

Abstract
The cells that line the mucosa of the human gastrointestinal tract (GI, that is, oral cavity, oesophagus, stomach, small intestine, large intestine, and rectum) are constantly challenged by adverse micro-environmental factors, such as different pH, enzymes, and bacterial flora. With exception of the oral cavity, these microenvironments also contain remnant cocktails of secreted enzymes and bacteria from upper organs along the tract. The density of the GI bacteria varies, from 103/mL near the gastric outlet, to 1010/mL at the ileocecal valve, to 1011 to 1012/mL in the colon. The total microbial population (ca. 1014) exceeds the total number of cells in the tract. It is, therefore, remarkable that despite the prima facie inauspicious mixture of harmful secretions and bacteria, the normal GI mucosa retains a healthy state of cell renewal. To counteract the hostile microenvironment, the GI epithelia react by speeding cell exfoliation (the GI mucosa has a turnover time of two to three days), by increasing peristalsis, by eliminating bacteria through secretion of plasma cell-immunoglobulins and by increasing production of natural antibacterial compounds, such as defensin-5 and lysozyme. Only recently, lysozyme was found up-regulated in Barrett’s oesophagitis, chronic gastritis, gluten-induced atrophic duodenitis (coeliac disease), collagenous colitis, lymphocytic colitis, and Crohn’s colitis. This up-regulation is a response directed to the special types of bacteria recently detected in these diseases. The aim of lysozyme up-regulation is to protect individual mucosal segments to chronic inflammation. The molecular mechanisms connected to the crosstalk between the intraluminal bacterial flora and the production of lysozyme released by the GI mucosae, are discussed. Bacterial resistance continues to exhaust our supply of commercial antibiotics. The potential use of lysozyme to treat infectious diseases is receiving much attention.
Keywords: lysozyme, chronic inflammation, oesophagus, stomach, duodenum, colon
1. Introduction
After birth, the cells that line the mucosa of the human gastrointestinal (GI) tract are increasingly threatened by adverse micro-environmental conditions, such as digestive juices of different pH, a wide variety of active enzymes, and enormous amounts of bacteria. Furthermore, cocktails of bacteria and secretions from upper organs challenge the epithelia of downstream organs along the tract.
1.1. Estimated Bacterial Flora in the GI Tract
The density of the bacterial flora in the GI tract is huge; it varies from 103/mL near the gastric outlet, to 1010/mL at the ileo-cecal valve, to 1011 to 1012/mL in the colon. The total microbial population (ca. 1014) [1] exceeds the total number of cells in the GI tract. About 500 to 1,000 different species exist, a biomass that weights about 1.5 kg [1]. Calculating an average genome size for 1,000 Escherichia coli species, the number of genes in this microbiome exceeds the total number of human genes by a factor of 100. These bacteria procreate in a luminal bolus that transports a concoction of secretions and mucus from various organs and in the glycocalix extracellular glycoproteins [2]. It is therefore remarkable that despite the prima facie inauspicious mixture of harmful secretions and bacteria, the normal GI mucosa retains a healthy state of cell renewal. To counteract the hostile microenvironment, the GI epithelia react by speeding cell exfoliation (the GI mucosa has a turnover time of two to three days), by increasing peristalsis, by eliminating bacteria through secretion of plasma cell-immunoglubulins and by increasing production of natural antibacterial compounds, such as defensin-5 and lysozyme.
1.2. The Discovery of Lysozyme
During a deliberate search for medical antibiotics, Alexander Fleming [3] discovered lysozyme, one of the natural defense substances against infection. Lysozyme, also known as muramidase or N-acetylmuramide glycanhydrolase, is a family of enzymes (EC 3.2.1.17), which damage bacterial cell walls by catalyzing hydrolysis of 1,4-beta-linkages between N-acetylmuramic acid and N-acetyl-d-glucosamine residues in a peptidoglycan, and between N-acetyl-d-glucosamine residues in chitodextrins [4]. Lysozyme, encoded by the LYZ gene [5], is an ancient enzyme whose origin goes back an estimated 400 to 600 million years [6]. Linkage studies indicate that the lysozyme M and P genes are present on the same chromosome; calculation from both partial protein sequence and phylogenetic data indicate that the duplication that gave rise to those genes occurred about 50 million years ago [5]. Lysozyme is only a generic name (v. gr. lysozyme c is a superfamily composed of 88 distinct lysozymes).
1.3. Lysozyme Is Up-Regulated in the GI Tract with Inflammation
In a series of immuno-histochemical studies lysozyme was found up-regulated in many organs of the GI undergoing chronic inflammation, such as Barrett’s oesophagitis, chronic gastritis, gluten-induced atrophic duodenitis (coeliac disease), collagenous colitis, lymphocytic colitis, ulcerative colitis (UC), and Crohn’s colitis [7,8,9,10], strongly suggesting that the associated bacterial flora plays an important role in the expression of this antimicrobial enzyme.
2. Barrett’s Oesophagus
Following protracted gastric reflux the normal squamous-cell epithelium of the distal oesophagus may undergo columnar-lined (metaplastic) transformation both in humans [11] and in non-human primates [12,13]. The metaplastic transformation in Barrett’s oesophagus includes accessory glands of oxyntic type and/or pyloric type with, or without, intercalated goblet cells [14]. The phenotype carrying goblet cells (GC), known as specialized epithelium or intestinal metaplasia (IM) is regarded, by the American College of Gastroenterology [15], a prerequisite for the histological diagnosis of Barrett’s esophagus (BE). More recently, The British Society of Gastroenterology (BSG) [16] defined Barrett’s oesophagus as a columnar lined oesophagus on biopsies taken from endoscopical areas suggestive of Barrett’s oesophagus. Thus, the presence of GC is not a sine qua non requirement for the diagnosis of Barrett’s oesophagus. This new definition by the BSG [16] has gained acceptance both in the rest of Europe [17] and in Asia [18]. Patients with gastro-oesophageal reflux often receive proton pump inhibitor medication [19]. The reduction of gastric acid secretion by this medication encourages bacterial growth in Barrett’s oesophagus and, hence, increased production of nitrosamines with secondary epithelial damage. In fact, oesophageal biopsies with of Barrett´s mucosa often show signs of on-going or past mucosal inflammation [20].
2.1. Bacteria in Barrett’s Oesophagus
Several workers demonstrated that special bacteria are more often present in oesophageal biopsies with Barrett’s oesophagus than in those without Barrett’s oesophagus [21,22,23]. Oesophageal microbiomes have been classified into two types: type I is dominated by the genus Streptococcus, concentrated in the phenotypically normal oesophagus, and type II contains a greater proportion of Gram-negative anaerobes/microaerophiles that are primarily found in oesophagitis and Barrett’s oesophagus [24]. In gastro-oesophageal reflux the residential bacterial populations contain 21 distinct species, including Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria [21]. In a more recent study with oesophageal biopsies and aspirates, McFarlane et al. [22] found in the mucosa of the Barrett’s oesophagus 46 bacterial species, belonging to 16 genera, with unique levels of Campylobacter consisus and C. rectus. Taken together, these microbiological findings denote a close association between the occurrence of columnar-lined oesophageal mucosa and the proliferation of abnormal bacteria in the oesophageal microenvironment. More recently, Liu et al. found Firmicutes (55%), Proteobacteria (20%), Bacteroidetes (14%), Fusobacteria (9%), and Actinobacteria (2%), in analysis of 138 16S rDNA sequences from 240 clones of six cases of Barrett’s oesophagus [23].
2.2. Lysozyme Is Up-Regulated in Barrett’s Oesophagitis
Increased lysozyme immunoreactivity was found In Barrett’s oesophagus; in the surface columnar epithelium, in the columnar epithelium of the pits of the glands (Figure 1), in goblet cells (Figure 2), as well as in Paneth cells in cases with intestinal metaplasia [24].
Figure 1.

Barrett’s mucosa (pyloric phenotype) showing lysozyme expression in surface epithelium, foveolar epithelium, and pyloric glands (lysozyme immunostain, ×4).
Figure 2.

Barrett’s mucosa (intestinal phenotype) showing lysozyme up-regulation, particularly in the luminal epithelial border and in goblet cells (lysozyme immunostain, ×40).
Intestinal metaplasia in Barrett’s oesophagus is not a mucosal transformation by intestinal cells, but a phenomenon of reconstruction by migrant stem cells of bone marrow origin that adapts to the hostile microenvironment [24]. Circulating bone-marrow cells would engraft into ulcerated mucosal areas affected by on-going chronic inflammation. In a murine model multi-potential progenitor cells of bone marrow origin were found to contribute to the intestinal metaplastic epithelium in the oesophagus [25]. Stem cells in the oesophagus might be instrumental in the molecular cross talk between intraluminal bacterial flora and the production of lysozyme [26]; signals released from the particular bacterial flora might induce stem cells in the Barrett’s oesophagus to generate differentiated cells rich in the antimicrobial enzyme lysozyme.
When compared to controls, lysozyme was up-regulated in all three Barrett’s mucosal phenotypes [24]. In some goblet cells, lysozyme was slightly expressed. This phenomenon might be due to a prior goblet cells-discharge of lysozyme-rich intracellular mucus into the lumen (Figure 3). Lysozyme was not expressed in parietal (oxyntic) cells, neither in Barrett’s oesophagus (Figure 4), nor in controls [24].
Figure 3.

Close-up view of Barrett’s mucosa of intestinal phenotype, showing faintly stained goblet cells and at arrow, marked lysozyme expression in the secreted mucus, in the lumen of the gland (lysozyme immunostain, ×40).
Figure 4.
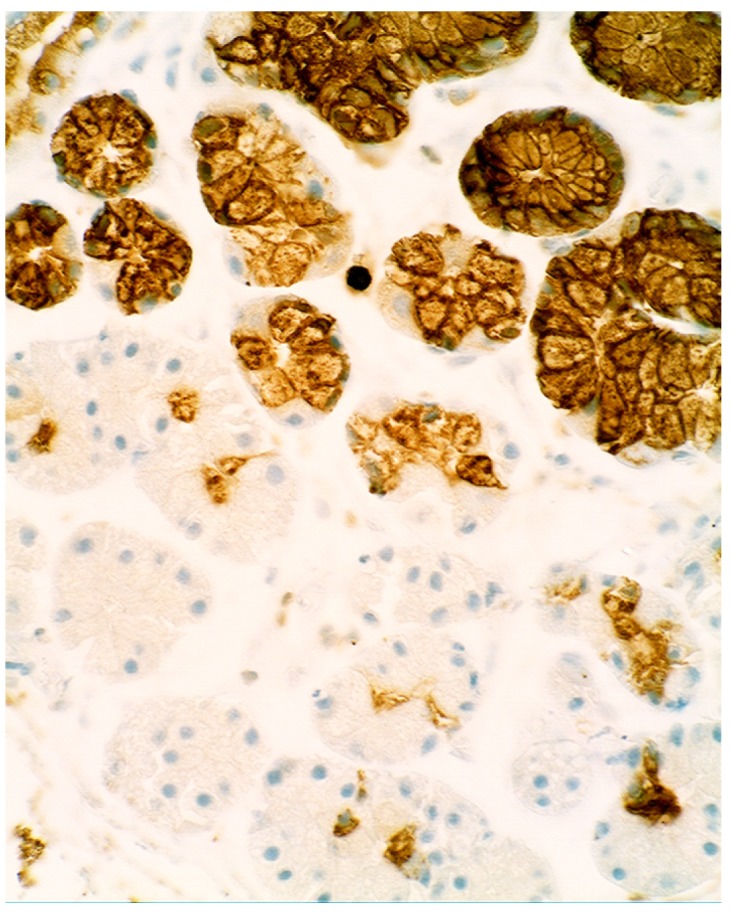
Barrett’s mucosa (oxyntic phenotype), showing lysozyme expression in neck glands. Note negative expression in oxyntic glands (×10).
3. Chronic Gastritis
Based on its topographic localization and aetiological cause(s), chronic gastritis is classified into antrum predominant gastritis (environmental or type B gastritis) and corpus predominant gastritis (autoimmune or type A gastritis) [27].
3.1. Antral Predominant Chronic Gastritis
In early work (1862), Crouvelhier [28] observed mucosal inflammation and gastric ulcers in the same stomach. Since then, many studies have been done to identify the pathogenesis of gastric mucosal inflammation and its sequels. Before 1983, ethanol, aspirin, radiation, virus, and fungi were known to induce antral chronic inflammation [29,30]. In the absence of those agents, it was observed that the most common form of gastric mucosal inflammation occurred in the lower social classes and in subjects with blue eyes [29]. However, in 1983, Warren and Marshal [31] discovered Helicobacter pylori, the aetiological agent of antral gastritis. Five years afterwards, Correa postulated that the H. pylori is the principal agent of the histological events which progress from chronic gastritis to gastric carcinoma through mucosal atrophy, intestinal metaplasia and epithelial dysplasia [32]. At the histological level, antral predominant chronic gastritis ( i.e.infiltrated by lymphocytes, plasma cells and eosinophils) is regarded as active, when infiltrated by neutrophils [33].
Approximately 50% of the world’s population are infected with these bacteria according to some studies [34].
3.2. Bacteria in Chronic Gastritis
Years ago, Giannella et al. [35] studied, in vitro, the bactericidal activity of the normal and achlorhydric gastric juice obtained from various patients. When the pH was less than 4.0, 99.9% of the bacteria were killed within 30 min, in vitro, indicating that the gastric bactericidal barrier is primarily pH-hydrochloric-acid dependent, with other constituents of gastric juice contributing little, if any, to the destruction of microorganism [35].
In H. pylori infected stomachs, patchy (multifocal) gastric mucosal inflammation is found, first at the incisura angularis [36] and subsequently in the antrum and less frequently in the corpus [34,36]. The host reacts to H. pylori by increasing the number of T- and B-lymphocytes, followed by infiltration of polymorphonuclear leucocytes. Bacterial adhesion molecules encourage attachment to foveolar cells, while bacteria proteases and ureases damage the gastric epithelium. H. pylori infection with strains that possess the cytotoxin-associated gene pathogenicity island secrete a number of bacterial products that cause a severe injury of the gastric mucosa [34,37].
3.3. Is Chronic Atrophic Gastritis Specifically Evoked by H. pylori and/or by Other Mucosal Insults?
The specific significance of H. pylori in the aetiology of chronic gastritis-atrophic gastritis has recently been questioned [38,39,40] as gastric mucosal inflammation might also be caused by factors other than H. pylori: viruses, Candida albicans, excessive alcohol use, chronic vomiting, retrograde bile reflux, autoantibodies, stress, aspirin, and anti-inflammatory drugs (v.gr. NSAID).
Corpus predominant (autoimmune) chronic gastritis is an inflammatory disease triggered by autoantibodies to parietal cells and intrinsic factor [41]. It may lead to the clinical entity, pernicious anemia. The prevalence of pernicious anemia resulting from autoimmune gastritis has been estimated as 127 cases/100,000 inhabitants in Northern Europe including United Kingdom, Denmark, and Sweden [42]. Autoimmune gastritis is characterized by the destruction of the specialized oxyntic glands of the body and fundus by specific lymphocytes. It is restricted to the gastric corpus and fundus, but a total destruction of the oxyntic mucosa only occasionally occurs. Histology reveals periglandular T-lymphocytic (CD3+) infiltration, glandular atrophy, and intestinal metaplasia restricted to the oxyntic mucosa. In advanced forms, the body mucosa is eventually replaced by pseudo-pyloric metaplasia (due to hyperplasia of the mucous neck cell). The mucosa of the antrum remains relatively spared. Autoimmune gastritis is accompanied by a linear or nodular neuroendocrine enterochromaffin-like cell hyperplasia in the corpus [43], or multiple carcinoid tumors.
3.4. Bacteria in Corpus Predominant, Autoimmune, Chronic Gastritis
The true role played by H. pylori in autoimmune gastritis remains controversial. Due to progressive mucosal atrophy microbial diversity increases with reduced acidity [44,45]. By applying temporal temperature gradient gel electrophoresis and 16S rRNA sequencing, Monstein et al. demonstrated particular microbes in the stomach other than H. pylori: Enterococcus, Pseudomonas, Streptococcus, Staphylococcus, and Stomatococcus [45]. Using large-scale 16S rRNA sequencing 128 phylotypes from eight phyla were identified [46], thus, confirming the complexity of microbiota in the gastric mucosa. In patients with antral gastritis, Li et al. found 133 phylotypes from eight bacterial phyla as well as 11 Streptococcus phylotypes, including Firmicutes phylum and Streptococcus genus from cultivated biopsies [47]. In the absence of H. pylori, other bacterial groups/species seem to be associated with corpus gastritis. Unfortunately, no patients with autoimmune chronic gastritis were included in Li et al. [47] studies.
3.5. Lysozyme Is Up-Regulated in Chronic Gastritis, Intestinal Metaplasia, Autoimmune Gastritis and Fundic-Gland Polyps
In chronic gastritis lysozyme is up-regulated in the neck region of the oxyntic mucosa, in antral pyloric glands (Figure 5) and in the surface-foveolar epithelium of the oxyntic mucosa [8]. In cases with in intestinal metaplasia, lysozyme is up-regulated in goblet cells (Figure 6), and in Paneth cells. In cases with autoimmune gastritis, lysozyme is up-regulated in pseudo-pyloric glands (Figure 7).
Figure 5.
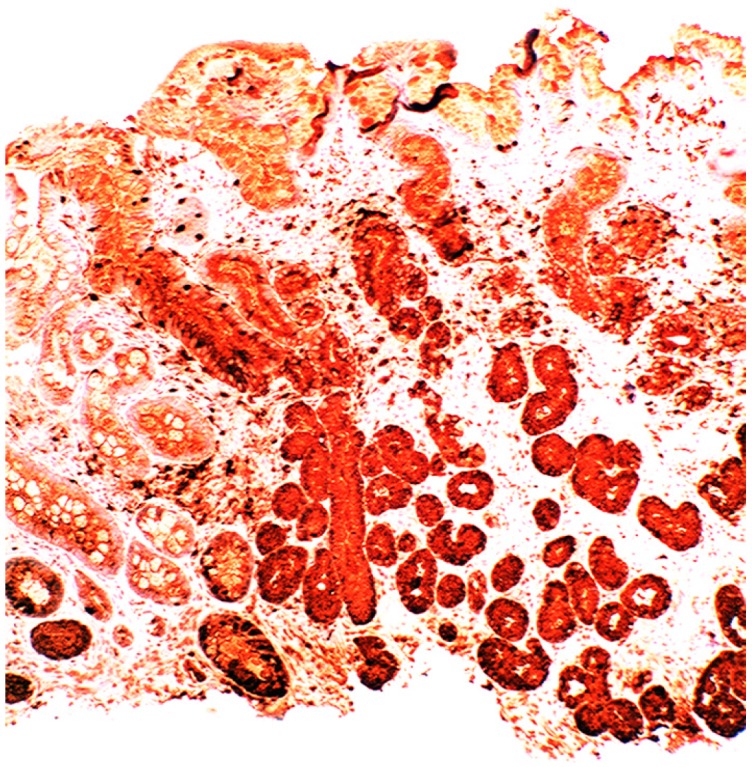
Chronic gastritis (antrum) showing lysozyme expression in surface epithelium, and in antro-pyloric glands (lysozyme immunostain, ×10).
Figure 6.

Chronic gastritis with intestinal metaplasia. Left panel: Oxyntic (corpus) mucosa showing lysozyme expression in goblet cells and in mucus neck cells. Note absence of lysoyme expression in parietal cells in the lower part of the picture (below arrows); Right panel: Antro-pyloric mucosa showing lysozyme expression in goblet cells and in Paneth cells at the bottom of the crypts (arrows, lysozyme immunostain, ×10).
Figure 7.

Autoimmune gastritis (corpus) showing lysozyme expression in pseudopyloric glands at arrows (lysozyme immuno-stain, ×4).
The increased lysozyme expression in the goblet cells of gastric intestinal metaplasia appears to be a phenomenon unrelated to the presence of Paneth cells, as lysozyme is similarly over expressed in cases with complete intestinal metaplasia (i.e., having Paneth cells) and with incomplete intestinal metaplasia (i.e., without Paneth cells). As pseudo-pyloric glands are generated by hyperplasia of the mucous neck cells it is not surprising that these cells retain the characteristics of the mucus neck cells, namely to express lysozyme [8]. Human defensin 5, secreted by Paneth cells in the small intestine, may also regulate and maintain microbial balance in the intestinal lumen [48]; in contrast the non-metaplastic atrophic gastric mucosa does not provide a similar defensive reaction. Intestinal metaplasia might evolve following a mucosal insult that affects the stem cells of the crypts of Lieberkhün [26]. Our studies [8,49] suggest that that gastric intestinal metaplasia and gastric atrophy are two different biological processes, atrophy being the result of the local destruction of glands by the chronic inflammation, and intestinal metaplasia, the consequence of an adaptive enzymatic up-regulation aimed to protect the mucosa from proliferating bacteria.
4. Intestinal Metaplasia in Disparate Geographical Regions
Years ago, Shousa et al. observed a significantly higher prevalence of intestinal metaplasia in gastric biopsies from British patients than in Yemeni patients [50]. In comparative studies of 1675 gastric biopsies and gastrectomy specimens having chronic gastritis we found intestinal metaplasia in 59% of the Japanese patients [51], in 50% of the Italian patients [52], in 32% of the Swedish patients [51], and in 13% of the Mexican patients [52] investigated, suggesting that environmental factors might trigger intestinal metaplasia in chronic gastritis. It has repeatedly been demonstrated that H. pylori is absent in areas with intestinal metaplasia or with pseudo-pyloric metaplasia. Hence, it appears safe to postulate that the aim of lysozyme overproduction in intestinal metaplasia and in pseudo-pyloric metaplasia might be to eradicate luminal proliferating bacteria in acid-deficient stomachs.
4.1. Fundic Gland Polyps
Fundic gland polyps are small (≤5 mm) nodules of the gastric mucosa characterized by microcysts lined with parietal, chief cells and occasional mucous foveolar cells [53]. Fundic gland polyps are usually found in patients with hereditary diseases, such as familial adenomatous polyposis/Gartner’s syndrome and juvenile polyposis, with non-hereditary (i.e., sporadic) gastric disorders or receiving proton-pump inhibitors. Somatic mutations in the adenomatous polyposis coli (APC) gene are frequent in fundic gland polyps from patients with familial adenomatous polyposis. In sporadic fundic gland polyps other mutations apply such as mutations of the β-catenin gene, a downstream target regulated by the APC protein. However, a common APC/β-catenin pathway seems to be involved in both syndromic and sporadic fundic gland polyps, through the targeting of different genes.
4.2. Absence of H. pylori in Fundic Gland Polyps
Several authors have noticed that H. pylori rarely proliferate in the stomach of patients with fundic gland polyps [53]. We also found absence of H. pylori infection in a series of fundic gland polyps [54]. The cause(s) for the absence of H. pylori infection might be related to lysozyme up-regulation (55]. In fact, lysozyme was found up-regulated in the surface epithelium, in the foveolar pits and in the cells that partly, or entirely, cover dilated glands of fundic gland polyps [55] (Figure 8). The overproduction of lysozyme by the fundic gland polyp epithelium concurred with the absence of H. pylori in these lesions [55].
Figure 8.

Fundic gland polyp. Marked lyozyme expression in the surface epithelium, in the foveolar pits and in the mucous-producing cells that partly cover the microcysts while the parietal cells were lysozyme negative (lysozyme immunostain, ×10).
4.3. Coeliac Disease
Coeliac disease is a common immune-mediated condition in the proximal small intestine, generated by a permanent intolerance to cereal gluten proteins in genetically predisposed individuals [56]. It often leads to mucosal atrophy. In most Western countries, the prevalence of diagnosed coeliac disease in children is 0.5%–1% [57]. In Sweden, the epidemiology of childhood coeliac disease has undergone dramatic changes over recent decade [57]; up to the early 1980s, the incidence of coeliac disease was about 1/1000, but during the mid and late 1980s, a sharp increase was observed at several paediatric clinics. Today, coeliac disease is the second most common chronic disease in Swedish children with an incidence of 3% [58].
Despite numerous sensitive and specific serological markers, duodenal biopsy remains the gold standard for diagnosing coeliac disease. Following diagnosis, a lifelong strict gluten-free diet is the treatment of choice to both restore mucosal normality and revert symptoms and possible complications.
4.4. Bacteria in Coeliac Disease
In later years, much attention has been focused on the abnormal microbiota present in the duodenum in patients with coeliac disease. Bifido bacterium, Bacteroides vulgatus, Escherichia coli, and rod-shape bacteria attached to the intestinal epithelium are higher in patients with coeliac disease than in controls whereas B. bacterium adolescentis/B. bacterium animalis lactis are more prevalent in patients with active coeliac disease than in patients with treated coeliac disease/control patients [59,60,61,62].
4.5. Lysozyme is Up-Regulated in Coeliac Disease
In the normal duodenal mucosa, Paneth cells produce lysozyme. In coeliac disease, lysozyme is up-regulated in goblet cells, in dilated crypts with mucus-metaplasia (Figure 9), a phenomenon more apparent in the bulbus [10] (Figure 10). It is not inconceivable that the lysozyme-rich mucus metaplasia mirror stem cell adaptation to the signals generated by the pathogenic bacteria present in the duodenal microenvironment [63].
Figure 9.

Chronic atrophic duodenitis (celiac disease). Villous atrophy showing lysozyme-rich mucus metaplasia in the lower part of the crypts. Note lysozyme expressing goblet cells (lysozyme immunostain, ×10).
Figure 10.

Chronic atrophic duodenitis (celiac disease). Villous atrophy showing extensive lysozyme-rich mucus metaplasia of the crypts. Note absence of Paneth cells (lysozyme immunostain, ×10).
5. Collagenous Colitis, Lymphocytic Colitis, Ulcerative Colitis and Crohn’s Colitis
In 1976, the Swedish pathologist CG Lindström reported the presence of a subepithelial amorphous band in the colonic mucosa in a patient having chronic watery diarrhoea and grossly normal colonoscopy [64]. Lindström called this setting collagenous colitis.
In 1989, Giardelo et al. [65] found increased number of intraepithelial lymphocytes in the superficial epithelium of the colon in patients having watery diarrhea and grossly normal colonoscopy, and proposed the term lymphocytic colitis for this type of microscopic colitis.
After an initial rise during the 1980s and early 1990s, the annual incidence of collagenous colitis and lymphocytic colitis has been stable during the past 15 years, in Sweden, at about 5/100,000 inhabitants for each disorder [66].
5.1. Bacteria in Microscopic Colitis
Our understanding of a possible altered bacteria flora in microscopic colitis is poor. In collagenous colitis Firmicutes and Bacteroidetes were found to dominate the microbiota with seven phylotypes among 50% of the clones: B. cellulosilyticus, B. caccae, B. thetaiotaomicron, B. uniformis, B. dorei, B. spp., and clones showing similarity to Clostridium clostridioforme [67]. More recently, Helal et al. found an association between E. coli and lymphocytic colitis [68].
5.2. Lysozyme Is Up-Regulated in Microscopic Colitis
In collagenous colitis lysozyme is up-regulated in the colonic crypts and in metaplastic Paneth cells [9] (Figure 11). In lymphocytic colitis, lysozyme is up-regulated in macrophages underlying the surface epithelium of the lamina propria [9] (Figure 12), as well as in the lower part of the crypts (Figure 13).
Figure 11.

Collagenous colitis. Marked lysozyme immunoreactivity in goblet cells in the crypts (lysozyme immunostain, ×40).
Figure 12.

Lymphocytic colitis showing marked lysozyme expression, in macrophages in the lamina propria juxtaposing the superficial epithelium (lysozyme immunostain, ×40).
Figure 13.

Lymphocyic colitis showing lysozyme expression, in the basal aspect of the crypts (arrow, lysozyme immunostain, ×20).
The increased production of lysozyme in collagenous colitis and in lymphocytic colitis supports a bacterial aetiology for these two diseases. The different mucosal cell types displaying increased production of lysozyme (epithelial vs. macrophages) substantiates the notion that collagenous colitis and lymphocytic colitis might be two different diseases.
Notably, collagenous colitis and lymphocytic colitis are also found in non-human primates having protracted intractable diarrhoea [69].
5.3. Bacteria in Inflammatory Bowel Disease
Our understanding of a possible altered bacteria flora in inflammatory bowel disease is less clear. In ulcerative colitis, bacterial diversity is reduced, including Clostridium groups IV, XIVa (Faecalibacterium prausnitzii) and Bifidobacteria and lactobacillia. On the other hand, C. difficile is increased [70,71,72,73,74,75,76,77,78,79,80,81].
In Crohn’s colitis, the number of mucosal bacteria such as Mycobacterium avium paratuberculosis, C. difficile, Ruminococcus gnavus, Enterobacteriaceae, and E. Coli are increased, while bacterial diversity, Clostridium groups IV, XIVa (F. prausnitzii), and Bifidobacteria and Lactobacillia, are reduced [76,77,78,79].
5.4. Lysozyme Up-Regulated in Inflammatory Bowel Disease
In active ulcerative colitis, lysozyme is up-regulated in metaplastic Paneth cells (left colon) and in the deep half of the crypts (Figure 14). In ulcerative colitis in remission, lysozyme is up-regulated in metaplastic Paneth cells [9] (left colon, Figure 15). No lysozyme expression is recorded in the crypts. In Crohn’s colitis lysozyme up-regulation is found in metaplastic Paneth cells (left colon), in the crypts as well as in the lamin propria mucosae [9] (Figure 16). The increased lysozyme production in the colonic mucosa in patients with inflammatory bowel disease may highlight an amplified mucosal protection against the pathogenic bacteria proliferating in the colonic microenvironment in these patients [70,71,72,73,74,75,76,77,78,79,80,81].
Figure 14.

Ulcerative colitis (active phase). Marked lysozyme immunoreactivity in the goblet cells in the crypts (lysozyme immunostain, ×10).
Figure 15.

Ulcerative colitis (in remission). Marked lysozyme immunoreactivity in hyperplastic- metaplastic Paneth cells in the bottom of the crypts of the left colon, (lysozyme immunostain, ×10).
Figure 16.

Crohn colitis: Marked lysozyme immunoreactivity in macrophages in the lamina propria and in epithelial cells at the bottom of the crypts (lysozyme immunostain, ×20).
6. Epilogue
Today, bacterial antibiotic resistance continues to exhaust our supply of effective antibiotics. The future challenge is how to solve the conundrum of bacterial resistance to antibacterial drugs. In his Presidential address, Alexander Fleming said, 80 years ago: “I choose lysozyme as the subject for this address for two reasons, firstly because I have a fatherly interest in the name and, secondly, because its importance in connection with natural immunity does not seem to be generally appreciated” [3]. Perhaps, the challenging legacy of Alexander Fleming, together with the more recent bacterial resistance to commercial antibiotics, offer the explanation for the boost in research on the potential use of lysozyme for the treatment of infectious diseases. This research includes not only laboratory and farmed animals, but also agricultural products [82,83,84,85,86,87,88,89,90,91].
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Kaper J.B., Sperandio V. Bacterial cell-to-cell signaling in the gastrointestinal tract. Infect. Immun. 2005;73:3197–3209. doi: 10.1128/IAI.73.6.3197-3209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dominguez-Bello M.G., Blaser M.J., Ley R.E., Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleming A. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. Sect. B. 1922;93:306–312. doi: 10.1098/rspb.1922.0023. [DOI] [Google Scholar]
- 4.Yoshimura K., Toibana A., Nakahama K. Human lysozyme: Sequencing of a cDNA, and expression and secretion by Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1988;150:794–801. doi: 10.1016/0006-291X(88)90461-5. [DOI] [PubMed] [Google Scholar]
- 5.Peters C., Kruse U., Pollwein R., Grzeschik K., Sippel T. The human lysozyme gene. Sequence organization and chromosomal localization. Eur. J. Biochem. 1989;182:507–512. doi: 10.1111/j.1432-1033.1989.tb14857.x. [DOI] [PubMed] [Google Scholar]
- 6.Sahoo N.R., Kumar P., Bhusan B., Bhattacharya T.K., Dayal S., Sahoo M. Lysozyme in livestock: A guide to selection for disease resistance: A review. J. Anim. Sci. Adv. 2012;2:347–360. [Google Scholar]
- 7.Rubio C.A., Lörinc E. Lysozyme is up-regulated in Barrett’s mucosa. Histopathology. 2011;58:796–799. doi: 10.1111/j.1365-2559.2011.03830.x. [DOI] [PubMed] [Google Scholar]
- 8.Rubio C.A., Befrits R. Increased lysozyme expression in gastric biopsies with intestinal metaplasia and pseudopyloric metaplasia. Int. J. Clin. Exp. Med. 2009;2:248–253. [PMC free article] [PubMed] [Google Scholar]
- 9.Rubio C.A. Lysozyme expression in microscopic colitis. J. Clin. Pathol. 2011;64:510–515. doi: 10.1136/jcp.2010.086850. [DOI] [PubMed] [Google Scholar]
- 10.Rubio C.A. Lysozyme-rich mucus metaplasia in duodenal crypts supersedes Paneth cells in celiac disease. Virchows Arch. 2011;459:339–346. doi: 10.1007/s00428-011-1129-3. [DOI] [PubMed] [Google Scholar]
- 11.Appelman H.D., Umar A., Orlando R.C., Sontag S.J., Nandurkar S., el-Zimaity H., Lanas A., Parise P., Lambert R., Shields H.M. Barrett’s esophagus: Natural history. Ann. N. Y. Acad. Sci. 2011;1232:292–308. doi: 10.1111/j.1749-6632.2011.06057.x. [DOI] [PubMed] [Google Scholar]
- 12.Rubio C.A., Dick E.J., Schlabritz-Loutsevitch N.E., Orrego A., Hubbard G.B. The columnar-lined mucosa at the gastroesophageal junction in non-human primates. Int. J. Clin. Exp. Pathol. 2009;2:481–488. [PMC free article] [PubMed] [Google Scholar]
- 13.Rubio C.A., Nilsson J.R., Owston M., Dick E.J., Jr. The length of the Barrett's mucosa in baboons, revisited. Anticancer Res. 2012;32:3115–3118. [PMC free article] [PubMed] [Google Scholar]
- 14.Spechler S., Goyal R.K. Barrett’s esophagus. N. Engl. J. Med. 1986;315:362–367. doi: 10.1056/NEJM198608073150605. [DOI] [PubMed] [Google Scholar]
- 15.Sampliner R.E. Practice guidelines on the diagnosis, surveillance, and therapy of Barrett’s esophagus. The Practice Parameters Committee of the American College of Gastroenterology. Am. J. Gastroenterol. 1998;93:1028–1032. doi: 10.1111/j.1572-0241.1998.00362.x. [DOI] [PubMed] [Google Scholar]
- 16.Playford R.J. New British Society of Gastroenterology (BSG) guidelines for the diagnosis and management of Barrett’s oesophagus. Gut. 2006;55:442–444. doi: 10.1136/gut.2005.083600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiocca R., Mastracci L., Milione M., Parente P., Savarino V. Gruppo Italiano Patologi Apparato Digerente (GIPAD) and Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP). Microscopic esophagitis and Barrett’s esophagus: The histology report. Dig. Liver Dis. 2011;43:S319–S330. doi: 10.1016/S1590-8658(11)60588-4. [DOI] [PubMed] [Google Scholar]
- 18.Takubo K., Vieth M., Aida J., Sawabe M., Kumagai Y., Hoshihara Y., Arai T. Differences in the definitions used for esophageal and gastric diseases in different countries: Endoscopic definition of the esophagogastric junction, the precursor of Barrett’s adenocarcinoma, the definition of Barrett’s esophagus, and histologic criteria for mucosal adenocarcinoma or high-grade dysplasia. Digestion. 2009;80:248–257. doi: 10.1159/000235923. [DOI] [PubMed] [Google Scholar]
- 19.Waldum H.L., Hauso Ø., Sandvik A.K. PPI-induced hypergastrinaemia and Barrett’s mucosa: The fog thickens. Gut. 2010;59:1157–1158. doi: 10.1136/gut.2010.213173. [DOI] [PubMed] [Google Scholar]
- 20.Yang L., Lu X., Nossa C.W., Francois F., Peek R.M., Pei Z., Yang L., Lu X., Nossa C.W., Francois F., et al. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137:588–597. doi: 10.1053/j.gastro.2009.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pei Z., Yang L., Peek R.M., Jr., Levine S.M., Pride D.T., Blaser M.J. Bacterial biota in reflux esophagitis and Barrett’s esophagus. World J. Gastroenterol. 2005;11:7277–7283. doi: 10.3748/wjg.v11.i46.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macfarlane S., Furrie E., Macfarlane G., Dillon J. Microbial colonization of the upper gastrointestinal tract in patients with Barrett’s esophagus. Clin. Infect. Dis. 2007;45:29–38. doi: 10.1086/518578. [DOI] [PubMed] [Google Scholar]
- 23.Liu N., Ando T., Ishiguro K., Maeda O., Watanabe O., Funasaka K., Nakamura M., Miyahara R., Ohmiya N., Goto H. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett’s esophagus. BMC Infect. Dis. 2013;13:130–136. doi: 10.1186/1471-2334-13-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubio C.A. Lysozyme is up-regulated in columnar-lined Barrett’s mucosa: A possible natural defence mechanism against Barrett’s esophagus-associated pathogenic bacteria. Anticancer Res. 2012;32:5115–5119. [PubMed] [Google Scholar]
- 25.Sarosi G., Brown G., Jaiswal K., Feagins L.A., Lee E., Crook T.W., Souza R.F., Zou Y.S., Shay J.W., Spechler S.J. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis. Esophagus. 2008;21:43–50. doi: 10.1111/j.1442-2050.2007.00744.x. [DOI] [PubMed] [Google Scholar]
- 26.Rubio C.A. Putative Stem Cells in Mucosas of the Esophago-Gastrointestinal Tract. In: Singh S.R., editor. Stem Cell, Regenerative Medicine and Cancer. Nova Science Publishers, Inc.; Haupauge, NY, USA: 2011. pp. 281–310. Chapter 10. [Google Scholar]
- 27.Misiewicz J., Tygat G., Goodwin C. The Sydney System: A new classification of gastritis. Work. Party Rep. 1990;1:10–11. [Google Scholar]
- 28.Cruveihier J. In: Traté d´anatomie pathologique genéràle. Bulletins de la Société d'anthropologie de Paris, editor. Billièr; Paris, France: 1862. p. 48. [Google Scholar]
- 29.MacDonald W., Rubin C. Gastric biopsy: A critical evaluation. Gastroenterology. 1967;53:143–170. [PubMed] [Google Scholar]
- 30.Edwards F., Coghill N. Aetiological factors in chronic atrophic gastritis. Br. Med. J. 1966;ii:1400–1415. doi: 10.1136/bmj.2.5527.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warren J., Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;I:1273–1275. [PubMed] [Google Scholar]
- 32.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:1319–1326. [PubMed] [Google Scholar]
- 33.Oh J.D., Kling-Backhed H., Giannakis M., Engstrand L.G., Gordon J.I. Interactions between gastric epithelial stem cells and Helicobacter pylori in the setting of chronic atrophic gastritis. Curr. Opin. Microbiol. 2006;9:21–27. doi: 10.1016/j.mib.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Dixon M.F., Genta R.M., Yardley J.H., Correa P. Classification and grading of gastritis: The updated Sydney System. International workshop on the histopathology of gastritis, Houston 1994. Am. J. Surg. Pathol. 1996;20:1161–1181. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Giannella R., Broitman S., Zamcheck N. Gastric acid barrier to ingested micro-organisms in man: Studies in vivo and in vitro. Gut. 1972;13:251–256. doi: 10.1136/gut.13.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubio C.A., Jaramillo E., Suzuki G., Lagergren P., Nesi G. Antralization of the gastric mucosa of the incisura angularis an its gastrin expression. Int. J. Clin. Exp. Pathol. 2009;2:65–70. [PMC free article] [PubMed] [Google Scholar]
- 37.Nakajima S., Nishiyama Y., Yamaoka M., Yasuoka T., Cho E. Changes in the prevalence of Helicobacter pylori infection and gastrointestinal diseases in the past 17 years. J. Gastroenterol. Hepatol. 2010;25:S99–S110. doi: 10.1111/j.1440-1746.2009.06214.x. [DOI] [PubMed] [Google Scholar]
- 38.Campbell D., Warren B., Thomas J., Figura N., Telford J., Sullivan P. The African enigma: Low prevalence of gastric atrophy, high prevalence of chronic inflammation in West African adults and children. Helicobacter. 2001;6:263–267. doi: 10.1046/j.1083-4389.2001.00047.x. [DOI] [PubMed] [Google Scholar]
- 39.Graham D.Y., Lu H., Yamaoka Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: Facts or medical myths. J. Dig. Dis. 2009;10:77–84. doi: 10.1111/j.1751-2980.2009.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saieva C., Rubio C.A., Nesi G., Zini E., Filomena A. Classification of gastritis in first-degree relatives of patients with gastric cancer in a high cancer-risk area in Italy. Anticancer Res. 2012;32:1711–1716. [PubMed] [Google Scholar]
- 41.Torbenson M., Abraham S.C., Boitnott J., Yardley J.H., Wu T. Autoimmune gastritis: Distinct histological and immuno-histochemical findings before complete loss of oxyntic glands. Mod. Pathol. 2002;15:102–109. doi: 10.1038/modpathol.3880499. [DOI] [PubMed] [Google Scholar]
- 42.Petersson F., Borch K., Franzén E. Prevalence of subtypes of intestinal metaplasia in the general population and in patients with autoimmune chronic atrophic gastritis. Scand. J. Gastroenterol. 2002;37:262–266. doi: 10.1080/003655202317284156. [DOI] [PubMed] [Google Scholar]
- 43.Rubio C.A., Kaufeldt A. Paucity of synaptophysin-expressing cells in Barrett’s mucosa. Histopathology. 2013;63:208–216. doi: 10.1111/his.12168. [DOI] [PubMed] [Google Scholar]
- 44.Guerre J., Vedel G., Gaudric M., Paul G., Cornuau J. Bacterial flora in gastric juice taken at endoscopy in 93 normal subjects. Pathol. Biol. 1986;34:57–60. [PubMed] [Google Scholar]
- 45.Monstein H.J., Tiveljung A., Kraft C.H., Borch K., Jonasson J. Profiling of bacterial flora in gastric biopsies from patients with Helicobacter pylori associated gastritis and histologically normal control individuals by temperature gradient gel electrophoresis and 16S rDNA sequence analysis. J. Med. Microbiol. 2000;49:817–822. doi: 10.1099/0022-1317-49-9-817. [DOI] [PubMed] [Google Scholar]
- 46.Bik E.M., Eckburg P.B., Gill S.R., Nelson K.E., Purdom E.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. USA. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X.X., Wong G.L., To K.F., Wong V.W., Lai L.H. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-Inflammatory drug use. PLoS ONE. 2009;4:e7985. doi: 10.1371/journal.pone.0007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen B., Porter E.M., Reynoso E., Shen C., Ghosh D., Connor J.T., Drazba J., Rho H.K., Gramlich T.L., Li R., et al. Human defensin 5 expression in intestinal metaplasia of the upper gastrointestinal tract. J. Clin. Pathol. 2005;58:687–691. doi: 10.1136/jcp.2004.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubio C.A. My approach to reporting a gastric biopsy. J. Clin. Pathol. 2007;60:160–166. doi: 10.1136/jcp.2006.039008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shousha S., el-Sherif A.M., el-Guneid A., Arnaout A.H. Helicobacter pylori and intestinal metaplasia: Comparison between British and Yemeni patients. Am. J. Gastroenterol. 1993;88:1373–1376. [PubMed] [Google Scholar]
- 51.Rubio C.A., Kato Y., Sugano H., Kitagawa T. Intestinal metaplasia of the stomach in Swedish and Japanese patients without ulcers or carcinoma. Jpn. J. Cancer Res. 1987;78:467–472. [PubMed] [Google Scholar]
- 52.Rubio C.A., Jessurum J., Kato Y. Low frequency of intestinal metaplasia in gastric biopsies from Mexican patients: A comparison with Japanese and Swedish patients. Jpn. J. Cancer Res. 1992;83:491–494. doi: 10.1111/j.1349-7006.1992.tb01954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shand A., Taylor A., Banerjee M., Lessels A., Coia J., Clark C., Haites N., Ghosh S. Gastric fundic gland polyps in south-east Scotland: Absence of adenomatous polyposis coli gene mutations and a strikingly low prevalence of Helicobacter pylori infection. J. Gastroenterol. Hepatol. 2002;17:1161–1164. doi: 10.1046/j.1440-1746.2002.02863.x. [DOI] [PubMed] [Google Scholar]
- 54.Rubio C.A. Plugs clog the glandular outlets in fundic gland polyps. Int. J. Clin. Exp. Pathol. 2009;3:69–74. [PMC free article] [PubMed] [Google Scholar]
- 55.Rubio C.A. Lysozyme overexpression in fundic gland polyps. Anticancer Res. 2010;30:1021–1024. [PubMed] [Google Scholar]
- 56.Dewar D.H., Ciclitira P.J. Clinical features and diagnosis of celiac disease. Gastroenterology. 2005;128:S19–S24. doi: 10.1053/j.gastro.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Ivarsson A., Högberg L., Stenhammar L., Swedish Childhood Coeliac Disease Working Group The swedish childhood coeliac disease working group after 20 years: History and future. Acta Paediatr. 2010;99:1429–1431. doi: 10.1111/j.1651-2227.2010.01832.x. [DOI] [PubMed] [Google Scholar]
- 58.Myléus A., Ivarsson A., Webb C., Danielsson L., Hernell O., Högberg L., Karlsson E., Lagerqvist C., Norström F., Rosén A., et al. Celiac disease revealed in 3% of Swedish 12-year-olds born during an epidemic. J. Pediatr. Gastroenterol. Nutr. 2009;49:170–176. doi: 10.1097/MPG.0b013e31818c52cc. [DOI] [PubMed] [Google Scholar]
- 59.Sánchez E., Donat E., Ribes-Koninckx C., Calabuig M., Sanz Y. Intestinal Bacteroides species associated with coeliac disease. J. Clin. Pathol. 2010;63:1105–1111. doi: 10.1136/jcp.2010.076950. [DOI] [PubMed] [Google Scholar]
- 60.Schippa S., Lebba V., Barbato M., di Nardo G., Totino V., Checchi M.P., Longhi C., Maiella G., Cucchiara S., Conte M.P. A distinctive “microbial signature” in celiac pediatric patients. BMC Microbiol. 2010;10:175–179. doi: 10.1186/1471-2180-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forsberg G., Fahlgren A., Hörstedt P., Hammarström S., Hernell O., Hammarström M.L. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am. J. Gastroenterol. 2004;99:894–904. doi: 10.1111/j.1572-0241.2004.04157.x. [DOI] [PubMed] [Google Scholar]
- 62.Ou G., Hedberg M., Hörstedt P., Baranov V., Forsberg G., Drobni M., Sandström O., Hammarström S. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am. J. Gastroenterol. 2009;104:3058–3067. doi: 10.1038/ajg.2009.524. [DOI] [PubMed] [Google Scholar]
- 63.Rubio C.A. Signaling pathways, gene regulation and duodenal neoplasias. In: Singh S.R., editor. Signaling, Gene Regulation and Cancer. Nova Science Publishers, Inc.; Haupauge, NY, USA: 2013. pp. 83–110. Chapter 6. [Google Scholar]
- 64.Lindström C.G. “Collagenous colitis” with watery diarrhoea: A new entity? Pathol Eur. 1976;11:87–89. [PubMed] [Google Scholar]
- 65.Giardiello F.M., Lazenby A.J., Bayless T.M., Levine E.J., Bias W.B., Ladenson P.W., Yardley J.H. Lymphocytic (microscopic) colitis. Clinico-pathologic study of 18 patients and comparison to collagenous colitis. Dig. Dis. Sci. 1989;34:1730–1738. doi: 10.1007/BF01540051. [DOI] [PubMed] [Google Scholar]
- 66.Wickbom A., Bohr J., Eriksson S., Udumyan R., Nyhlin N., Tysk C. Stable incidence of collagenous colitis and lymphocytic colitis in Örebro, Sweden, 1999–2008: A continuous epidemiologic study. Inflamm. Bowel Dis. 2013;19:2387–2393. doi: 10.1097/MIB.0b013e31829ed8cd. [DOI] [PubMed] [Google Scholar]
- 67.Gustafsson R.J., Ohlsson B., Benoni C., Jeppsson B., Olsson C. Mucosa-associated bacteria in two middle-aged women diagnosed with collagenous colitis. World J. Gastroenterol. 2012;18:1628–1634. doi: 10.3748/wjg.v18.i14.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helal T.E., Ahmed N.S., el Fotoh O.A. Lymphocytic colitis: A clue to bacterial etiology. World J. Gastroenterol. 2005;11:7266–7271. doi: 10.3748/wjg.v11.i46.7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rubio C.A., Hubbard G.B. Chronic colitis in baboons: Similarities with chronic colitis in humans. In Vivo. 2001;15:109–116. [PubMed] [Google Scholar]
- 70.Khalil N.A., Walton G.E., Gibson G.R., Tuohy K.M., Andrews S.C. In vitro batch cultures of gut microbiota from healthy and ulcerative colitis (UC) subjects suggest that sulphate-reducing bacteria levels are raised in UC and by a protein-rich diet. Int. J. Food Sci. Nutr. 2014 doi: 10.3109/09637486.2013.825700. in press. [DOI] [PubMed] [Google Scholar]
- 71.Kumari R., Ahuja V., Paul J. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. World J. Gastroenterol. 2013;19:3404–3408. doi: 10.3748/wjg.v19.i22.3404. [DOI] [PubMed] [Google Scholar]
- 72.Varela E., Manichanh C., Gallart M., Torrejón A., Borruel N., Casellas F., Guarner F., Antolin M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013;38:151–156. doi: 10.1111/apt.12365. [DOI] [PubMed] [Google Scholar]
- 73.Pilarczyk-Zurek M., Chmielarczyk A., Gosiewski T., Tomusiak A., Adamski P., Zwolinska-Wcislo M., Mach T., Heczko P., Strus M. Possible role of Escherichia coli in propagation and perpetuation of chronic inflammation in ulcerative colitis. BMC Gastroenterol. 2013 doi: 10.1186/1471-230X-13-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yukawa T., Ohkusa T., Shibuya T., Tsukinaga S., Mitobe J., Takakura K., Takahara A., Odahara S., Tajiri H. Nested culture method improves detection of Fusobacterium from stool in patients with ulcerative colitis. Jpn. J. Infect. Dis. 2013;66:109–111. doi: 10.7883/yoken.66.109. [DOI] [PubMed] [Google Scholar]
- 75.Kabeerdoss J., Sankaran V., Pugazhendhi S., Ramakrishna B.S. Clostridium leptum group bacteria abundance and diversity in the fecal microbiota of patients with inflammatory bowel disease: A case-control study in India. BMC Gastroenterol. 2013;13:20–26. doi: 10.1186/1471-230X-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gałecka M., Szachta P., Bartnicka A., Łykowska-Szuber L., Eder P., Schwiertz A. Faecalibacterium prausnitzii and Crohn’s disease—Is there any connection? Pol. J. Microbiol. 2013;62:91–95. [PubMed] [Google Scholar]
- 77.Kale-Pradhan P.B., Zhao J., Palmer J., Wilhelm S.M. The role of antimicrobials in Crohn’s disease. Expert. Rev. Gastroenterol. Hepatol. 2013;7:281–288. doi: 10.1586/egh.13.6. [DOI] [PubMed] [Google Scholar]
- 78.Nickerson K., McDonald C. Crohn’s disease-associated adherent-invasive Escherichia coli adhesion is enhanced by exposure to the ubiquitous dietary polysaccharide maltodextrin. PLoS One. 2012;7:e52132. doi: 10.1371/journal.pone.0052132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Erickson A., Cantarel B., Lamendella R., Darzi Y., Mongodin E., Pan C., Shah M., Halfvarson J., Jansson J.K. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L., Sharma Y., Anderson C., et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shanahan F. The microbiota in inflammatory bowel disease: Friend, bystander, and sometime-villain. Nutr. Rev. 2012;70:S31–S37. doi: 10.1111/j.1753-4887.2012.00502.x. [DOI] [PubMed] [Google Scholar]
- 82.Muriel-Galet V., Talbert J.N., Hernandez-Munoz P., Gavara R., Goddard J.M. Covalent immobilization of lysozyme on ethylene vinyl alcohol films for nonmigrating antimicrobial packaging applications. J. Agric. Food Chem. 2013;61:6720–6727. doi: 10.1021/jf401818u. [DOI] [PubMed] [Google Scholar]
- 83.Ibrahim H.R., Imazato K., Ono H. Human lysozyme possesses novel antimicrobial peptides within its N-terminal domain that target bacterial respiration. J. Agric. Food Chem. 2011;59:10336–10345. doi: 10.1021/jf2020396. [DOI] [PubMed] [Google Scholar]
- 84.Oliver W.T., Wells J.E. Lysozyme as an alternative to antibiotics improves growth performance and small intestinal morphology in nursery pigs. J. Anim. Sci. 2013;91:3129–3136. doi: 10.2527/jas.2012-5782. [DOI] [PubMed] [Google Scholar]
- 85.Teneback C., Scanlon T., Wargo M., Bement J., Griswold K., Leclair L.W. Bioengineered lysozyme reduces bacterial burden and inflammation in a murine model of mucoid Pseudomonas aeruginosa lung infection. Antimicrob. Agents Chemother. 2013;57:5559–5564. doi: 10.1128/AAC.00500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pridgeon J.W., Klesius P.H., Dominowski P.J., Yancey R.J., Kievit M.S. Chicken-type lysozyme in channel catfish: Expression analysis, lysozyme activity, and efficacy as immunostimulant against Aeromonas hydrophila infection. Fish Shellfish Immunol. 2013;35:680–688. doi: 10.1016/j.fsi.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 87.Cegielska-Radziejewska R., Szablewski T. Effect of modified lysozyme on the microflora and sensory attributes of ground pork. J. Food Prot. 2013;76:338–342. doi: 10.4315/0362-028X.JFP-12-075. [DOI] [PubMed] [Google Scholar]
- 88.Lamppa J.W., Tanyos S.A., Griswold K.E. Engineering Escherichia coli for soluble expression and single step purification of active human lysozyme. J. Biotechnol. 2013;164:1–8. doi: 10.1016/j.jbiotec.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rosu V., Bandino E., Cossu A. Unraveling the transcriptional regulatory networks associated to mycobacterial cell wall defective form induction by glycine and lysozyme treatment. Microbiol. Res. 2013;168:153–164. doi: 10.1016/j.micres.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 90.Scanlon T.C., Teneback C.C., Gill A., Bement J.L., Weiner J.A., Lamppa J., Leclair L.W., Griswold K.E. Enhanced antimicrobial activity of engineered human lysozyme. ACS Chem. Biol. 2010;5:809–818. doi: 10.1021/cb1001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bhavsar T., Liu M., Hardej D., Liu X., Cantor J. Aerosolized recombinant human lysozyme ameliorates Pseudomonas aeruginosa-induced pneumonia in hamsters. ACS Chem. Biol. 2010;5:809–818. doi: 10.1021/cb1001119. [DOI] [PubMed] [Google Scholar]