

Abstract
Two new studies report that triglyceride (TG)-lowering mutations in APOC3 reduce coronary heart disease (CHD) (Crosby et al., 2014; Jørgensen et al., 2014). Here, we explore limitations of using Mendelian randomization to evaluate CHD risk, including potential confounding by the widespread use of statin therapy.
The status of plasma TG levels as a risk factor for CHD has been debated for decades. In most studies, plasma TG levels are associated with CHD, but adjusting for confounding variables (e.g., smoking, insulin resistance, and diabetes) substantially attenuates the association (Di Angelantonio et al., 2009). Two recent studies take a genetic approach to untangle the Gordian knot between TG levels and CHD (Crosby et al., 2014; Jørgensen et al., 2014).
Instead of stratifying individuals based on plasma TG levels, the studies divide participants into two groups according to their APOC3 genotypes. Plasma APOC3 and TG levels are highly correlated. Stratifying by APOC3 genotype rather than plasma level of TG circumvents confounding by factors that affect both plasma TG levels and CHD, an approach referred to as Mendelian randomization (Katan, 1986).
One study, led by Sekar Kathiresan, identified four rare variants in APOC3 that were associated with a 39% reduction in plasma TG levels (Crosby et al., 2014). The variants were then tested for association with CHD in 110,097 individuals from 15 different studies. Mutation carriers had a 40% reduction in CHD compared to noncarriers. The other study, led by Anne Tybjærg-Hansen, used a similar strategy (Jørgensen et al., 2014). They found three APOC3 variants that were associated with a 44% reduction in plasma TG levels. In a cohort of 75,725 Danes, carriers of these variants had a 41% reduction in CHD. Taken together, these findings provide compelling evidence that reducing APOC3 expression will reduce CHD risk. The question remains as to whether the reduced CHD risk in APOC3 variant carriers is due to lower plasma TG levels or to other associated factors, such as lower plasma levels of LDL cholesterol (LDL-C), APOC3, or remnant lipoproteins, or to increased levels of HDL-C.
Reductions in LDL-C are consistently associated with reduced CHD. Figure 1A plots the reduction in CHD as a function of the reduction in LDL-C in four studies in which subjects were treated for 5 years with a cholesterol-lowering statin (green line). The blue line shows the reduction in CHD in subjects with DNA variations that lower LDL-C levels. For each percentage reduction in LDL-C, the LDL-C-lowering variants produce a much greater reduction in CHD than seen in the statin trials. Presumably this reflects the fact that DNA variants lower LDL-C levels from birth, whereas statin treatment is initiated when atherosclerotic plaques have already developed.
Figure 1. Genetic and Pharmacological Reduction in LDL-C and Coronary Heart Disease.
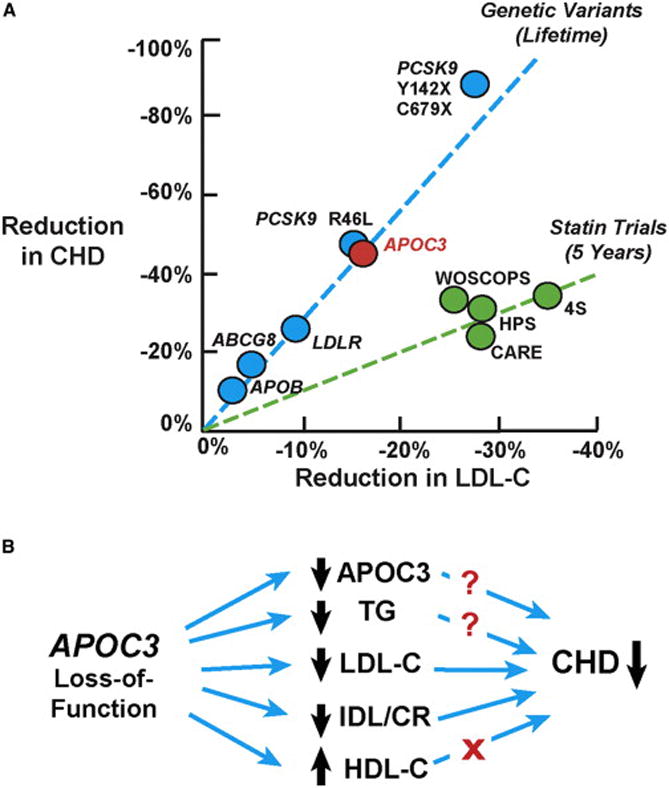
(A) Reduction in CHD risk associated with genetic variants (blue circles) and pharmacological agents (green circles) that lower plasma levels of LDL-C. Genetic variations that reduce plasma LDL-C levels are associated with a greater reduction in CHD compared to that seen in statin trials. The sources of the data shown in this figure are as follows: APOB rs754523 (PMID: 18193043), LDLR rs2228671 (PMID: 18714375), ABCG8 rs4245791 (PMID: 24657701), and PCSK9 (PMID: 16554528). The red circle represents the CHD reduction (~46%) that is predicted for a loss-of-function mutation in APOC3 (R19X) (Crosby et al., 2014; Pollin et al., 2008). WOSCOPS, The West of Scotland Coronary Prevention Study (PMID: 7566020); CARE, Cholesterol and Recurrent Events Trial (PMID: 8801446); HPS, Heart Protection Study (PMID: 12114036); 4S, The Scandinavian Simvastatin Survival Study (PMID: 7968073).
(B) Effects of APOC3 loss-of-function variants on circulating lipid and lipoprotein levels and on CHD. Proven causal links are indicated by blue arrows. Question marks indicate where causality has not been established. The red X indicates no causal relationship. IDL, intermediate density lipoprotein; CR, chylomicron remnant.
Can the reduction in CHD in the APOC3 mutant carriers be explained by a reduction in LDL-C levels? The effects of APOC3 inactivation on LDL-C levels remain inconclusive (Pollin et al., 2008; Tachmazidou et al., 2013). Pollin et al. identified a nonsense mutation (R19X) in APOC3 that is common in the Amish and is associated with a 17% reduction in plasma LDL-C levels (Pollin et al., 2008). Based on prior genetic studies, mutations that lower LDL-C by 17% should decrease CHD by ~46% (Figure 1A), which is similar to the reduction observed in the two APOC3 studies (40% and 41%).
The APOC3 carriers in the discovery cohort of the Kathiresan study (Crosby et al., 2014) had a 16% reduction in LDL-C level, which is similar to that observed in the Amish, but corresponding data were provided for only a subset of the cohorts in the CHD association study. In the Tybjærg-Hansen study (Jørgensen et al., 2014), the mean plasma LDL-C level was only 3% lower in APOC3 carriers than in noncarriers. This modest reduction in LDL-C cannot account for the dramatic reduction in CHD associated with the APOC3 variants.
A factor that may mask the contribution of plasma LDL-C levels to the reduction in CHD in APOC3 carriers is statin treatment. Statins are more likely to be prescribed to individuals who have higher plasma LDL-C levels, multiple CHD risk factors, or established CHD. If APOC3 noncarriers have higher plasma LDL-C levels and/or more CHD, they would be more likely to be treated with statins. This would lower their LDL levels and obscure differences in LDL-C levels between APOC3 carriers and noncarriers.
Could an excess of statin use among noncarriers mask the effect of APOC3 variants on LDL-C levels in these two studies? Statin use was not described in the Kathiresan study. In the Tybjærg-Hansen study, statin use was more prevalent among noncarriers than carriers, although the difference did not reach statistical significance. Given the large effects of LDL-C on CHD risk and the association between APOC3 mutations and LDL-C levels, it is premature to conclude that the reduction in CHD in APOC3 variant carriers is independent of plasma LDL-C levels. Resolution of this issue will require additional studies in statin-naive individuals or in which pre-statin LDL-C levels are used in the analysis.
Alternatively, factors other than LDL-C levels may contribute to the reduction in CHD. APOC3 has pleiotropic effects (Figure 1B). It is possible that APOC3 itself promotes atherosclerosis (Ginsberg and Brown, 2011), or alternatively, that APOC3 retards clearance of atherogenic lipoprotein remnants. If the lower levels of TG in APOC3 carriers are atheroprotective, studies using other variants that lower TG levels without affecting other CHD risk factors should replicate the association. Carriers of the APOC3 variants also have higher plasma levels of HDL-C, which are inversely associated with CHD. Ironically, Tybjærg-Hansen and Kathiresan performed the key genetic studies that showed plasma HDL-C levels are not causally related to CHD risk (Frikke-Schmidt et al., 2008; Voight et al., 2012). Thus, it is unlikely that HDL-C is conferring the cardioprotective effect of the APOC3 mutations.
The identification of other sequence variations that lower plasma TG levels without altering other risk factors would bolster the contention that TG lowering is causally linked to reduction in CHD. APOC3 may be an excellent therapeutic target for patients with severe hypertriglyceridemia. These patients are at risk of developing pancreatitis, a potentially life-threatening disorder, and the armamentarium of drugs to treat severe hypertriglyceridemia is extremely limited. The high circulating levels of APOC3 (10–20 mg/dl) may limit the efficacy of targeting APOC3 using antibody-based therapies, but strategies that target hepatic APOC3 mRNA may prove efficacious.
Homozygotes for loss-of-function mutations can provide important clues as to the safety, as well as the efficacy, of therapies targeting a specific protein. Neither study identified any individuals with total APOC3 deficiency. Identification of such individuals would provide reassurance that extreme pharmacological inhibition of APOC3 would not have unforeseen detrimental effects.
As these two studies attest, human genetic studies have fueled a resurgence of interest in lipid-modifying therapies for CHD prevention. Studies using a Mendelian randomization approach hold the promise of identifying new therapeutic agents for CHD, but they must address problems associated with pleiotropy and account for effects of statin treatment on plasma LDL-C levels. Short-term reductions in LDL-C levels due to statin therapy do not reflect lifetime exposure to this atherogenic lipoprotein. Mendelian randomization, although a powerful approach, does not eliminate all confounding factors.
References
- Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, et al. TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute N Engl J Med. 2014;371:22–31. [Google Scholar]
- Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, et al. Emerging Risk Factors Collaboration JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, Schnohr P, Grande P, Tybjaerg-Hansen A. JAMA. 2008;299:2524–2532. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- Ginsberg HN, Brown WV. Arterioscler Thromb Vasc Biol. 2011;31:471–473. doi: 10.1161/ATVBAHA.110.221846. [DOI] [PubMed] [Google Scholar]
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. N Engl J Med. 2014;371:32–41. doi: 10.1056/NEJMoa1308027. [DOI] [PubMed] [Google Scholar]
- Katan MB. Lancet. 1986;1:507–508. doi: 10.1016/s0140-6736(86)92972-7. [DOI] [PubMed] [Google Scholar]
- Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, Post W, McLenithan JC, Bielak LF, Peyser PA, et al. Science. 2008;322:1702–1705. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachmazidou I, Dedoussis G, Southam L, Farmaki AE, Ritchie GR, Xifara DK, Matchan A, Hatzikotoulas K, Rayner NW, Chen Y, et al. UK10K consortium Nat Commun. 2013;4:2872. doi: 10.1038/ncomms3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Hólm H, Ding EL, Johnson T, et al. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]