Mayer et al. this issue). Note that this equilibrium partitioning (EqP) approach does not imply that exposure is limited to uptake from porewater, only that the porewater concentration is a better reflection of the chemical activity in sediment that controls chemical uptake through all routes of exposure and ultimate equilibrium distribution between sediment and organism lipid.
Use of PSMs in tiered sediment assessments
One example of a tiered assessment that uses PSMs is presented in the US Environmental Protection Agency's (USEPA) guidance document on Equilibrium Partitioning Sediment Benchmarks (ESBs) (USEPA 2012a). The first tier of this approach uses ESBs to assess the likelihood of toxicity to the benthos using an additive toxic unit (TU) model (Figure 5). Concentrations of contaminants in sediment that do not exceed the benchmark (i.e., a sum of TUs of 1) are considered protective of sensitive aquatic organisms and require no further consideration based on this line of evidence. Sediments with concentrations that exceed the benchmark may pose a risk to aquatic organisms and may require further consideration or remediation. In the second tier, passive samplers can be used to measure concentrations of freely dissolved contaminants in porewater. As in the first tier, concentrations in porewater that do not exceed the benchmark are considered protective of benthic aquatic organisms, but concentrations that exceed the benchmark may pose a risk and may require further consideration. Sediment toxicity testing can be conducted in the third tier to verify the findings of the first 2 tiers. However, it should be recognized that if a whole sediment toxicity test finds significant toxicity, the cause or causes may be toxic chemicals other than those measured in Tiers 1 and 2 (e.g., specific pesticides or ammonium) or by confounding factors such as inappropriate O2 levels or pH. Results of porewater toxicity identification evaluations can be used in conjunction with other lines of evidence (e.g., chemical analysis, site history information, benthic community analysis) to identify the contaminants of concern at a field site and help focus on the selection of the best remedial alternatives (USEPA 2005). An example of the use of PSMs in developing a site-specific exposure-effect relationship in support of a contaminated sediment site assessment is provided in the Supplemental Data.
Figure 5.
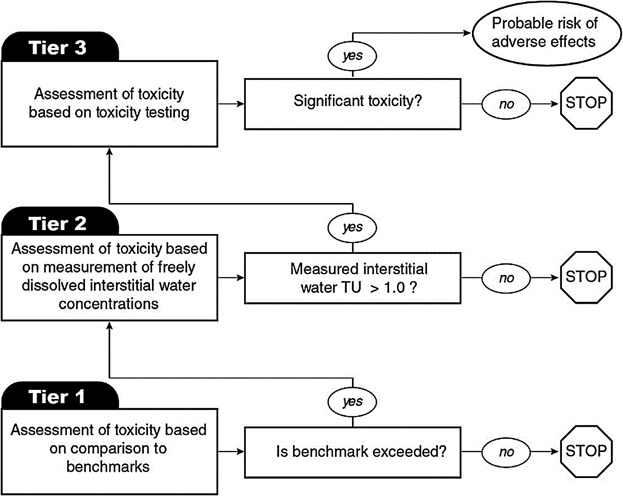
Schematic of a tiered assessment that uses PSMs to measure Cfree for organic chemicals (based on USEPA 2012a).
Conclusions and recommendations
The workgroup concluded that the science of using PSMs for measuring Cfree is sufficiently mature that relatively specific guidance can be provided for their routine use in contaminated sediment site assessments. The use and interpretation of PSMs presents a departure from conventional methods of site assessments and therefore requires the involvement of trained personnel familiar with the science reviewed in detail in Lydy et al. (this issue) and Mayer et al. (this issue). By following this guidance, an environmental scientist or engineer familiar with contaminated sediment management should be able to apply PSMs to incorporate the assessment of Cfree as part of their investigation. This endorsement of using PSMs in contaminated sediment site assessments is given while recognizing that there remain a range of scientific issues that need to be addressed in the future to improve the application of PSMs. These improvements will result in the collection of better quality and more robust data, solidifying the scientific basis for environmental management decisions.
Recommendations for future work include:
Further interlaboratory tests to build greater confidence in the precision of the methods when used by different laboratories
Development of SRMs that will allow for routine checks of method accuracy by any laboratory
Further development of the nonequilibrium PSMs in the field and further validation of PRC use in static sediment environments
Expansion of the list of organic compounds for which Kpw values are reliably measured or predicted from chemical structure
Peer-reviewed publications of more case study examples where PSMs have been used in site assessments and management decisions
Continued studies to further demonstrate the predictive capability of passive samplers for toxicity and bioaccumulation assessments.
Acknowledgments
Support for the workshop is gratefully acknowledged from ExxonMobil Corporation, Southern California Coastal Water Research Project (SCCWRP), the US Department of Defense Strategic Environmental Research and Development Program, Department of Environmental Sciences, University of California, Riverside, and the Society of Environmental Toxicology and Chemistry. The participants thank Nikki Mayo, Greg Schiefer, Angelica Bajza, Maribel Gonzalez, and Stephen Weisberg of SCCWRP for assistance in workshop planning. We also thank Wenjian Lao, Abigail Joyce, Mallory Pirogovsky, and Kai Zhang for their assistance during the workshop. The authors thank Dr Loretta A Fernandez (Northeastern University, Boston, MA) for her calculations of adjusted of KPSM values for different temperature and salinity conditions presented in Supplemental Data Figure S1. U Ghosh thanks NIEHS Superfund Research Program for partial support through grant 1R01ES020941. S Kane Driscoll thanks the Department of Defense ESTCP program for support through project ER-201216. D Reible would like to acknowledge partial support through ESTCP project ER-0624.
Supplemental Data
All Supplemental Data may be found in the online version of this article.
Provisional values for (Kpw) for selected PAHs.
Table S2. Provisional values for (Kpw) for selected PCBs.
Temperature, salinity, and pressure correction of Kpw.
Calculation of the mass of polymer required to achieve a known detection limit.
Example of the use of PSMs in support of site assessment and management.
Figure S1. Log KPEW versus (a) temperature, calculated following Lohmann (2012) for compounds with an excess enthalpy of solution in water of 39 kJ mol-1. The assumed excess enthalpy of solution is the average of 13 compounds (4 PAHs, 6 PCBs, and 3 DDTs), derived from data compiled by Mackay et al. (2006); and (b) salinity, calculated following Lohmann (2012), using log KOW-dependent Setchenow constants, as described by Ni and Yalkowsky (2003).
Figure S2. Amphipod survival in a 28-day sediment toxicity test versus (a) concentration of total PAH in bulk sediment; (b) ESB-based Sum-TU for PAHs in bulk sediment, and (c) Sum-TU for PAHs based on Cfree measurements.
References
- Adams RG, Lohmann R, Fernandez LA, MacFarlane JK, Gschwend PM. Polyethylene devices: passive samplers for measuring dissolved hydrophobic organic compounds in aquatic environments. Environ Sci Technol. 2007;41:1317–1323. doi: 10.1021/es0621593. [DOI] [PubMed] [Google Scholar]
- [ASTM] American Society for Testing and Materials. 2007. Standard test method for determination of parent and alkyl polycyclic aromatics in sediment porewater using solid-phase microextraction and gas chromatography/mass spectrometry in selected ion monitoring mode. Available from: http://www.astm.org/Standards/D7363.htm.
- Beckingham B, Ghosh U. Polyoxymethylene passive samplers to monitor changes in bioavailability and diffusive flux of PCBs after activated carbon amendment to sediment in the field. Chemosphere. 2013;91:1401–1407. doi: 10.1016/j.chemosphere.2012.12.074. [DOI] [PubMed] [Google Scholar]
- Booij K, Hofmans HE, Fischer CV, van Weerlee EM. Temperature-dependent uptake rates of nonpolar organic compounds by semipermeable membrane devices and low-density polyethylene membranes. Environ Sci Technol. 2003;37:361–366. doi: 10.1021/es025739i. [DOI] [PubMed] [Google Scholar]
- Burgess RM, Berry WJ, Mount DR, Di Toro DM. Mechanistic sediment quality guidelines based on contaminant bioavailability: equilibrium partitioning sediment benchmarks (ESBs) Environ Toxicol Chem. 2013;32:102–114. doi: 10.1002/etc.2025. [DOI] [PubMed] [Google Scholar]
- Choi Y, Cho Y-M, Gala WR, Luthy RG. Measurement and modeling of activated carbon performance for the sequestration of parent- and alkylated-polycyclic aromatic hydrocarbons in petroleum-impacted sediments. Environ Sci Technol. 2013;47:1024–1032. doi: 10.1021/es303770c. [DOI] [PubMed] [Google Scholar]
- Cornelissen G, Pettersen A, Broman D, Mayer P, Breedveld GD. Field testing of equilibrium passive samplers to determine freely dissolved native polycyclic aromatic hydrocarbon concentrations. Environ Toxicol Chem. 2008;27:499–508. doi: 10.1897/07-253.1. [DOI] [PubMed] [Google Scholar]
- Di Toro DM, Zarba CS, Hansen DJ, Berry WJ, Swartz RC, Cowan CE, Pavlou SP, Allen HE, Thomas NA, Paquin PR. Technical basis for establishing sediment quality criteria for nonionic organic chemicals by using equilibrium partitioning. Environ Toxicol Chem. 1991;10:1541–1583. [Google Scholar]
- DiFilippo EL, Eganhouse RP. Assessment of PDMS-water partition coefficients: Implications for passive environmental sampling of hydrophobic organic compounds. Environ Sci Technol. 2010;44:6917–6925. doi: 10.1021/es101103x. [DOI] [PubMed] [Google Scholar]
- Fagervold SK, Chai Y, Davis JW, Wilken M, Cornelissen G, Ghosh U. Bioaccumulation of polychlorinated dibenzo-p-dioxins/dibenzofurans in E. fetida from floodplain soils and the effect of activated carbon amendment. Environ Sci Technol. 2010;44:5546–5552. doi: 10.1021/es9027138. [DOI] [PubMed] [Google Scholar]
- Fernandez LA, MacFarlane JK, Tcaciuc AP, Gschwend PM. Using performance reference compounds in polyethylene passive samplers to deduce sediment porewater concentrations for numerous target chemicals. Environ Sci Technol. 2009a;43:8888–8894. doi: 10.1021/es901877a. [DOI] [PubMed] [Google Scholar]
- Fernandez LA, MacFarlane JK, Tcaciuc AP, Gschwend PM. Measurement of freely dissolved PAH concentrations in sediment beds using passive sampling with low-density polyethylene strips. Environ Sci Technol. 2009b;43:1430–1436. doi: 10.1021/es802288w. [DOI] [PubMed] [Google Scholar]
- Fernandez L, Lao W, Maruya K, White C, Burgess RM. Passive sampling to measure baseline dissolved persistent organic pollutant concentrations in the water column of the Palos Verdes Shelf Superfund site. Environ Sci Technol. 2012;46:11937–11947. doi: 10.1021/es302139y. [DOI] [PubMed] [Google Scholar]
- Friedman CL, Burgess RM, Perron MM, Cantwell MG, Ho KT, Lohmann R. 2009;43:2865–2870. doi: 10.1021/es803695n. Comparing polychaete and polyethylene uptake to assess sediment resuspension effects on PCB bioavailability. Environ Sci Technol. [DOI] [PubMed] [Google Scholar]
- Ghosh U, Hawthorne S. Particle-scale measurement of PAH aqueous equilibrium partitioning in impacted sediments. Environ Sci Technol. 2010;44:1204–1210. doi: 10.1021/es902215p. [DOI] [PubMed] [Google Scholar]
- Ghosh U, Weber AS, Jensen JN, Smith JR. Relationship between PCB desorption equilibrium, kinetics, and availability during land biotreatment. Environ Sci Technol. 2000;34:2542–2548. [Google Scholar]
- Gidley P, Kwon S, Yakirevich A, Magar V, Ghosh U. Advection dominated transport of polycyclic aromatic hydrocarbons in amended sediment caps. Environ Sci Technol. 2012;46:5032–5039. doi: 10.1021/es202910c. [DOI] [PubMed] [Google Scholar]
- Greenberg MS, Chapman PM, Allan IJ, Anderson KA, Apitz SE, Beegan C, Bridges TS, Brown SS, Cargill JG, IV, McCulloch MC. Passive sampling for assessment of contaminated sediments: Risk management. Integr Environ Assess Manag. 2014;10:224–236. doi: 10.1002/ieam.1511. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwend PM, MacFarlane JK, Reible DD, Lu X, Hawthorne SB, Nakles DV, Thompson T. Comparison of polymeric samplers for accurately assessing PCBs in pore waters. Environ Toxicol Chem. 2011;30:1288–1296. doi: 10.1002/etc.510. [DOI] [PubMed] [Google Scholar]
- Hale SE, Kwon S, Ghosh U, Werner D. Polychlorinated biphenyl sorption to activated carbon and the attenuation caused by sediment. Global NEST J. 2010;12:318–326. [Google Scholar]
- Hawker DW, Connell DW. Octanol-water partition coefficients of polychlorinated biphenyl congeners. Environ Sci Technol. 1988;22:382–387. [Google Scholar]
- Hawthorne SB, Miller DJ, Grabanski CB. Measuring low picogram per liter concentrations of freely dissolved polychlorinated biphenyls in sediment pore water using passive sampling with polyoxymethylene. Anal Chem. 2009;81:9472–9480. doi: 10.1021/ac9019413. [DOI] [PubMed] [Google Scholar]
- Hawthorne SB, Jonker MT, van der Heijden SA, Grabanski CB, Azzolina NA, Miller DJ. Measuring picogram per liter concentrations of freely dissolved parent and alkyl PAHs (PAH-34), using passive sampling with polyoxymethylene. Anal Chem. 2011;83:6754–6761. doi: 10.1021/ac201411v. [DOI] [PubMed] [Google Scholar]
- Hilal SH, Karickhoff SW, Carreira LA. Sparc On-Line Calculator 4.5—Predicting water solubility and log Kow—Based on “prediction of the solubility, activity coefficient and liquid/liquid partition coefficient of organic compounds. Qsar Comb Sci. 2004;23:709–720. ”. [Google Scholar]
- Hong L, Ghosh U, Mahajan T, Zare RN, Luthy RG. PAH Sorption mechanism and partitioning behavior in lampblack-impacted soils from former oil-gas plant sites. Environ Sci Technol. 2003;37:3625–3634. doi: 10.1021/es0262683. [DOI] [PubMed] [Google Scholar]
- Hsieh MK, Fu CT, Wu SC. Simultaneous estimation of glass-water distribution and PDMS-water partition coefficients of hydrophobic organic compounds using simple batch method. Environ Sci Technol. 2011;45:7785–7791. doi: 10.1021/es201040j. [DOI] [PubMed] [Google Scholar]
- Huckins JN, Manuweera GK, Petty JD, Mackay D, Lebo JA. Lipid-containing semipermeable-membrane devices for monitoring organic contaminants in water. Environ Sci Technol. 1993;27:2489–2496. [Google Scholar]
- Huckins JN, Petty JD, Booij K. Monitors of organic chemicals in the environment. New York (NY): Springer; 2006. [Google Scholar]
- Huckins JN, Petty JD, Lebo JA, Almeida FV, Booij K, Alvarez DA, Cranor WL, Clark RC, Mogensen BB. Development of the permeability/performance reference compound approach for in situ calibration of semipermeable membrane devices. Environ Sci Technol. 2002;36:85–91. doi: 10.1021/es010991w. [DOI] [PubMed] [Google Scholar]
- Huckins JN, Petty JD, Orazio CE, Lebo JA, Clark RC, Gibson VL, Gala WR, Echols KR. Determination of uptake kinetics (sampling rates) by lipid-containing semipermeable membrane devices (SPMDs) for polycyclic aromatic hydrocarbons (PAHs) in water. Environ Sci Technol. 1999;33:3918–3923. [Google Scholar]
- Jahnke A, Mayer P, McLachlan M. Sensitive equilibrium sampling to study polychlorinated biphenyl disposition in Baltic Sea sediment. Environ Sci Technol. 2012;46:10114–10122. doi: 10.1021/es302330v. [DOI] [PubMed] [Google Scholar]
- Jonker MTO, Koelmans AA. Polyoxymethylene solid phase extraction as a partitioning method for hydrophobic organic chemicals in sediment and soot. Environ Sci Technol. 2001;35:3742–3748. doi: 10.1021/es0100470. [DOI] [PubMed] [Google Scholar]
- Kane Driscoll SB, Amos CB, McArdle ME, Menzie CA, Coleman A. Predicting sediment toxicity at former manufactured gas plants using equilibrium partitioning benchmarks for PAH mixtures. Soil Sed Contam. 2009;18:307–319. [Google Scholar]
- Kreitinger JP, Newhauser EF, Doherty FG, Hawthorne SB. Greatly reduced bioavailability and toxicity of polycyclic aromatic hydrocarbons to Hyalella azteca in sediments from manufactured gas plant sites. Environ Toxicol Chem. 2007;26:1146–1157. doi: 10.1897/06-207r.1. [DOI] [PubMed] [Google Scholar]
- Lampert D, Lu X, Reible D. Long-term PAH monitoring results from the Anacostia River active capping demonstration using polydimethylsiloxane (PDMS) fibers. Environ Sci Process Impacts. 2013;15:554–562. doi: 10.1039/c3em30826j. [DOI] [PubMed] [Google Scholar]
- Lohmann R. Critical review of low-density polyethylene's partitioning and diffusion coefficients for trace organic contaminants and implications for its use as a passive sampler. Environ Sci Technol. 2012;46:606–618. doi: 10.1021/es202702y. [DOI] [PubMed] [Google Scholar]
- Lu X, Drake B, Skwarski A, Reible D. Predicting bioavailability of PAHs and PCBs with porewater concentrations measured by solid-phase microextraction fibers. Environ Toxicol Chem. 2011;30:1109–1116. doi: 10.1002/etc.495. [DOI] [PubMed] [Google Scholar]
- Lydy M, Landrum PF, Oen A, Allinson M, Smedes F, Harwood A, Li H, Maruya K, Liu J. Passive sampling methods for contaminated sediments: state of the science for organic contaminants. Integr Environ Assess Manag. 2014;10:167–178. doi: 10.1002/ieam.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäenpää K, Leppänen M, Reichenberg F, Figueiredo K, Mayer P. Equilibrium sampling of persistent and bioaccumulative compounds in soils and sediment—Comparison of two approaches to determine equilibrium partition concentrations in lipids. Environ Sci Technol. 2011;45:1041–1047. doi: 10.1021/es1029969. [DOI] [PubMed] [Google Scholar]
- Maruya KA, Zeng EY, Tsukada D, Bay SM. A passive sampler based on solid-phase microextraction for quantifying hydrophobic organic contaminants in sediment pore water. Environ Toxicol Chem. 2009;28:733–740. doi: 10.1897/08-322R.1. [DOI] [PubMed] [Google Scholar]
- Mayer P, Parkerton TF, Adams RG, Cargill JG, Gan J, Gouin T, Gschwend PM, Hawthorne SB, Helm P, Witt G. Passive sampling in contaminated sediment assessment: Scientific rationale supporting use of freely dissolved concentrations. Integr Environ Assess Manag. 2014;10:197–209. doi: 10.1002/ieam.1508. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle ME, Kane Driscoll SB, Booth PN. An ecological risk-based cleanup strategy for contaminated sediments in a freshwater brook. Int J Soil Sediment Water. 2010;3 : Article 4. [Google Scholar]
- McCoy CA, Corbett DR. Review of submarine groundwater discharge (SGD) in coastal zones of the Southeast and Gulf regions of the United States with management implications. J Environ Manage. 2009;90:644–651. doi: 10.1016/j.jenvman.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Muijs B, Jonker MTO. Temperature-dependent bioaccumulation of polycyclic aromatic hydrocarbons. Environ Sci Technol. 2009;43:4517–4523. doi: 10.1021/es803462y. [DOI] [PubMed] [Google Scholar]
- Muijs B, Jonker MTO. Assessing the bioavailability of complex petroleum hydrocarbon mixtures in sediments. Environ Sci Technol. 2011;45:3554–3561. doi: 10.1021/es103855a. [DOI] [PubMed] [Google Scholar]
- Muijs B, Jonker MTO. Does equilibrium passive sampling reflect actual in situ bioaccumulation of PAHs and petroleum hydrocarbon mixtures in aquatic worms. Environ Sci Technol. 2012;46:937–944. doi: 10.1021/es202951w. [DOI] [PubMed] [Google Scholar]
- [NRC] The National Research Council. Bioavailability of contaminants in soils and sediments. Washington DC: National Academies Press; 2003. p. 432. p. [Google Scholar]
- Oen AMP, Janssen EML, Cornelissen G, Breedveld GD, Eek E, Luthy RG. In situ measurement of PCB pore water concentration profiles in activated carbon-amended sediment using passive samplers. Environ Sci Technol. 2011;45:4053–4059. doi: 10.1021/es200174v. [DOI] [PubMed] [Google Scholar]
- Perron MM, Burgess RM, Ho KT, Pelletier MC, Friedman CL, Cantwell MG, Shine JP. Development and evaluation of reverse polyethylene samplers for marine phase II whole-sediment toxicity identification evaluations. Environ Toxicol Chem. 2009;28:749–758. doi: 10.1897/08-229.1. [DOI] [PubMed] [Google Scholar]
- Perron MM, Burgess RM, Suuberg EM, Cantwell MG, Pennell KG. Performance of passive samplers for monitoring estuarine water column concentrations 1. Contaminants of concern. Environ Toxicol Chem. 2013a;32:2182–2189. doi: 10.1002/etc.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron MM, Burgess RM, Suuberg EM, Cantwell MG, Pennell KG. Performance of passive samplers for monitoring estuarine water column concentrations 2. Emerging contaminants. Environ Toxicol Chem. 2013b;32:2190–2196. doi: 10.1002/etc.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reible DD, Lotufo G. 2012. Final technical report. Demonstration and evaluation of solid phase microextraction for the assessment of bioavailability and contaminant mobility. Environmental Restoration Project ER-0624 May 2012. Alexandria (VA): SERDP.
- Reichenberg F, Mayer P. Two complementary sides of bioavailability: Accessibility and chemical activity of organic contaminants in sediments and soils. Environ Toxicol Chem. 2006;25:1239–1245. doi: 10.1897/05-458r.1. [DOI] [PubMed] [Google Scholar]
- Schwarzenbach RP, Gschwend PM, Imboden DM. Environmental organic chemistry. 2nd ed. New York (NY): Wiley; 2003. p. 1000. p. [Google Scholar]
- Smedes F, Geertsma RW, van der Zande T, Booij K. Polymer-water partition coefficients of hydrophobic compounds for passive sampling: application of cosolvent models for validation. Environ Sci Technol. 2009;43:7047–7054. doi: 10.1021/es9009376. [DOI] [PubMed] [Google Scholar]
- Tomaszewski JE, Luthy RG. Field deployment of polyethylene devices to measure PCB concentrations in pore water of contaminated sediment. Environ Sci Technol. 2008;42:6086–6091. doi: 10.1021/es800582a. [DOI] [PubMed] [Google Scholar]
- [USEPA] US Environmental Protection Agency. Contaminated sediment remediation guidance for hazardous waste sites. Washington DC: Office of Solid Waste and Emergency Response; 2005. EPA/540/R-05/012. [Google Scholar]
- [USEPA] US Environmental Protection Agency. Equilibrium partitioning sediment benchmarks (ESBs) for the protection of benthic organisms: Procedures for the determination of the freely dissolved interstitial water concentrations of nonionic organics. Washington DC: Office of Research and Development; 2012a. EPA-600-R-02-012. [Google Scholar]
- [USEPA] US Environmental Protection Agency. Guidelines for using passive samplers to monitor organic contaminants at Superfund sediment sites. Washington DC: Office of Superfund Remediation and Technology Innovation and Office of Research and Development; 2012b. OSWER Directive 9200.1-110 FS December 2012. [Google Scholar]
- Van der Heijden SA, Jonker MTO. 2009;43:3757–3763. doi: 10.1021/es803329p. PAH bioavailability in field sediments: Comparing different methods for predicting in situ bioaccumulation. Environ Sci Technol. [DOI] [PubMed] [Google Scholar]
- Vinturella AE, Burgess RM, Coull BA, Thompson KM, Shine JP. The use of passive samplers to mimic uptake of polycyclic aromatic hydrocarbons by benthic polychaetes. Environ Sci Technol. 2004;38:1154–1160. doi: 10.1021/es034706f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Provisional values for (Kpw) for selected PAHs.
Table S2. Provisional values for (Kpw) for selected PCBs.
Temperature, salinity, and pressure correction of Kpw.
Calculation of the mass of polymer required to achieve a known detection limit.
Example of the use of PSMs in support of site assessment and management.
Figure S1. Log KPEW versus (a) temperature, calculated following Lohmann (2012) for compounds with an excess enthalpy of solution in water of 39 kJ mol-1. The assumed excess enthalpy of solution is the average of 13 compounds (4 PAHs, 6 PCBs, and 3 DDTs), derived from data compiled by Mackay et al. (2006); and (b) salinity, calculated following Lohmann (2012), using log KOW-dependent Setchenow constants, as described by Ni and Yalkowsky (2003).
Figure S2. Amphipod survival in a 28-day sediment toxicity test versus (a) concentration of total PAH in bulk sediment; (b) ESB-based Sum-TU for PAHs in bulk sediment, and (c) Sum-TU for PAHs based on Cfree measurements.