

Abstract
Background
Progressive left ventricular (LV) dysfunction can be a major late complication in patients with chronic right ventricular (RV) pressure overload (e.g., tetralogy of Fallot). We therefore examined LV function (serial echocardiography and ex vivo Langendorff) and histology in a model of infant pressure-load RV hypertrophy (RVH).
Methods and Results
Ten-day-old rabbits (N=6 per time point, total = 48) that underwent pulmonary artery banding (PAB) were sacrificed at 2–8 weeks after PAB, and comparisons were made with age-matched sham controls. LV performance (myocardial performance index, MPI) decreased during the progression of RVH although the LV ejection fraction (EF) was maintained. In addition, RVH caused significant septal displacement, reduced septal contractility, and decreased LV end-systolic (LVDs) and diastolic (LVDd) dimensions, resulting in LV diastolic dysfunction with the appearance of preserved EF. Significant septal and LV free wall apoptosis (myocyte-specific TUNEL and activated caspase-3), fibrosis (Masson’s trichrome stain), and reduced capillary density (CD31 immunostaining) occurred in the PAB group after 6–8 wks (all p<0.05).
Conclusion
This is the first study showing that pressure overload of the RV resulting in RVH causes LV diastolic dysfunction while preserving EF through mechanical and molecular effects upon the septum and LV myocardium. In particular, the development of RVH is associated with septal and LV apoptosis and reduced LV capillary density.
Keywords: right ventricular hypertrophy, heart failure with preserved ejection fraction, diastolic dysfunction
Introduction
Despite intensive basic science and clinical investigations, heart failure remains a leading cause of hospitalization and mortality worldwide.1–4 In patients with congenital heart disease (CHD), left ventricular (LV) dysfunction is an important cause of heart failure as well as an important prognostic factor.5, 6 Furthermore, progressive LV dysfunction can be a major problem in patients with chronic RV pressure load such as tetralogy of Fallot (TOF) as they age.6, 7 Several late complications, including sudden death, have been recognized after 2-stage TOF repair and may be related not only to surgical factors (e.g. ischemia-reperfusion injury and myocardial scarring), but also to time-dependent events (e.g. pressure and/or volume overload, cyanosis) existing before and after repair that permanently damage the myocardium.8 Several investigators have suggested that these pathological changes lead to permanent heart damage and have found a lower incidence of ventricular arrhythmias among children who have undergone repair at younger ages. It is therefore generally agreed that long periods of pressure load should be avoided. However, underlying mechanistic factors have not been identified.
The concept of heart failure with preserved ejection fraction (HFpEF) has been recently discussed in some detail9–14 and it appears that the phenomenon is related to diastolic LV dysfunction. In HFpEF, EF is maintained within normal limits despite the development of the signs and symptoms of heart failure. Some children with CHD suffer from HF despite preservation of EF, but HFpEF in CHD has not been investigated. Relevant to the aforementioned difficulties in long-term outcome in patients with lesions such as tetralogy of Fallot, there is compelling evidence that optimal RV systolic function requires a substantial contribution from the LV, most likely via the interventricular septum.15
One potential cellular mechanism of progressive myocardial dysfunction is apoptosis, which is a tightly regulated and enzymatically-driven process in which cells die in response to internal and/or external stimuli.1, 2, 16–18 It has hitherto been thought that terminally differentiated cells such as cardiomyocytes and neuronal cells have limited or no potential for regeneration by mitosis and are correspondingly less likely to undergo apoptosis.17, 19, 20 More recently, activation of cardiomyocyte apoptotic pathways have been recognized to contribute to decreases in cardiomyocyte number and potentially to altered cardiac function in several situations such as postnatal maturation, hypertension, diabetes, aging, pressure overload, and ischemia/reperfusion injury.1, 17, 21–27 Insufficient capillary angiogenesis may be an important contributing factor in hypertrophic situations because the increased myocardial mass requires greater blood supply to maintain contractile processes, especially in the face of increased afterload.28, 29 Altered structural responses by the microvessels to the increased pressure load in the RV is a possible mechanism for chamber-specific dysfunction.30 It is unknown, however, whether capillary density is altered in the LV as a result of RVH.
Understanding the effects and mechanisms of RV pressure overload upon LV function are important not only to patients with various forms of congenital hearts disease (e.g. TOF) but also to children and adults with pulmonary hypertension from a variety of causes. Taking these aspects into consideration, we hypothesized that RV pressure overload would induce mechanical and resultant cellular changes in the interventricular septum, which could then be conferred upon the LV.
Materials and Methods
Experimental Model
Ten-day-old New Zealand White rabbits weighing 160±33 g were used. All animals were cared for in accordance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication No. 86–23, revised 1996). The protocols used in this study were approved by the Institutional Animal Care and Use Committee at Children’s Hospital, Boston.
The rabbits were anesthetized with intravenous injections of ketamine (50 mg/kg) and xylazine (2 mg/kg). After bupivacaine (0.1–0.2 ml) was injected subcutaneously, a left thoracotomy was performed through the 2nd or 3rd intercostal space. The pericardium was incised and the main pulmonary artery was exposed. The pulmonary artery was banded with 2–0 silk, constricting it by 10–25% of its diameter. A 20 gauge catheter was placed in the left chest cavity and the wound was closed. After as much air as possible had been evacuated via the catheter, it was removed. Sham-operated controls underwent an identical procedure except for pulmonary artery banding (PAB). Buprenorphine (0.01 to 0.05 mg/kg) was administered for pain every 8 to 12 hours, or as needed. All animals were observed for up to 2, 4, 6 and 8 weeks after the operation.
Experimental Groups
A total of 48 rabbits were assigned, 24 to the control group and the 24 to the PAB group. In each group, 6 rabbits were sacrificed at 2, 4, 6 and 8 weeks after the operation. Immediately after the rabbits were deeply anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg), their hearts were harvested. Once removed, they were attached to a perfusion apparatus (Langendorff system) via the aortic arch.31 For blood washout, a Krebs-Ringer solution (100 mmol/L NaCl, 4.7 mmol/L KCl, 1.1 mmol/L KH2PO4,1.2 mmol/L MgSO4, 25 mmol/L NaHCO3, 1.7mmol/L CaCl2, 11.5 mmol/L Glucose D(+), pH 7.4) with insulin (10 U/L) was infused for several minutes, after which hearts were divided and blotted dry for wet weight measurement and subsequent analyses. The perfusion was performed at 37°C while O2 95% and CO2 5% were bubbled through the medium.
Langendorff Perfusion
A separate group (N=6 each) of hearts from sham and banded animals at 8 weeks were perfused with a Krebs-Ringer solution (see above) at 120 cm H2O for 30 minutes. Next, a balloon-tipped Millar micromanometry pressure catheter was inserted into the LV through the mitral valve for LV pressure-volume measurement. Balloon volume was adjusted by increments of 0.1 mL using a calibrated syringe and both LV developed pressure and diastolic pressure were recorded; tau (the half-time of LV pressure decline) was derived from these P-V recordings using PVAN (AD Instruments) software.
Echocardiography
Transthoracic echocardiography was performed every week after surgery until the rabbits were euthanized. Rabbits were anesthetized with intravenous injections of ketamine (50 mg/kg) and xylazine (2 mg/kg). Measurements of wall thickness at end-systole and end-diastole were made in M mode, and the LV dimensions at end-systole (LVDs) and end-diastole (LVDd) were also measured. To estimate tricuspid regurgitation (TR) and the pressure gradient at the PAB site, the peak pressure difference was estimated using spectral continuous wave Doppler mode. Ejection fraction (EF) and MPI (TEI index) were determined. To calculate MPI (TEI index), two periods were measured using the pulsed Doppler mode: the ejection time (c) and the time interval (d) between the end of LV inflow and onset. MPI was then calculated by the following formula:
MPI= [(d-c)/c]×100, and was determined three times for each animal and recorded as the average of the three measurements. All studies were recorded on VHS videotape with simultaneous ECG.
Apoptosis
A) TUNEL staining
Hearts were fixed in 4% paraformaldehyde, paraffin-embedded, and sectioned. TUNEL staining was performed in deparaffinized and rehydrated sections using the FragEL DNA fragmentation detection kit (EMD, Biosciences Inc., San Diego, CA). The sections were incubated with terminal deoxynucleotidyl transferase and fluorescein-labeled dUTP. Then, mouse desmin monoclonal antibody (Sigma-Aldrich, St. Louis, MO) was used for identification of cardiomyocytes, followed by incubation with a secondary anti-mouse immunoreagent conjugated to the red-fluorescent Alexa-594TM fluorophore at a concentration of 1:200 (Molecular Probes, Eugene, OR). All nuclei were stained with blue fluorescent DAPI nucleic acid stain (Molecular Probes, Eugene, OR). Slides were visualized using a microscope with a Nikon 20× objective and a 10× eyepiece. Both the total number of nuclei and the number of TUNEL-positive nuclei in the LV and the interventricular septum (IVS) were counted in 10 random fields of vision per tissue section using Metamorph software (Universal Imaging Corporation, West Chester, PA), and the results were expressed as the number of TUNEL-positive cardiomyocyte nuclei (i.e. simultaneously DAPI/TUNEL/desmin positive) per 1000 total cardiomyocyte nuclei.
B) Western Immunoblotting
After the hearts were harvested and flushed free of blood, specimens of LV and IVS were frozen in liquid nitrogen and stored at −80°C. These tissues were homogenized in cold lysis solution containing 150 mM NaCl, 20 mM Tris HCl (pH 7.6), 1 mM EDTA, 0.5% sodium deoxycholate, 70 mM NaF, 1% Nonidet P-40, complete protease inhibitor cocktail (Boehringer Mannheim), 200 mM sodium orthovanadate, and 2 mM phenylmethylsulfonyl fluoride. After 10 min of incubation on ice with intermittent, brief vortexing, debris was pelleted in the microcentrifuge, and the supernatants were stored at −80°C. Protein concentrations were determined using the bicinchoninic acid (BCA) protein determination kit (Pierce). SDS-PAGE and transfer to nitrocellulose were performed, and identical gels were stained with Coomassie brilliant blue R250 to confirm equal protein loading. The nitrocellulose membranes were rinsed in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20 (TBS-T), and were blocked in 5% nonfat milk/TBS-T for 1 h at 22°C on a rocking platform. Membranes were rinsed four times in TBS-T and then incubated overnight at 4°C on an orbital shaker with primary antibodies diluted in TBS-T containing 5% BSA (Sigma). Anti-active caspase 3 polyclonal antibody (Upstate), anti-caspase 9 polyclonal antibody (Upstate), anti-Bax polyclonal antibody (Upstate), and anti-Bcl-2 monoclonal antibody (Santa Cruz Biotech.) were used at a concentration of 0.1 mg/ml. Excess primary antibodies were washed from the nitrocellulose membranes with three 10-min washes in TBS-T, and then these membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-rabbit secondary antibodies (Amersham) diluted 1:2,000 in 5% milk/TBS-T. After three further 10-min washes in TBS-T, bound antibodies were detected with the enhanced chemiluminescence kit (Amersham) and exposed to Kodak X-Omat AR film. Densitometric determination of mean integrated areas under the curve from unsaturated autoradiograms of immunoblots was performed using Scion Image software (National Institutes of Health).
Microvascular Density
Microvascular density was determined from paraffin-embedded cross-sections of the heart, which were deparaffinized and stained with anti-CD-31 antibody (monoclonal mouse anti-human CD31 Endothelial Cell, clone JC70A, Dako Corporation, Carpentaria, CA) using a red fluorescent secondary antibody (Alexa-594TM fluorophore; Molecular Probes, Eugene, OR) and then cover-slips were applied with fluorescent mounting medium (Dako Corporation, Carpentaria, CA). To confirm this result, microvascular density was also determined on histological sections obtained from hearts perfused with fluorescein-isothiocyanate-conjugated (FITC) Lycopersicon esculentum lectin (Sigma-Aldrich, St. Louis, MO). Slides were examined using a microscope with a Nikon 20× objective and a 10× eyepiece. The number of microvessels appearing was counted in 5 random fields of vision per region of each tissue section (LV and IVS) using Metamorph software, and the results were expressed as the number of microvessels per 1.27×10−7m2.
Fibrosis
Separate deparaffinized tissue sections were stained with Masson-Trichrome, which results in fibrotic (collagen-enriched) areas appearing blue, whereas cellular elements appear red. In this way, the ratio between the blue and red areas (fibrosis vs. myocardium) serves as an estimate of the progression of the fibrosis. Slides were visualized using a microscope with a Nikon 10× objective and a 10× eyepiece. Blue and red areas were measured in 5 random fields of vision per each part of the tissue section (LV and IVS) using Metamorph software, and the results were expressed as the ratio of the blue to red areas.
Statistical Analysis
Data are expressed as the mean ± standard deviation. Statistical analyses was performed using the Stat View software package (version 5.0, SAS Institute Inc.,NC). Differences between the groups were tested for significance by one-way ANOVA using Bonferroni’s correction for multiple comparisons. After normal distribution was confirmed and equal variance testing was done, the Student t test was used to compare individual data sets. A two-tailed probability value of less than 0.05 was considered statistically significant.
Results
Morphology
The RVs of the PAB group became substantially larger and dilated (Figure 1A). Dilatation started around 4 weeks after PAB, so that the RV became about 3 times the size of that of the control group at 8 weeks after PAB (Figure 1B). The body weight and the wet weights of the LV of both groups were almost the same (data not shown), but the RV wet weight was almost 3 times that of the control group (p<0.01) (Figure1B). Reflecting RV hypertrophy, the RV/LV weight ratio in the PAB group increased significantly as the animals grew, while those of the control group remained essentially unchanged (p<0.01) (Figure 1C). At 8 weeks after PAB, the average RV/LV weight ratio exceeded 1.5.
Figure 1. Fresh-cut sections at mid-ventricle level of whole rabbit hearts.


In Part A, the upper row images show representative control sections, and the lower row, those from PAB hearts. RV thickness and chamber size increased in PAB hearts.
There were significant differences in both absolute (B) and LV-normalized (C) RV mass within 4 weeks of PAB. †P<0.01 versus the control group.
Echocardiography
A) Peak pressure difference across PAB and the tricuspid valve
As the animals grew, the estimated peak pressure difference across the PAB site increased, reaching a plateau after 7 weeks (37.9±5.6 mmHg) (Figure 2A). Likewise, the estimated peak pressure difference across the tricuspid valve, which was calculated from the peak flow velocity of tricuspid regurgitation (TR), rose by the 6th week after PAB, but remained at almost the same level thereafter (51.7±7.0 mmHg at 8 weeks). (Figure 2B)
Figure 2. Echocardiographic estimation of RV pressure.

The peak pressure difference across the PAB site (A) and the tricuspid valve (B) were estimated from the peak flow velocity (spectral continuous-wave Doppler). By both measures, RV pressure progressively increased, stabilizing at approximately 7 weeks postoperatively.
B) LV dimensions and LV function
Although there was no significant difference in LV free wall thickness, (Figure 3A), the septum in the PAB group at systole was significantly thinner than that of the control group (p<0.05) (Figure 3B). As RV pressure increased, the septum became flat, potentially impairing septal contractility in the PAB group.
Figure 3. Echocardiographic assessments of LV and septal dimensions and function.



Thickness of the left ventricle (LV; A) and the septum (B): no significant difference was found in LV wall thickness, but that of the septum in the PAB group was significantly thinner than that of the controls. LV chamber dimensions (C) were significantly less in the PAB group than the control group. In the key, (s) refers to end-systole and (d), to end-diastole. The myocardial performance index (MPI), shown in D, increased consistent with progression of LV dysfunction. Ejection fraction (EF) is shown in E, but was likely to be “artifactually” affected by the deformity of the LV as shown in C (see text also). * P<0.05, and †P<0.01 versus the control group.
LV function was assessed using EF and MPI (TEI index) (Figures 3D, E). Although EF appeared to increase in the PAB group (Figure 3E), this was likely to be an overestimation of actual LV systolic function because LVDd and LVDs were smaller in the PAB group than in the control group (Figure 3C). For example, at 6 wks (Figure 3C), LVDs and Dd were 12.6 mm and 8.8 mm, respectively, in the control group, and 10.8 mm and 7.1 mm in the PAB group. The difference between LVDd and LVDs was almost the same (3.8 mm and 3.7 mm) in both groups, but the EF calculated was correspondingly higher the PAB group than the control group. On the other hand, the MPI (TEI index) of the PAB group increased slowly after 3 weeks and was significantly higher than that of the control group by 6 weeks after PAB (Figure 3D), indicating reduced LV function in the PAB group.
Langendorff Perfusion
The developed pressure-LV balloon volume response appeared to be attenuated in the isolated, Langendorff-perfused LV from PAB animals (Figure 4A). The slope (compliance) of the LV diastolic P-V curve appeared to be somewhat steeper (reduced compliance) in the LV from PAB animals, especially at higher LV volumes (Figure 4B). Impaired LV diastolic function in PAB animals was also suggested by the fact that LV tau was prolonged by 84% (P<0.01) in PAB animals compared to control.
Figure 4. LV systolic and diastolic function estimated in the ex vivo isovolumic Langendorff mode.

The graph on the left shows peak developed LV pressure as a function of LV balloon volume. The graph on the right shows the LV diastolic pressure-balloon volume relationship. The response of LV peak developed pressure tended to be blunted while diastolic compliance, especially at higher LV volumes, was reduced in PAB animals.
Apoptosis
A) Cardiomyocyte-specific TUNEL staining
More TUNEL-positive nuclei were found in the PAB group than in the control group (Figure 5A). In each myocardial portion of the control group, the number of TUNEL-positive nuclei was at a basal level (0.9–1.5/1000 nuclei), but the number of TUNEL-positive nuclei increased significantly at 4, 6 and 8 weeks after PAB in both the LV and the septum in the PAB group (Figure 5B).
Figure 5. Assessment of apoptosis by TUNEL staining.

A1: TUNEL-positive cells (green fluorescence). A2: DAPI-positive nuclei (blue fluorescence). A3: desmin-positive cells (red fluorescence). A4: color-combined image (all from heart 6 weeks after PAB). Magnification ×200. Arrows indicate TUNEL-positive nuclei. The white scale bar represents 100 microns.
B: Graphs summarize the number of TUNEL-positive nuclei per 1000 cardiomyocyte nuclei, and represent the average from 10 different fields selected randomly. In the septum, more TUNEL-positive nuclei were found in the PAB group than in the control group, especially at 6 weeks after PAB. * P<0.05, and †P<0.01 versus the control group.
B) Apoptotic Proteins
The expression of Bax was significantly higher in the PAB group after 6 weeks than in the control group in the septum. As a result, the ratio of Bax to Bcl-2 was significantly higher in the PAB group (p<0.01) (Figure 6A). The expression of caspase-3 and 9 in the PAB group was also higher than that of the control group, especially 6 weeks after PAB (p<0.01 at 6 weeks, p<0.05 at 8 weeks) (Figure 6B, 6C). These results are consistent with a more pro-apoptotic milieu in the septum and LV of PAB animals as opposed to control.
Figure 6. Expression of selected apoptosis proteins (Bax, Bcl-2, caspase-3, and caspase-9) by Western immunoblotting.


A; In the PAB group, the expression of pro-apoptotic Bax was significantly greater than in the control group at 8 weeks, beginning at 4 weeks after PAB. The ratio of Bcl-2/Bax was significantly lower in the PAB group after 6 and 8 weeks.
B,C; The expression of active caspase-3 and total caspase-9 was also higher in the PAB group after 6 and 8 weeks. *p<0.05, and †p<0.01 versus the control group.
Fibrosis
Representative areas of fibrosis and myocardium are shown in blue and red, respectively (Figure 7A). The degree of fibrosis is shown as the ratio between blue and red areas in the myocardium (Figure 7B). Although fibrosis progressed gradually as the animals grew in both groups, within 4 weeks there was significantly more septal fibrosis in the PAB group than in the controls (at 4 weeks, p<0.05; at 6 and 8 weeks, p<0.01) (Figure 5B). No difference in overall fibrosis was detected in the LV free wall.
Figure 7. Assessment of myocardial fibrosis.


Representative Masson-Trichrome stained sections of infant rabbit myocardium. In (A), the upper two images are from week 6 hearts and the lower two from week 8 samples. Those on the left are from control, those on the right, from PAB. Magnification ×100. The white bars represent 100 microns. The graphs in (B) show the percentage of the tissue occupied by fibrosis, based upon the average of 10 randomly selected areas from each heart. There was significant septal (as well as RV) fibrosis in PAB hearts compared to that in controls after 4 weeks, but no significant change was found in LV free wall. *P<0.05, and †P<0.01 versus the control group.
Microvascular density
As the right side of Figure 8B shows, microvessels were well-stained by CD31 antibody (red color) as well as by lectin in Figure 6C (green color). Generally, microvascular density decreased in both groups as the animals matured. By 6 weeks, however, the decrease of microvascular density in the septum and LV of the PAB group was significantly greater than that in the control. (p<0.05) (Figure 8A). In the septum, the difference was also significant at 8 weeks (p<0.05)
Figure 8. Assessment of alterations in microvascular density.
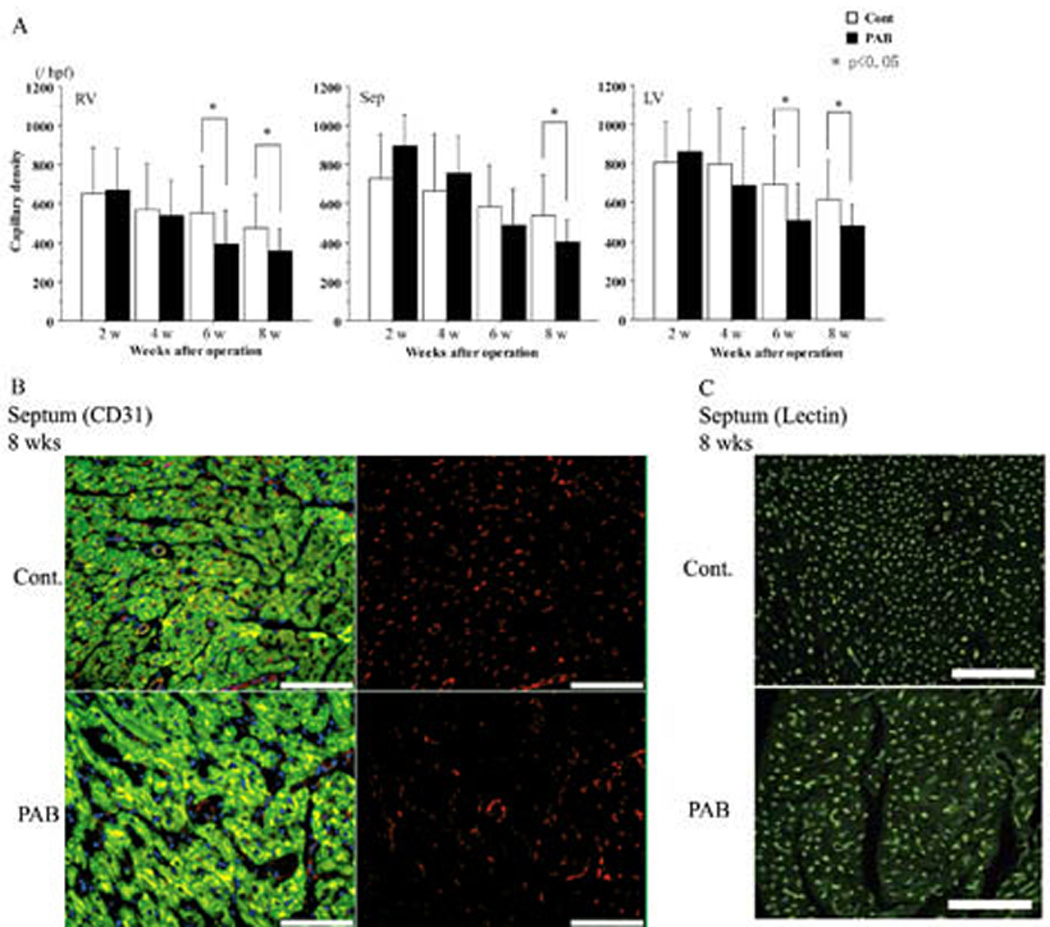
The graphs in (A) portray differences in the numbers of microvessels stained by CD31 antibody. In the septum at 8 weeks and the LV and the RV at 6 and 8 weeks, microvascular density decreased significantly in the PAB hearts. *p<0.05 versus the control group.
Representative images of CD31 staining are shown in (B); CD 31 staining. Microvessels reacting with antibody to the endothelial cell-specific epitope CD 31 appear in red (on the right); the merged images stained with DAPI (DNA, nuclei, blue) and desmin (muscle, green) are in Panel B on the left. Examples of lectin staining of capillaries in isolated-perfused control (top image) and PAB (bottom image) hearts, visualized as relatively punctuate areas of bright green fluorescence, are shown in (C). For both (B) and (C), magnification ×200; the white scale indicates 100 microns.
Discussion
Recently, HFpEF has been the subject of increased discussion. HFpEF can be indicative of LV diastolic function, and is a potential complication of prolonged hypertension.9–14 Although there is extensive literature pertaining to LV hypertrophy and failure due to chronic pressure overload and other abnormalities, there is little information about LV dysfunction caused by the development of RVH.1, 4, 19, 31–34 In this study, we showed not only that RVH influenced LV function, particularly diastolic compliance, after PAB, but also that septal and LV cardiomyocyte apoptosis may contribute to the LV dysfunction. Since we performed PAB in 10-day-old rabbits, the development and time course of LV dysfunction in conjunction with the progression of RVH could be demonstrated relatively clearly.
Of course, it is difficult to measure LV function using imaging techniques when the shape of the LV is deformed, as it was here by the progression of RVH.35 We therefore also used tissue Doppler to determine the MPI (TEI index), which is measured as the sum of isovolumic contraction and relaxation durations divided by ejection time 35–37. The tissue Doppler-derived MPI is a simple, semi-quantitative, non-geometric index of ventricular function that is applicable to the study of ventricular function in the presence of distorted ventricular morphologies as occurs frequently in congenital heart disease.35 In HFpEF, EF is usually maintained within normal limits, but LV function, particularly diastolic function, is impaired. The results of the Langendorff system, as well as the MPI analyses (where most, if not all of the MPI abnormality was due to the diastolic duration measurement) indicated that LV relaxation was impaired by RVH; this appeared to be, at least in part, due to reduced compliance of the septum.
In our study, PAB served to cause gradual constriction, leading to RVH, as the animals grew. We therefore consider this a good experimental model due the gradual onset of the abnormal pressure load and subsequent development and progression of RVH. At 6 weeks after PAB, RV dilatation was almost complete, and the wet weight of the RV of the PAB group was three times that of the control group. We found, as a part of this process, that RVH deformed the septum which flattened and then deviated into the left side of the heart (Figure1, 3). Both increased apoptosis and fibrosis appeared to be closely related, at least temporally, to the development of cardiac dysfunction. This dysfunction is on the one hand “macroscopic”, arising from physical compression of the LV due to RV enlargement and septal shift. Based upon our results, we speculate that these local abnormalities of wall stress and deformation contribute to activation of apoptotic pathways (Figures 5 and 6), and potentially to dysregulation of the pro- and anti-angiogenic factors that regulate angiogenesis and fibrosis (Figures 7 and 8). The degeneration of cardiomyocytes and inadequate microvascular development contributed to decreased compliance of the LV, further promoting LV dysfunction.
Although normal maturation of the RV was associated with a modest reduction in microvascular density, it appears that RVH markedly decreased the number of small capillaries relative to the amount of myocardial tissue. Whether this is in fact the result of deficient production (angiogenesis) and/or increased destruction, as well as the mechanisms involved, require prior study. Interestingly, we have previously found that the process of LV pressure overload hypertrophy is able to effectively inhibit hypoxia-induced pro-angiogenic signaling; one likely mechanism is an alteration in the balance of endogenous pro- and anti-angiogenic regulatory molecules.38 In addition, it will be important to determine whether the apparent “mismatch” of muscle mass to capillary supply is sufficient to impair oxygen and substrate delivery and thereby be a primary cause of cardiomyocyte damage, as well as activation of pro-fibrotic pathways, as well as whether these effects overall are sufficient to result in ventricular failure.39–41
Considering the results of this study, it appears that RV pressure overload in the infant is relatively poorly tolerated and needs to be relieved early, not only to prevent deterioration of RV function, but also to prevent pathologic remodeling of the interventricular septum and LV as well. This study also shows that pressure overload resulting in RVH causes LV diastolic dysfunction with preservation of EF by exerting both mechanical and molecular effects upon the septum and the LV myocardium. In particular, the development of RVH is associated with septal and LV apoptosis, pathologic LV remodeling, and reduced LV capillary density. Potential mediators and mechanisms (e.g. paracrine vs. circulating stimuli) merit further investigation.
Heart failure with preserved ejection fraction (HFpEF) is closely related to the diastolic left ventricular (LV) dysfunction. Some children with congenital heart disease (CHD) suffer from HFpEF, because right ventricular hypertrophy (RVH) causes the diastolic LV dysfunction. LV dysfunction is the principal physiologic disorder leading to HF, and progressive LV dysfunction can be a major late complication in patients with chronic RV pressure overload. In order to investigate this phenomenon, we have created a baby rabbit model that showed progressive RVH. We performed pulmonary artery banding (PAB) in 10-day-old rabbits. PAB gradually constricted the PA, resulting in RVH. Our model enabled us to investigate LV function accurately in relation to physical development and demonstrate clearly how RVH influenced LV function, especially diastolic dysfunction, after PAB in relation to physical development in infant RVH. This study for the first time shows that a pressure overload resulting in RVH causes LV diastolic dysfunction with preservation of EF by exerting both mechanical and molecular effects upon the septum and the LV myocardium. In particular, the development of RVH is associated with septal and LV apoptosis, pathologic LV remodeling, and reduced LV capillary density. Our overall results indicated that apoptosis occurred immediately soon after PAB, before PAB caused a marked degree of constriction of PA, and apoptosis increased during the progress of LV dysfunction. Following this, apoptosis progressed as the rabbits grew older until LV dysfunction was almost complete, and finally resulted in LV failure due to excessive fibrosis.
Acknowledgments
Funding Sources: Supported in part by National Institutes of Health grants HL066186 and HL07434 (to FXM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: none
References
- 1.Chen C, Ma L, Linfert DR, Lai T, Fallon JT, Gillam LD, Waters DD, Tsongalis GJ. Myocardial cell death and apoptosis in hibernating myocardium. J Am Coll Cardiol. 1997;30:1407–1412. doi: 10.1016/s0735-1097(97)00309-4. [DOI] [PubMed] [Google Scholar]
- 2.Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure focus on cardial hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol. 2006;48(Suppl):A56–A66. [Google Scholar]
- 4.Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- 5.Marcu CB, Beek AM, Van Rossum AC. Cardiovascular magnetic resonance imaging for the assessment of right heart involvement in cardiac and pulmonary disease. Heart Lung Circ. 2006;15:362–370. doi: 10.1016/j.hlc.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Tzemos N, Harris L, Carasso S, Subira LD, Greutmann M, Provost Y, Redington AN, Rakowski H, Siu SC, Silversides CK. Adverse left ventricular mechanics in adults with repaired tetralogy of Fallot. Am J Cardiol. 2009;103:420–425. doi: 10.1016/j.amjcard.2008.09.101. [DOI] [PubMed] [Google Scholar]
- 7.Urashima T, Zhao M, Wagner R, Fajardo G, Farahani S, Quertermous T, Bernstein D. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol. 2008;295:H1351–H1368. doi: 10.1152/ajpheart.91526.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pigula FA, Khalil PN, Mayer JE, del Nido PJ, Jonas RA. Repair of tetralogy of Fallot in neonates and young infants. Circulation. 1999;100(Suppl):II157–II161. doi: 10.1161/01.cir.100.suppl_2.ii-157. [DOI] [PubMed] [Google Scholar]
- 9.Bursi F, Weston SA, Redfield MM, Jacobsen SJ, Pakhomov S, Nkomo VT, Meverden RA, Roger VL. Systolic and diastolic heart failure in the community. JAMA. 2006;296:2209–2216. doi: 10.1001/jama.296.18.2209. [DOI] [PubMed] [Google Scholar]
- 10.Klapholz M, Maurer M, Lowe AM, Messineo F, Meisner JS, Mitchell J, Kalman J, Phillips RA, Steingart R, Brown EJ, Jr, Berkowitz R, Moskowitz R, Soni A, Mancini D, Bijou R, Sehhat K, Varshneya N, Kukin M, Katz SD, Sleeper LA, Le Jemtel TH. Hospitalization for heart failure in the presence of a normal left ventricular ejection fraction: results of the New York Heart Failure Registry. J Am Coll Cardiol. 2004;43:1432–1438. doi: 10.1016/j.jacc.2003.11.040. [DOI] [PubMed] [Google Scholar]
- 11.Masoudi FA, Havranek EP, Smith G, Fish RH, Steiner JF, Ordin DL, Krumholz HM. Gender, age, and heart failure with preserved left ventricular systolic function. J Am Coll Cardiol. 2003;41:217–223. doi: 10.1016/s0735-1097(02)02696-7. [DOI] [PubMed] [Google Scholar]
- 12.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 13.Senni M, Tribouilloy CM, Rodeheffer RJ, Jacobsen SJ, Evans JM, Bailey KR, Redfield MM. Congestive heart failure in the community: trends in incidence and survival in a 10-year period. Arch Intern Med. 1999;159:29–34. doi: 10.1001/archinte.159.1.29. [DOI] [PubMed] [Google Scholar]
- 14.Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, Levy D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population-based cohort. J Am Coll Cardiol. 1999;33:1948–1955. doi: 10.1016/s0735-1097(99)00118-7. [DOI] [PubMed] [Google Scholar]
- 15.Damiano RJ, Jr, La Follette P, Jr, Cox JL, Lowe JE, Santamore WP. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261:H1514–H1524. doi: 10.1152/ajpheart.1991.261.5.H1514. [DOI] [PubMed] [Google Scholar]
- 16.Hetts SW. To die or not to die: an overview of apoptosis and its role in disease. JAMA. 1998;279:300–307. doi: 10.1001/jama.279.4.300. [DOI] [PubMed] [Google Scholar]
- 17.MacLellan WR, Schneider MD. Death by design. Programmed cell death in cardiovascular biology and disease. Circ Res. 1997;81:137–144. doi: 10.1161/01.res.81.2.137. [DOI] [PubMed] [Google Scholar]
- 18.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda S, Hamada M, Hiwada K. Cardiomyocyte apoptosis with enhanced expression of P53 and Bax in right ventricle after pulmonary arterial banding. Life Sci. 1999;65:925–933. doi: 10.1016/s0024-3205(99)00322-7. [DOI] [PubMed] [Google Scholar]
- 20.Kitahori K, Takamoto S, Takayama H, Suematsu Y, Ono M, Motomura N, Morota T, Takeuchi K. A novel protocol of retrograde cerebral perfusion with intermittent pressure augmentation for brain protection. J Thorac Cardiovasc Surg. 2005;130:363–370. doi: 10.1016/j.jtcvs.2004.11.059. [DOI] [PubMed] [Google Scholar]
- 21.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- 22.Hamet P, Richard L, Dam TV, Teiger E, Orlov SN, Gaboury L, Gossard F, Tremblay J. Apoptosis in target organs of hypertension. Hypertension. 1995;26:642–648. doi: 10.1161/01.hyp.26.4.642. [DOI] [PubMed] [Google Scholar]
- 23.Piot CA, Padmanaban D, Ursell PC, Sievers RE, Wolfe CL. Ischemic preconditioning decreases apoptosis in rat hearts in vivo. Circulation. 1997;96:1598–1604. doi: 10.1161/01.cir.96.5.1598. [DOI] [PubMed] [Google Scholar]
- 24.Roy N, Friehs I, Cowan DB, Zurakowski D, McGowan FX, del Nido PJ. Dopamine induces postischemic cardiomyocyte apoptosis in vivo: an effect ameliorated by propofol. Ann Thorac Surg. 2006;82:2192–2199. doi: 10.1016/j.athoracsur.2006.06.086. [DOI] [PubMed] [Google Scholar]
- 25.Takeda K, Yu ZX, Nishikawa T, Tanaka M, Hosoda S, Ferrans VJ, Kasajima T. Apoptosis and DNA fragmentation in the bulbus cordis of the developing rat heart. J Mol Cell Cardiol. 1996;28:209–215. doi: 10.1006/jmcc.1996.0020. [DOI] [PubMed] [Google Scholar]
- 26.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–281. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- 27.Yue TL, Ma XL, Wang X, Romanic AM, Liu GL, Louden C, Gu JL, Kumar S, Poste G, Ruffolo RR, Jr, Feuerstein GZ. Possible involvement of stress-activated protein kinase signaling pathway and Fas receptor expression in prevention of ischemia/reperfusion-induced cardiomyocyte apoptosis by carvedilol. Circ Res. 1998;82:166–174. doi: 10.1161/01.res.82.2.166. [DOI] [PubMed] [Google Scholar]
- 28.Friehs I, Stamm C, Cao-Danh H, McGowan FX, del Nido PJ. Insulin-like growth factor-1 improves postischemic recovery in hypertrophied hearts. Ann Thorac Surg. 2001;72:1650–1656. doi: 10.1016/s0003-4975(01)03098-3. [DOI] [PubMed] [Google Scholar]
- 29.Marcus ML, Harrison DG, Chilian WM, Koyanagi S, Inou T, Tomanek RJ, Martins JB, Eastham CL, Hiratzka LF. Alterations in the coronary circulation in hypertrophied ventricles. Circulation. 1987;75:I19–I25. [PubMed] [Google Scholar]
- 30.Ohuchi H, Beighley PE, Dong Y, Zamir M, Ritman EL. Microvascular development in porcine right and left ventricular walls. Pediatr Res. 2007;61:676–680. doi: 10.1203/pdr.0b013e31805365a6. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi K, McGowan FX, Jr, Glynn P, Moran AM, Rader CM, Cao-Danh H, del Nido PJ. Glucose transporter upregulation improves ischemic tolerance in hypertrophied failing heart. Circulation. 1998;98(Suppl):II234–II239. discussion II240–231. [PubMed] [Google Scholar]
- 32.Braun MU, Szalai P, Strasser RH, Borst MM. Right ventricular hypertrophy and apoptosis after pulmonary artery banding: regulation of PKC isozymes. Cardiovasc Res. 2003;59:658–667. doi: 10.1016/s0008-6363(03)00470-x. [DOI] [PubMed] [Google Scholar]
- 33.Rouleau JL, Kapuku G, Pelletier S, Gosselin H, Adam A, Gagnon C, Lambert C, Meloche S. Cardioprotective effects of ramipril and losartan in right ventricular pressure overload in the rabbit: importance of kinins and influence on angiotensin II type 1 receptor signaling pathway. Circulation. 2001;104(8):939–944. doi: 10.1161/hc3401.093149. [DOI] [PubMed] [Google Scholar]
- 34.Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, Tremblay J, Schwartz K, Hamet P. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest. 1996;97:2891–2897. doi: 10.1172/JCI118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eidem BW, O'Leary PW, Tei C, Seward JB. Usefulness of the myocardial performance index for assessing right ventricular function in congenital heart disease. Am J Cardiol. 2000;86:654–658. doi: 10.1016/s0002-9149(00)01047-x. [DOI] [PubMed] [Google Scholar]
- 36.Oberhansli I, Branden G, Girod M, Friedli B. Estimation of pulmonary artery pressure by ultrasound. A study comparing simultaneously recorded pulmonary valve echogram and pulmonary arterial pressures. Pediatr Cardiol. 1982;2:123–130. doi: 10.1007/BF02424947. [DOI] [PubMed] [Google Scholar]
- 37.Tei C, Nishimura RA, Seward JB, Tajik AJ. Noninvasive Doppler-derived myocardial performance index: correlation with simultaneous measurements of cardiac catheterization measurements. J Am Soc Echocardiogr. 1997;10:169–178. doi: 10.1016/s0894-7317(97)70090-7. [DOI] [PubMed] [Google Scholar]
- 38.Choi YH, Cowan DB, Nathan M, Poutias D, Stamm C, del Nido PJ, McGowan FX., Jr Myocardial hypertrophy overrides the angiogenic response to hypoxia. PLoS One. 2008;3:e4042. doi: 10.1371/journal.pone.0004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamahara K, Min KD, Tomoike H, Kangawa K, Kitamura S, Nagaya N. Pathological role of angiostatin in heart failure: an endogenous inhibitor of mesenchymal stem-cell activation. Heart. 2009;95:283–289. doi: 10.1136/hrt.2008.152223. [DOI] [PubMed] [Google Scholar]
- 41.Friehs I, Barillas R, Vasilyev NV, Roy N, McGowan FX, del Nido PJ. Vascular endothelial growth factor prevents apoptosis and preserves contractile function in hypertrophied infant heart. Circulation. 2006;114(Suppl):I290–I295. doi: 10.1161/CIRCULATIONAHA.105.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]