

Abstract
Alterations in vascular structure and function are a central component of demyelinating disease. In addition to blood-brain barrier (BBB) breakdown, which occurs early in the course of disease, recent studies have described angiogenic remodeling, both in multiple sclerosis tissue and in the mouse demyelinating model, experimental autoimmune encephalomyelitis (EAE). As the precise timing of vascular remodeling in demyelinating disease has yet to be fully defined, the purpose of the current study was to define the time-course of these events in the MOG35-55 EAE model. Quantification of endothelial cell proliferation and vessel density revealed that a large part of angiogenic remodeling in cervical spinal cord white matter occurs during the pre-symptomatic phase of EAE. At the height of vascular remodeling, blood vessels in the cervical spinal cord showed strong transient upregulation of fibronectin and the α5β1 integrin. In vitro experiments revealed that α5 integrin inhibition reduced brain endothelial cell proliferation under inflammatory conditions. Interestingly, loss of vascular integrity was evident in all vessels during the first 4–7 days post-immunization, but after 14 days, was localized predominantly to venules. Taken together, our data demonstrate that extensive vascular remodeling occurs during the pre-symptomatic phase of EAE and point to a potential role for the fibronectin-α5β1 integrin interaction in promoting vascular remodeling during demyelinating disease.
Keywords: experimental autoimmune encephalomyelitis (EAE), vascular remodeling, angiogenesis, blood-brain barrier (BBB), endothelial cells, α5 integrin, fibronectin
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory disease resulting in demyelination and degeneration of axons in the central nervous system (CNS). This results in disrupted nerve conduction, leading to physical and cognitive disabilities (ffrench-Constant 1994; Lassmann 1998). While the precise trigger of MS remains elusive, it is characterized pathologically by multiple inflammatory lesions of white matter that are separated in time and space. Though most of the damage is caused by infiltrating leukocytes, evidence suggests that alterations in blood vessel properties play a central role in the initiation and/or maintenance of this pathology (Roscoe et al. 2009; Seabrook et al. 2010). Blood-brain barrier (BBB) breakdown occurs at an early stage of MS (Gay and Esiri 1991; Kirk et al. 2003), and is thought to be mediated in part by the action of proteases such as matrix metalloproteinase-9 (MMP-9) (Benveniste 1997). These proteases degrade vascular basement membrane extracellular matrix (ECM) proteins such as laminins, collagen IV and fibronectin, as well as inter-endothelial tight junction proteins including claudin-5, ZO-1, and occludin (Rosenberg 2002; Yong et al. 2001). BBB breakdown results in influx of inflammatory leukocytes and leakage of serum proteins such as fibrinogen, fibronectin and vitronectin, which together further amplify the inflammatory response via their activating influence on CNS-resident microglia (Adams et al. 2007; Milner et al. 2007).
Active angiogenic remodeling has been described both in MS (Holley et al. 2010; Ludwin et al. 2001) and in the animal model of MS, experimental autoimmune encephalomyeltitis (EAE) (Kirk et al. 2004; Roscoe et al. 2009; Seabrook et al. 2010), resulting in increased vascular density. However, it is currently unknown whether these new vessels play a beneficial or harmful role in MS progression. One school of thought is that vascular remodeling is an integral part of the pathogenic process, whereby an abnormal vascular remodeling response leads to the formation of leaky angiogenic cerebral blood vessels (Holley et al. 2010; Roscoe et al. 2009; Seabrook et al. 2010).
However, counter to this argument, recent studies have described hypoxic-like injury within MS lesions (Lassmann 2003; Trapp and Stys 2009), suggesting that hypoxia induces new vessel formation, which may be part of an endogenous protective response similar to that described in other neurodegenerative diseases, such as ischemic stroke (Greenberg and Jin 2005; Wei et al. 2001) and Alzheimer’s disease (Wang et al. 2011). In animal models of ischemic stroke, hypoxic preconditioning promotes a strong angiogenic response (LaManna et al. 1992), and protects against ischemic infarct (Stowe et al. 2011). This raises the notion that angiogenic remodeling may be a general protective mechanism that acts to limit subsequent neurological insults. In support of this idea, cerebral vessels in hypoxic pre-conditioned mice show elevated expression of tight junction proteins (Li et al. 2010a) and reduced leukocyte adherence post-stroke (Stowe et al. 2011), suggesting that angiogenic remodeling may promote tightening of the BBB and reduce leukocyte extravasation. More recently, Dore-Duffy and colleagues showed that chronic mild hypoxic reduced clinical severity of EAE and leukocyte infiltration into the CNS (Dore-Duffy et al. 2011).
It is a high priority to define the molecular mechanisms mediating vascular remodeling in EAE, as this could accelerate the design of clinical strategies aimed at manipulating vascular remodeling in MS. Vascular endothelial growth factor (VEGF) is elevated in MS (Proescholdt et al. 2002), and VEGF blockade inhibited angiogenesis and improved clinical score in EAE while attenuating demyelination and inflammation (Roscoe et al. 2009). Extracellular matrix (ECM) proteins also play a major role in vascular remodeling (Astrof and Hynes 2009; Silva et al. 2008). This idea is supported by our recent finding that the fibronectin-α5β1 integrin interaction drives cerebral angiogenesis by promoting brain endothelial cell proliferation during cerebral hypoxia (Li et al. 2012b). Our current study had two goals. First, characterize the precise time-course of angiogenic remodeling at different stages of EAE, defining changes in vascular density, endothelial cell proliferation, and BBB integrity. Second, as angiogenic vessels in the hypoxic and ischemic CNS show strong upregulation of fibronectin and the α5β1 integrin (Li et al. 2012a; Milner et al. 2008a), determine whether this also occurs in EAE.
MATERIALS AND METHODS
Animals
The studies described have been reviewed and approved by The Scripps Research Institute Institutional Animal Care and Use Committee. Wild-type C57Bl/6 was maintained under pathogen-free conditions in the closed breeding colony of The Scripps Research Institute.
Experimental autoimmune encephalomyelitis (EAE)
EAE was performed using a protocol and materials provided by Hooke Laboratories (Lawrence, MA). Briefly, 8–10 week old C57Bl/6 female mice were immunized sub-cutaneously with 100 μl of 1 mg/ml MOG33-35 peptide emulsified in complete Freud’s adjuvant (CFA) containing 2 mg/ml Mycobacterium tuberculosis in both the base of the tail and upper back. In addition, on days 0 and 1, mice also received an intraperitoneal injection of 275 ng pertussis toxin. Control mice received CFA not containing MOG peptide. This protocol leads to robust induction of clinical EAE on days 12–14 following immunization. Animals were monitored daily for clinical signs and scored as follows: 0-no symptoms; 1-flaccid tail; 2-paresis of hind limb; 3-paralysis of hind limbs; 4-quadriplegia; 5-death. Mice were euthanized at different time-points of 0, 4, 7, 14, 21 (acute symptomatic) and 35 (chronic symptomatic) days post-injection.
Immunohistochemistry and Antibodies
Immunohistochemistry was performed as described previously (Milner and Campbell, 2002), on 10 μm frozen sections of cold PBS-perfused brain and cervical spinal cord. The following monoclonal antibodies were obtained from BD Pharmingen (La Jolla, CA): rat monoclonal antibodies reactive for CD31-FITC conjugated (clone MEC13.3), MHC class II (clone M5/114.15.2) and the α5 integrin subunit (clone 5H1027 (MFR5)). Other antibodies used included: rabbit anti-fibronectin, rabbit anti-laminins, and a mouse monoclonal anti-α-smooth muscle actin (SMA)-Cy3 conjugate (clone 1A4) (from Sigma, St. Louis, MO), rabbit anti-fibrinogen (Calbiochem, Billerica, MA), rabbit anti-Ki67 (Vector Laboratories, Burlingame, CA), rat anti-MBP (Chemicon, Temecula, CA), and hamster anti-CD31 (clone 2H8; Abcam, Cambridge, MA). Secondary antibodies used included goat anti-Armenian hamster-DayLight 594 (Biolegend, San Diego, CA), anti-rat Alexa Fluor 488 (Invitrogen, Carlsbad, CA) and anti-rabbit and anti-rat Cy3 (Jackson Immunoresearch, Baltimore, PA).
Image analysis
Images were taken using a 20X objective on a Zeiss Imager M1.m (Thornwood, NY). Analysis was performed in the cervical spinal cord region. Three images were taken per region and the mean value calculated for each subject. All data analysis was performed using Perkin Elmer Volocity software (Waltham, MA). This includes quantification counts of α5 integrin and Ki67, sum area of CD31-positive vessels, and relative intensity of vascular fibronectin at different time points of EAE. Each experiment was performed with three different animals per condition, and the results expressed as the mean ± SEM. Statistical significance was assessed by using the Student’s t test, in which p < 0.05 was defined as statistically significant.
Brain endothelial cell culture
Pure cultures of mouse brain endothelial cells (BEC) were obtained as described previously (Milner et al. 2008b), with the modification that puromycin (4μg/ml, Alexis GmbH, Grunberg, Germany) was included in culture media between days 1–3 to remove contaminating cell types. Endothelial cell purity was >99% as determined by CD31 in flow cytometry. BEC were used only for the first passage.
Proliferation assays
Glass coverslips were coated with fibronectin (Sigma, 10 μg/ml) for 2 hours, then washed, and BEC plated and cultured until cells reached ~50% confluence. TNF-α was added (10 ng/ml, Genentech, San Francisco, CA) in the presence or absence of 5 μg/ml anti-α5 integrin function-blocking antibody (clone HMα5-1, BD Pharmingen) or isotype control, and BEC were cultured overnight in the presence of BrdU (Invitrogen, Carlsbad, CA), then fixed in acid/alcohol and processed for BrdU immunocytochemistry according to the manufacturer’s instructions. BEC proliferation was assessed by quantifying the number of BrdU-positive cells as a percentage of the total number of cells (Hoechst staining), and the results expressed as the mean ± SEM of four experiments. Statistical significance was assessed by using Student’s t test, in which p < 0.05 was defined as statistically significant.
RESULTS
Extensive vascular remodeling occurs in the spinal cord of pre-symptomatic EAE mice
EAE was induced in C57Bl/6 mice following immunization with MOG35-55 peptide. In keeping with findings from this lab and others, mice began developing symptoms 14 days post-immunization, clinical severity peaked around 21 days (acute symptomatic phase), and improved slightly thereafter, but never completely recovered at the experimental endpoint of 35 days (chronic symptomatic phase) (Figure 1A) (Crocker et al. 2006; Maier et al. 2002; Milner et al. 2007). To examine the vascular changes that occur in the spinal cord at the different stages of EAE, we performed immunofluorescent (IF) staining with the endothelial cell marker CD31 on frozen sections of mouse cervical spinal cord, at 0, 4 7, 14, 21 and 35 days post-immunization. By quantifying the total area of CD31+ vessels, this showed that total vascular area in the cervical spinal cord increased as early as 4 days post-immunization, and by 7 days was significantly elevated (126.0 ± 4.0% of the value of control mice, p < 0.01). This trend continued and appeared to plateau between 21–35 days, with the total vascular area at 35 days increased to 153.4 ± 18.4% of the control value (p < 0.05) (Figures 1B–C). Over the same time period, changes in vascular area were not observed in CFA control mice. This demonstrates that EAE is associated with a pronounced angiogenic remodeling response in the cervical spinal cord, culminating in increased numbers of blood vessels and a greater vessel area. Interestingly, a considerable part of this vascular remodeling occurs during the pre-symptomatic phase of EAE.
Figure 1.

Characterization of vascular remodeling in EAE. A. Clinical progression of EAE, scored as described in Materials and Methods. This experiment included 9 EAE and 3 CFA control mice. Data are expressed as the mean ± SD. B. Quantification of change in total vascular area in cervical spinal cord during development of EAE, as measured by CD31 IF and Volocity software. Experiments were performed with 3 mice per time-point, and the results expressed as the mean ± SEM. * p < 0.05, ** p < 0.01. Note that total vascular area was significantly increased 7 days post-immunization, continued to increase up to 21 days, and thereafter was maintained at greater than 50% higher than control levels. C. Representative CD31 IF images demonstrating increasing vascular density during EAE development. Scale bar =100 μm.
Cerebrovascular remodeling is associated with endothelial cell proliferation
To determine if the increased vascular area is the result of an active angiogenic response we performed dual-IF staining with the endothelial cell marker, CD31 and the cell proliferation marker Ki67. This revealed the presence of proliferating CD31-positive endothelial cells in remodeling vessels in the cervical spinal cord of EAE mice, but an absence of CD31/Ki67 dual-positive cells in control mice (0 day) (Figure 2A–B). Quantification revealed that the number of CD31/Ki67 dual-positive cells increased during the development of EAE, reached a peak 14 days post-EAE induction (6.3 ± 0.7 CD31/Ki67 dual-positive cells/field compared to 0 in control mice, p < 0.001), and then diminished during the acute and chronic phases (Figure 2C). These findings demonstrate an active angiogenic response in the EAE spinal cord, and reveal that the major angiogenic/endothelial proliferative response in EAE occurs prior to the development of symptoms. This results in an increased and sustained blood vessel density during the acute and chronic phases of EAE. To determine whether endothelial proliferation occurs in white or grey matter, we analyzed two adjacent sections, and stained one for myelin basic protein (MBP), and the other for CD31/Ki67. As shown in Figure 3A–B, this revealed that CD31/Ki67 dual-positive proliferating endothelial cells were found predominantly in MBP-positive white matter (quantified in Figure 3E).
Figure 2.

Endothelial cell proliferation during EAE. A. Dual IF for CD31 (Alexa Fluor-488, green) and Ki67 (Cy3, red) was performed on frozen sections of EAE cervical spinal cord taken from 0, 4, 7, 14, and 21 days post-immunization. Scale bar =100 μm. Note that control tissue contains no Ki67+ cells, but these gradually increase with development of EAE, reaching a peak after 14 days. B. CD31/Ki67 dual IF high power images from the 7 and 14 day time-points, showing endothelial cell proliferation within remodeling blood vessels (see arrows). Scale bar = 25 μm. C. Quantification of endothelial cell proliferation during EAE development. Data points represent the mean ± SEM of 3 experiments. Note that endothelial cell proliferation peaks at 14 days post-immunization, then declines. * P<0.05, *** p < 0.01.
Figure 3.

EAE-induced angiogenesis occurs in myelinated regions of the cervical spinal cord. Adjacent sections of 7 day post-immunized cervical spinal cord were stained with either: (A) MBP (Alexa Fluor-488, green) or (B) CD31 (Alexa Fluor-488, green)/Ki67 (Cy3, red) dual-IF. Scale bar = 100 μm. C shows a magnified inset (scale bar = 50 μm) taken from panel B. D. Further examples of Ki67-positive proliferating endothelial cells within angiogenic blood vessels. Scale bar = 25 μm Note that MBP staining demarcates the white matter (WM), and that within the cervical spinal cord, endothelial cell proliferation occurs predominantly in the myelinated region. E. Quantification of endothelial cell proliferation within the grey and white matter at different stages of EAE development. Data points represent the mean ± SEM of 3 experiments. Note that endothelial cell proliferation occurs predominantly in the white matter, and increases up to the 14 day time-point. * P<0.05, ** p < 0.01.
BBB integrity is lost early in EAE
As BBB breakdown is an early event in MS (Gay and Esiri 1991; Kirk et al. 2003), we next examined the time-course of vascular leakage in EAE by performing dual-IF for CD31 and fibrinogen. As shown in Figures 4A and 4D, fibrinogen staining in CFA control mice (0 day) showed a perfectly overlapping pattern with the endothelial marker CD31, indicating that all fibrinogen was intravascular. However, as early as 4 and 7 days post-immunization, a disconnect appeared between CD31 and fibrinogen staining in EAE mice, in that fibrinogen staining extended beyond that of CD31, demonstrating loss of BBB integrity. Interestingly, in the spinal cord of 4 and 7 day post-immunized mice, fibrinogen leak was evident in all size vessels, but with increasing time (14 days onwards), the fibrinogen leak became more restricted to larger vessels. This is well illustrated in Figure 4, which shows that at the 14 day, 21 day (acute), and 35 day (chronic) time-points, many large vessels showed fibrinogen leakage, indicating BBB breakdown, but the integrity of smaller vessels remained intact. Dual-IF with fibrinogen and another vascular marker, laminin, which demarcates the glia limitans, confirmed these findings (Figure 4B). To determine which vessels leak in the acute and chronic symptomatic phases of EAE, we next performed dual-IF with fibrinogen and alpha-smooth muscle actin (α-SMA), as α-SMA is expressed at high levels in arterioles, and but is barely detectable in venules (Figure 5A). This analysis revealed that both in the acute and chronic symptomatic stages of EAE (21 and 35 days respectively), diffuse extravascular fibrinogen staining was more strongly associated with venules (arrowheads) than arterioles (arrows) (Figure 5B and quantified in Figure 5C). Furthermore, dual-IF with laminin and MHC class II, a marker of infiltrating leukocytes, showed that in chronic EAE (day 35), infiltrating leukocytes were closely associated with post-capillary venules (Figure 6). Interestingly, leukocyte accumulation in these vessels was associated with marked reduction in laminin staining of vessel walls (see arrow and arrowhead), consistent with the notion that leukocytes secrete MMPs to degrade vascular basement membrane proteins as part of the extravasation process (Gidday et al. 2005; Kelly et al. 2006)
Figure 4.

Time-course of blood-brain barrier (BBB) disruption during EAE development. A. Dual IF for CD31 (Alexa Fluor-488, green) and fibrinogen (Cy3, red) was performed on frozen sections of cervical spinal cord taken from 0, 4, 7, 14, 21, and 35 days post-immunization. B. Dual IF for laminin (Alexa Fluor-488, green) and fibrinogen (Cy3, red). C. Dual IF for CD31 (Alexa Fluor-488, green) and MHC II (Cy3, red). Scale bar (in A–C) = 100 μm. D. Higher power (magnified) images taken from panels A and B. Scale bar = 50 μm. Note that no extravascular fibrinogen was evident in control tissue, and that while in 4 and 7 day EAE spinal cord, fibrinogen leak was evident in all size vessels, with increasing time (14 days onwards), fibrinogen leak was largely restricted to bigger vessels (see arrows). In contrast to fibrinogen leakage, leukocyte infiltration was not evident until day 14 post-immunization (see arrows in C).
Figure 5.

Fibrinogen leakage in the acute and chronic stages of EAE is largely restricted to venules. A. Dual IF for CD31 (Alexa Fluor-488, green) and α-SMA (Cy3, red) was performed on frozen sections of cervical spinal cord. Scale bar =100 μm. Note that α-SMA strongly labels arterioles (labeled A) but is barely detectable in venules (labelled V). B. Dual IF for fibrinogen (Alexa Fluor-488, green) and α-SMA (Cy3, red) was performed on frozen sections of cervical spinal cord taken from 21 (acute) and 35 day (chronic) EAE tissue. Scale bar =100 μm. Note that in the acute and chronic stages of EAE, diffuse extravascular fibrinogen leakage was predominantly associated with venules (arrowheads), rather than arterioles (arrows). C. Quantification of fibrinogen leakage from arterioles and venules at different stages of EAE development. Data points represent the mean ± SEM of 3 experiments. Note that fibrinogen leakage occurs predominantly from venules, particularly in chronic EAE (35 day time-point). * P<0.05, ** p < 0.01.
Figure 6.
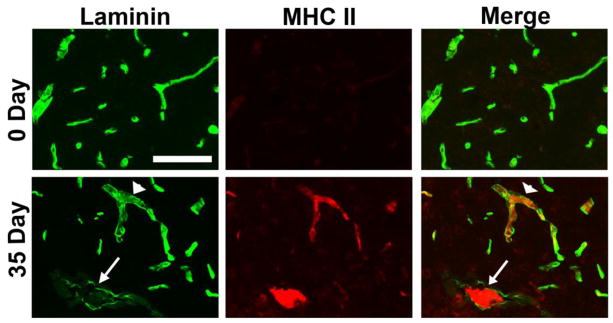
Perivascular cuffing and laminin degradation in post-capillary venules in EAE. A. Dual IF for laminin (Alexa Fluor-488, green) and MHC class II (Cy3, red) was performed on frozen sections of cervical spinal cord taken from 0 and 35 days post-immunization. Scale bar = 100 μm. Note that in chronic EAE (day 35), infiltrating leukocytes were closely associated with post-capillary venules, and that at sites of leukocyte accumulation, vascular laminin staining was greatly reduced (see arrow and arrowhead), suggestive of ongoing proteolysis of basement membrane laminin.
Blood vessels in the spinal cord of pre-symptomatic EAE mice show increased expression of fibronectin and the α5β1 integrin
In previous studies, we showed that angiogenic vessels in the hypoxic and ischemic CNS strongly upregulate expression of fibronectin and its receptor α5β1 integrin (Li et al. 2010b; Milner et al. 2008a). Now, we addressed whether this holds true for remodeling vessels in the spinal cord of EAE mice. Consistent with previous findings, vessels in the normal CNS (CFA controls; 0 day) expressed low levels of fibronectin (Figure 7A). By contrast, as early as 4 days post-immunization, vessels in the spinal cord of pre-symptomatic mice showed markedly elevated levels of fibronectin expression. Quantification of fluorescent intensity revealed that vascular fibronectin expression levels increased to reach a peak level 14 day post-immunization (146.7 ± 8.8% of control value, p < 0.05), and gradually decreased thereafter, such that fibronectin levels in the chronic phase were similar to CFA controls (quantified in Figure 7B). In a parallel manner, while endothelial cells in the CFA control spinal cord expressed only low levels of α5 integrin, this fibronectin receptor was strongly upregulated on vessels in the pre-symptomatic spinal cord, with the number of α5 integrin-positive vessels per field increasing after 4, 7 and 14 days EAE, to 184 ± 5.8% (p < 0.005), 228 ± 51.8% (p < 0.01), and 260 ± 47.3% (p < 0.005) of the control value, respectively. In a similar manner to the temporal expression pattern of fibronectin, the number of α5 integrin-positive vessels in the chronic phase of EAE had largely returned to CFA control levels (quantified in Figure 7C). This demonstrates that remodeling blood vessels in the spinal cord transiently upregulate fibronectin and the α5β1 integrin during the pre-symptomatic phase of EAE.
Figure 7.
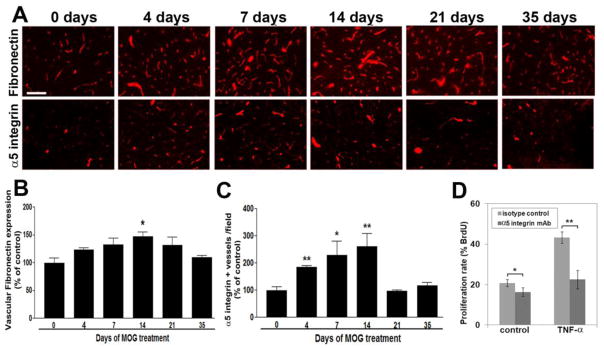
Upregulation of fibronectin and the α5 integrin on remodeling vessels during EAE. A. IF staining of fibronectin and α5 integrin on frozen sections of cervical spinal cord taken from 0, 4, 7, 14, 21, and 35 days post-immunization. Scale bar =100 μm. B. Quantification of vascular fibronectin staining was performed using Volocity software as described in Materials and Methods. Data points represent vascular expression relative to control levels, expressed as the mean ± SEM of 3 experiments. * p < 0.05. C. Quantification of α5 integrin-positive vessels was performed using Volocity software. Data points represent the number of α5 integrin-positive vessels relative to control levels, expressed as the mean ± SEM of 3 experiments. * p < 0.01, ** p < 0.005. Note that vascular expression of fibronectin and the number of α5 integrin-positive vessels increased during EAE development, to reach a peak 14 days post-immunization, and gradually decreased thereafter. D. BEC proliferation plated on fibronectin was examined over 16 hours, and expressed as the % of BEC that incorporated BrdU. All points represent the mean ± SEM of four experiments. Note that TNF-α promoted BEC proliferation, and that functional blockade of the α5 integrin reduced BEC proliferation. * p < 0.05, ** p < 0.005.
α5 integrin blockade inhibits TNF-α-driven brain endothelial cell proliferation
Our previous studies described increased endothelial expression of α5 integrin on cerebral vessels, both in mild hypoxia and following cerebral ischemia (Li et al. 2012a; Milner et al. 2008a). Functional analysis of a transgenic mouse lacking endothelial α5 integrin expression (α5-EC-KO mouse) revealed delayed and attenuated endothelial proliferation following mild hypoxia, suggesting an important pro-angiogenic role for endothelial α5β1 integrin (Li et al. 2012b). In light of our current findings, showing similar upregulation of endothelial α5 integrin on remodeling cerebral blood vessels during the early stages of EAE, we next examined in vitro whether α5 integrin mediates the BEC proliferative response to a pro-inflammatory stimulus. Previously, we found that TNF-α strongly stimulates BEC proliferation (Welser et al. 2010), and our current experiments, using bromodeoxyuridine (BrdU) incorporation, confirmed this finding, with TNF-α increasing proliferation (from 20.7 ± 1.8% under control conditions to 43.3 ± 2.9%, p < 0.001; see Figure 7D). Furthermore, incubation with an α5 integrin function-blocking antibody significantly reduced brain endothelial cell proliferation, both under non-stimulated conditions (from 20.7 ± 1.8% to 16.3 ± 2.1%, p < 0.05) and in response to TNF-α (from 43.3 ± 2.9% to 22.5 ± 4.6%, p < 0.005). This demonstrates that α5 integrin mediates endothelial cell proliferation, suggesting that this is an important mechanism supporting angiogenic remodeling in EAE tissue
DISCUSSION
Alterations in vascular structure and function are a central component of demyelinating disease (Gay and Esiri 1991; Kirk et al. 2003; Zlokovic 2008). MRI studies in MS patients and studies of animal models demonstrate that BBB breakdown occurs early in the course of disease, (Gay and Esiri 1991; Kirk et al. 2003). More recently, angiogenic remodeling and increased vascular density have been described in MS (Holley et al. 2010; Ludwin et al. 2001) and EAE (Kirk et al. 2004; Roscoe et al. 2009; Seabrook et al. 2010). However, to date, the precise timing of the vascular remodeling events in demyelinating disease has not been fully defined. In the current study we examined these events in the MOG35-55 mouse model of EAE. Our main findings were: (i) extensive angiogenic remodeling in the cervical spinal cord occurs during the pre-symptomatic phase of EAE, with brain endothelial cell proliferation peaking at 14 days, resulting in increased blood vessel density at later time-points, (ii) vascular remodeling changes occurred predominantly in the myelinated regions, (iii) angiogenic blood vessels showed increased expression of fibronectin and the α5β1 integrin, (iv) inhibition of the α5 integrin reduced brain endothelial proliferation in a pro-inflammatory environment (TNF-α) in vitro, and (v) loss of vascular integrity was evident in all vessels during the first 4–7 days post-immunization, but after 14 days was localized specifically to venules. Taken together, our studies demonstrate that vascular remodeling occurs early in the development of EAE, and point to a potential role for the fibronectin-α5β1 interaction in driving this process.
The timing of vascular changes in demyelinating disease
Alterations in blood vessel form and function have been documented for over 140 years. In his histological studies, Rindfleisch observed that MS lesions were commonly associated with abnormal-looking blood vessels (Rindfleisch 1872). More recently, within the last ten years, MRI studies have described enhanced cerebral blood flow within relapsing-remitting and secondary-progressive MS lesions (Rashid et al. 2004), and one of these studies documented increased blood flow three weeks prior to gadolinium enhancement of the vessels, suggesting a potential role for vascular changes early in lesion evolution (Wuerfel et al. 2004). At the histological level, Ludwin described an increased number and size of vessels within acute MS lesions, as well as evidence of endothelial proliferation at this stage (Ludwin et al. 2001). A recent study confirmed these findings, showing increased vessel density at all stages of the MS lesion, and the highest levels of endothelial cell proliferation in normal appearing white matter in MS-affected brains (Holley et al. 2010). Consistent with these findings, our results show that extensive vascular remodeling occurs in myelinated regions during the pre-symptomatic phase of EAE, and thus precede development of the MS lesion. However, a recent study in an alternative relapsing-remitting EAE model in the rat, challenged this consensus view by showing that the predominant vascular remodeling response in this particular model occurs during the relapsing phase of disease (Seabrook et al. 2010). This reinforces the need for caution when interpreting data from studies using different species and/or different animal models.
Is angiogenic remodeling in demyelinating disease a good or a bad thing?
Despite overwhelming evidence that vascular remodeling is an integral part of demyelinating disease in MS (Holley et al. 2010; Ludwin et al. 2001), and EAE (Kirk et al. 2004; Roscoe et al. 2009; Seabrook et al. 2010), it has yet to be determined whether this is harmful or beneficial to the patient. The current consensus leans strongly towards the idea that vascular remodeling in demyelinating disease is destructive and part of the pathogenesis of demyelinating lesions (Holley et al. 2010; Kirk et al. 2004; Roscoe et al. 2009). Early in the evolution of lesions, a hypoxic-like state exists as a result of loss of efficient saltatory conduction leading to increased energy demand (Lassmann 2003; Trapp and Stys 2009), which in turn leads to release of angiogenic growth factors, such as VEGF (Roscoe et al. 2009). Significantly, VEGF blockade inhibited angiogenesis and improved clinical score in EAE while attenuating demyelination and inflammation (Roscoe et al. 2009), thus providing the first evidence that vascular remodeling contributes to demyelinating pathogenesis, and that blockade of angiogenic factors could be beneficial in the treatment of MS. Our finding, that vascular remodeling precedes the development of acute demyelinated lesions is certainly consistent with a pathogenic role for vascular remodeling. However, evidence from other neurological diseases suggests that a state of hypoxic-induced angiogenic remodeling might actually be beneficial. For instance, in mice models of ischemic stroke, hypoxic pre-conditioning promotes a strong angiogenic response, which results in smaller ischemic infarcts (Miller et al. 2001; Stowe et al. 2011). Taken with the finding that VEGF-induced angiogenesis ameliorates cognitive decline in a mouse model of Alzheimer’s disease (Wang et al. 2011), this suggests that at least in these conditions, angiogenic remodeling protects against subsequent neurological insult. Interestingly, recent experiments in the EAE model showed that hypoxic pre-conditioning reduced clinical severity of EAE as well as reducing leukocyte infiltration (Dore-Duffy et al. 2011). Thus, while the timing of vascular remodeling, early in demyelinating disease, is consistent with a causative role in disease pathogenesis, at the current time, it cannot be excluded that certain aspects of this remodeling might be protective. It is most likely that vascular remodeling during demyelinating disease encompasses both harmful (e.g.: BBB breakdown) and beneficial (e.g.: formation of new blood vessels) components. We have shown in the chronic hypoxia model of cerebrovascular remodeling, that angiogenesis proceeds in the absence of BBB breakdown, resulting in increased tight junction protein expression (Li et al. 2010a). This demonstrates that angiogenesis is not inextricably linked to BBB breakdown. Most likely the outcome of vascular remodeling depends on the overall balance of vasculo-modulatory factors, and that in comparison to the mild hypoxic situation, the acutely demyelinating region contains an excess of pro-inflammatory mediators and angiogenic factors that results in the production of incompletely assembled, leaky vessels. If this proves to be correct, then perhaps future therapeutic efforts should be based less on total blockade of vascular remodeling, and more on titrating the response to produce functional new vessels with high vascular integrity.
Upregulation of fibronectin and the α5β1 integrin on angiogenic vessels in EAE spinal cord
One of the main goals of this study was to determine whether remodeling vessels in the EAE spinal cord showed altered expression of fibronectin and the α5β1 integrin, and indeed we found this to be true. These findings are consistent with our recent work describing elevated expression levels of fibronectin and the α5β1 integrin on angiogenic vessels under conditions of cerebral hypoxia or ischemia (Li et al. 2012a; Milner et al. 2008a), though interestingly, the time-course of expression varies somewhat between the different models. In the chronic mild hypoxia model, levels of fibronectin and the α5β1 integrin peak after 4 days hypoxia and then decline (Li et al. 2010a), while in the EAE model, these levels are extended over a longer time course, peaking after 14 days, before declining. Interestingly, in both models there is a tight correlation between the peak of fibronectin/α5β1 integrin expression, and the maximum rate of endothelial proliferation. This demonstrates that in EAE, a large part of the angiogenic remodeling occurs prior to the onset of neurological symptoms.
Fibronectin is a major inducer of angiogenic remodeling in many systems, both during development, and in physiological and pathological remodeling in the adult (Astrof and Hynes 2009; Kim et al. 2000; Risau and Lemmon 1988; Tonnesen et al. 1985). Recently we demonstrated the importance of the fibronectin-α5β1 axis in cerebrovascular remodeling by showing that mutant mice lacking endothelial α5 integrin, showed a delayed and attenuated angiogenic response to cerebral hypoxia (Li et al. 2012b). Based on this finding, we suggested that manipulation of the fibronectin-α5β1 axis might provide a therapeutic approach to enhance cerebral angiogenesis in the ischemic CNS, and by inference, a similar approach could be used to regulate vascular remodeling in demyelinating disease. However, as it is currently unclear whether angiogenic remodeling has beneficial or deleterious effects in demyelinating disease, it is impossible to predict if such a strategy would involve stimulation or inhibition of α5 integrin-mediated angiogenesis. In light of the urgency of this question, we plan to address this critical gap in knowledge by examining vascular changes and EAE progression in mice lacking endothelial α5 integrin expression (α5-EC-KO transgenic mice), which show a markedly attenuated angiogenic response to cerebral hypoxia (Li et al. 2012b). Using this approach, we should be able to shed light on two key questions: (i) is α5 integrin required for mediating angiogenic changes in EAE, and if, as predicted, this turns out to be correct, (ii) use α5-EC-KO mice to delay the angiogenic process, in order to answer the question: what effect does blocking angiogenesis have on clinical progression in EAE?
Highlights.
Examined vascular remodeling in the spinal cord of the MOG35-55 EAE model
Found increased vascularity and endothelial proliferation in pre-symptomatic phase
Vascular remodeling changes occurred predominantly in myelinated regions
Remodeling vessels showed increased expression of fibronectin and α5β1 integrin
Suggests that α5β1 integrin drives endothelial division and angiogenesis in EAE
Acknowledgments
This work was supported by a Postdoctoral Fellowship Award from the American Heart Association (AB) and by the NIH RO1 grant NS060770 (RM). This is manuscript number 23012 from The Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–82. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrof S, Hynes RO. Fibronectins in vascular development. Angiogenesis. 2009;12:165–175. doi: 10.1007/s10456-009-9136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Whitmire JK, Frausto RF, Chertboonmuang P, Soloway PD, Whitton JL, Campbell IL. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am J Pathol. 2006;169:2104–16. doi: 10.2353/ajpath.2006.060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore-Duffy P, Wencel M, Katyshev V, Cleary K. Chronic mild hypoxia ameliorates chronic inflammatory activity in myelin oligodendrocyte glycoprotein (MOG) peptide induced experimental autoimmune encephalomyelitis (EAE) Adv Exp Med Biol. 2011;701:165–173. doi: 10.1007/978-1-4419-7756-4_23. [DOI] [PubMed] [Google Scholar]
- ffrench-Constant C. Pathogenesis of multiple sclerosis. Lancet. 1994;343:271–275. doi: 10.1016/s0140-6736(94)91118-5. [DOI] [PubMed] [Google Scholar]
- Gay D, Esiri M. Blood-Brain Barrier Damage in Acute Multiple Sclerosis Plaques. Brain. 1991;114:557–572. doi: 10.1093/brain/114.1.557. [DOI] [PubMed] [Google Scholar]
- Gidday JM, gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–68. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Holley JE, Newcombe J, Whatmore JL, Gutowski NJ. Increased blood vessel density and endothelial cell proliferation in multiple sclerosis cerebral white matter. Neuosci Lett. 2010;470:65–70. doi: 10.1016/j.neulet.2009.12.059. [DOI] [PubMed] [Google Scholar]
- Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol. 2006 doi: 10.1016/j.expneurol.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am J Pathol. 2000;156:1345–1362. doi: 10.1016/s0002-9440(10)65005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junction abnormality in multiple sclerosis white matter affects all calibres of vessel and is asscoaiated with blood-brain barrier leakage and active demyelination. J Pathol. 2003;201:319–327. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- Kirk S, Frank JA, Karlik SJ. Angiogenesis in multiple sclerosis: is it good, bad or an epiphenomenon? J Neurol Sci. 2004;217:125–130. doi: 10.1016/j.jns.2003.10.016. [DOI] [PubMed] [Google Scholar]
- LaManna JC, Vendel LM, Farrell RM. Brain adaptation to chronic hypobaric hypoxia in rats. J Appl Physiol. 1992;72:2238–2243. doi: 10.1152/jappl.1992.72.6.2238. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Mutiple sclerosis pathology. In: Compston A, editor. McAlpine’s multiple sclerosis. 3. Churchill Livingstone; 1998. pp. 323–58. [Google Scholar]
- Lassmann H. Hypoxia-like tissue injury as a component of multiple sclerosis lesions. J Neurol Sci. 2003;206:187–191. doi: 10.1016/S0022-510X(02)00421-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu F, Welser-Alves JV, McCullough LD, Milner R. Upregulation of fibronectin and the α5β1 and αvβ3 integrins on blood vessels within the cerebral ischemic penumbra. Exp Neurol. 2012a;233:283–291. doi: 10.1016/j.expneurol.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Welser-Alves JV, van der Flier A, Boroujerdi A, Hynes RO, Milner R. An angiogenic role for the α5β1 integrin in promoting endothelial cell proliferation during cerebral hypoxia. Exp Neurol. 2012b;237:46–54. doi: 10.1016/j.expneurol.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Welser JV, Dore-Duffy P, Del Zoppo GJ, LaManna JC, Milner R. In the hypoxic central nervous system, endothelial cell proliferation is followed by astrocyte activation, proliferation, and increased expression of the α6β4 integrin and dystroglycan. Glia. 2010a;58:1157–1167. doi: 10.1002/glia.20995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Welser JV, Milner R. Absence of the αvβ3 integrin dictates the time-course of angiogenesis in the hypoxic central nervous system: accelerated endothelial proliferation correlates with compensatory increases in α5β1 integrin expression. J Cereb Blood Flow Metab. 2010b;30:1031–1043. doi: 10.1038/jcbfm.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwin SK, Henry JM, McFarland HF. Vascular proliferation and angiogenesis in MS: clinical and pathogenic implications. J Neuropath Exp Neurol. 2001;60:505. [Google Scholar]
- Maier J, Kincaid CL, Pagenstecher A, Campbell IL. Regulation of signal transducer and activator of transcription (STAT) and suppressor of cytokine signaling (SOCS) gene expression in the brain of mice with astrocyte-targeted production of IL-12 and experimental autoimmune encephalomyelitis. Am J Pathol. 2002;160:271–88. doi: 10.1016/S0002-9440(10)64371-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport. 2001;12:1663–9. doi: 10.1097/00001756-200106130-00030. [DOI] [PubMed] [Google Scholar]
- Milner R, Crocker SJ, Hung S, Wang X, Frausto RF, Del Zoppo GJ. Fibronectin- and Vitronectin-Induced Microglial Activation and Matrix Metalloproteinase-9 Expression Is Mediated by Integrins α5β1 and αvβ5. J Immunol. 2007;178:8158–8167. doi: 10.4049/jimmunol.178.12.8158. [DOI] [PubMed] [Google Scholar]
- Milner R, Hung S, Erokwu B, Dore-Duffy P, LaManna JC, del Zoppo GJ. Increased expression of fibronectin and the α5β1 integrin in angiogenic cerebral blood vessels of mice subject to hypobaric hypoxia. Mol Cell Neurosci. 2008a;38:43–52. doi: 10.1016/j.mcn.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner R, Hung S, Wang X, Berg G, Spatz M, del Zoppo G. Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke. 2008b;39:191–197. doi: 10.1161/STROKEAHA.107.486134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proescholdt MA, Jacobson S, Tresser N, Oldfield EH, Merrill MJ. Vascular endothelial growth factor is expressed in multiple sclerosis plaques and can induce inflammatory lesions in experimental allergic encephalomyelitis rats. J Neuropath Exp Neurol. 2002;61:914–925. doi: 10.1093/jnen/61.10.914. [DOI] [PubMed] [Google Scholar]
- Rashid W, Parkes LM, Ingle GT, Chard DT, Toosy AT, Altmann DR, Symms MR, Tofts PS, Thompson AJ, Miller DH. Abnormalities of cerebral perfusion in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2004;75:1288–93. doi: 10.1136/jnnp.2003.026021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindfleisch E. In: Pathological Histology: an introduction to the study of pathological anatomy. Klonan WC, Miles FT, translators. London: Trubner and Co; 1872. pp. 652–8. [Google Scholar]
- Risau W, Lemmon V. Changes in the Vascular Extracellular Matrix during Embryonic Vasculogenesis and Angiogenesis. Dev Biol. 1988;125:441–450. doi: 10.1016/0012-1606(88)90225-4. [DOI] [PubMed] [Google Scholar]
- Roscoe WA, Welsh ME, Carter DE, Karlik SJ. VEGF and angiogenesis in acute and chronic MOG (35–55) peptide induced EAE. J Neuroimmunol. 2009;209:6–15. doi: 10.1016/j.jneuroim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Littlewood-Evans A, Brinkmann V, Pollinger B, Schnell C, Hiestand PC. Angiogenesis is present in experimental autoimmune encephalomyelitis and pro-angiogenic factors are increased in multiple sclerosis lesions. J Neuroinflammation. 2010;7:95. doi: 10.1186/1742-2094-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R, D’Amico G, Hodivala-Dilke KM, Reynolds LE. Integrins: the keys to unlocking angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1703–13. doi: 10.1161/ATVBAHA.108.172015. [DOI] [PubMed] [Google Scholar]
- Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol. 2011;69:975–985. doi: 10.1002/ana.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnesen MG, Jenkins D, Siegal SL, Lee LA, Huff JC, Clark RA. Expression of fibronectin, laminin, and factor VIII-related antigen during development of the human cutaneous microvasculature. J Invest Dermatol. 1985;85:564–568. doi: 10.1111/1523-1747.ep12277410. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- Wang P, Xie ZH, Guo YJ, Zhao CP, Jiang H, Song Y, Zhu ZY, Lai C, Xu SL, Bi JZ. VEGF-induced angiogenesis ameliorates the memory impairment in APP transgenic mouse model of Alzheimer’s disease. Biochem Biophys Res Comm. 2011;411:620–626. doi: 10.1016/j.bbrc.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Wei L, Erinjeri JP, Rovainen CM, Woolsey TA. Collateral growth and angiogenesis around cortical stroke. Stroke. 2001;32:2179–2184. doi: 10.1161/hs0901.094282. [DOI] [PubMed] [Google Scholar]
- Welser J, Li L, Milner R. Microglial activation state exerts a biphasic influence on brain endothelial cell proliferation by regulating the balance of TNF and TGF-β1. J Neuroinflammation. 2010;7:89. doi: 10.1186/1742-2094-7-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuerfel J, Bellmann-Strobl J, Brunecker P, Aktas O, McFarland HF, Villringer A, Zipp F. Changes in cerebral perfusion precede plaque formation in multiple sclerosis: a longitudinal perfusion MRI study. Brain. 2004;127:111–9. doi: 10.1093/brain/awh007. [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]