

Summary
A period of mild brain overgrowth with an unknown etiology has been identified as one of the most common phenotypes in autism. Here, we test the hypothesis that maternal inflammation during critical periods of embryonic development can cause brain overgrowth and autism-associated behaviors as a result of altered neural stem cell function. Pregnant mice treated with low-dose lipopolysaccharide at embryonic day 9 had offspring with brain overgrowth, with a more pronounced effect in PTEN heterozygotes. Exposure to maternal inflammation also enhanced NADPH oxidase (NOX)-PI3K pathway signaling, stimulated the hyperproliferation of neural stem and progenitor cells, increased forebrain microglia, and produced abnormal autism-associated behaviors in affected pups. Our evidence supports the idea that a prenatal neuroinflammatory dysregulation in neural stem cell redox signaling can act in concert with underlying genetic susceptibilities to affect cellular responses to environmentally altered cellular levels of reactive oxygen species.
Graphical Abstract
Highlights
-
•
Mild maternal inflammation produces brain overgrowth and autistic behaviors in pups
-
•
Maternal inflammation increases stem cell division, ROS levels, and PI3K activation
-
•
Genetic susceptibility produces even greater brain overgrowth when combined with MIR
-
•
Overgrowth and some associated abnormal behaviors can be rescued by inhibition of NOX
In this article, Kornblum and colleagues show that exposure to maternal inflammation and an altered cellular redox balance during critical periods of embryonic development can promote brain overgrowth and autism-associated behaviors as a result of altered neural stem cell function. Additionally, they demonstrate that PTEN heterozygosity can act as a genetic susceptibility to produce a more pronounced brain overgrowth phenotype.
Introduction
Macrocephaly, which is defined as having a head circumference greater than the 97th percentile on normal growth curves, can be caused by alterations in a number of genes, including the tumor-suppressor genes PTEN and TSC. Patients with these relatively rare mutations have a high risk of autism spectrum disorders (ASD) (Fidler et al., 2000; Klein et al., 2013; Zhou and Parada, 2012). The conditional deletion of PTEN in mice produces brain overgrowth due in part to a sustained increase in neural stem cell self-renewal and neurogenesis in the subventricular zone (SVZ) (Groszer et al., 2006; Gregorian et al., 2009). Both PTEN and TSC are negative regulators of the PI3K/AKT/mTOR signaling pathway, which is a central regulator of cellular growth and survival. In addition to overt macrocephaly, a more mild and pervasive brain overgrowth in ASD was recently identified in retrospective studies of head circumference (Courchesne et al., 2003; Schumann et al., 2010). Although direct evidence of a relationship between brain overgrowth and the onset of ASD pathology has yet to be demonstrated, induced brain overgrowth in some animal models has been shown to result in abnormal autism-associated behaviors (Fatemi et al., 2002; Bauman et al., 2013).
In addition to strong evidence for the genetic transmission of autism, studies have shown that certain perinatal factors increase the risk for ASD and brain overgrowth (Nordahl et al., 2013; Bill and Geschwind, 2009). These environmental factors, through interactions with genetic susceptibility during critical periods of development, have the potential to play a significant role in the etiology of ASD (Voineagu et al., 2011; Hallmayer et al., 2011). One such environmental risk factor for ASD is the maternal inflammatory response (MIR) (Patterson, 2011). Clinical studies have identified a positive association between brain overgrowth and maternal inflammation, suggesting a pathogenic link between the immune system and brain growth in ASD (Sacco et al., 2007; Nordahl et al., 2013). Although the MIR has been linked to both autism and brain overgrowth, the mechanisms underlying its potential pathological effects remain undefined.
Here, we propose a hypothesis that explains how a mild MIR can contribute to brain overgrowth by enhancing neural stem and progenitor cell proliferation through the dysregulation of cellular redox signaling. This hypothesis is based on evidence that reactive oxygen species (ROS) at nontoxic levels can increase stem cell self-renewal and neurogenesis through the reversible inactivation of PTEN protein and subsequent enhancement of PI3K pathway activation (Le Belle et al., 2011). The MIR stimulates the generation of ROS through the actions of various cytokines and activation of the NADPH oxidase (NOX) enzyme, which enhances signal transduction for many growth and trophic factors that are important for normal brain development (Beloosesky et al., 2012; Clement et al., 2010; Chen et al., 2011). We hypothesize that the MIR activates NOX, which in turn elevates cellular ROS levels and leads to increased stem and progenitor proliferation during early brain development, resulting in macrocephaly and autism. This pathological process would be expected to be enhanced by interaction with certain genetic susceptibilities, such as heterozygous PTEN mutations.
Results
Exposure to Low-Dose Lipopolysaccharide-Induced MIR Produces Brain Overgrowth in Most Offspring
Pregnant dams given a single injection with a low dose of lipopolysaccharide (LPS) on embryonic day 9 (E9) gave birth to pups with mild brain overgrowth (Figure 1A; p < 0.05). Although at birth the pups had smaller body weights, their brain sizes were not similarly smaller. Because body and brain weight track together (e.g., see Rebello and Ramos, 2006), brain size is commonly normalized to body weight (brain wet weight/body weight). Using this normalization, we found that at birth and at postnatal day 20 (P20) there was an approximate 15% increase in brain weight in mice exposed to MIR (P20; Figure 1A; p < 0.05). However, by P60 the overgrowth was no longer significant. On the other hand, if one considers the gross brain wet weight alone, the brains of MIR-exposed pups were not larger at birth, but became larger than those of controls by P20. Exposure to low-level MIR resulted in a slightly larger litter size, which led us to repeat key experiments after normalizing the litter size at birth through cross-fostering, which did not alter any of the outcome measures (Figure S1A available online). We also tested the effects of embryonic MIR exposure on postnatal brain weight in PTEN heterozygous (Het) and wild-type (WT) littermate mice. We found that Het pups from LPS-treated dams had 34%–58% greater brain weights than their WT littermates at birth, whereas Het brains were only approximately 8% larger than WT brains from vehicle-treated dams. This demonstrates a dramatic effect from the interaction of a genetic susceptibility with an environmental factor such as the MIR (Figure 1B; p < 0.01).
Figure 1.
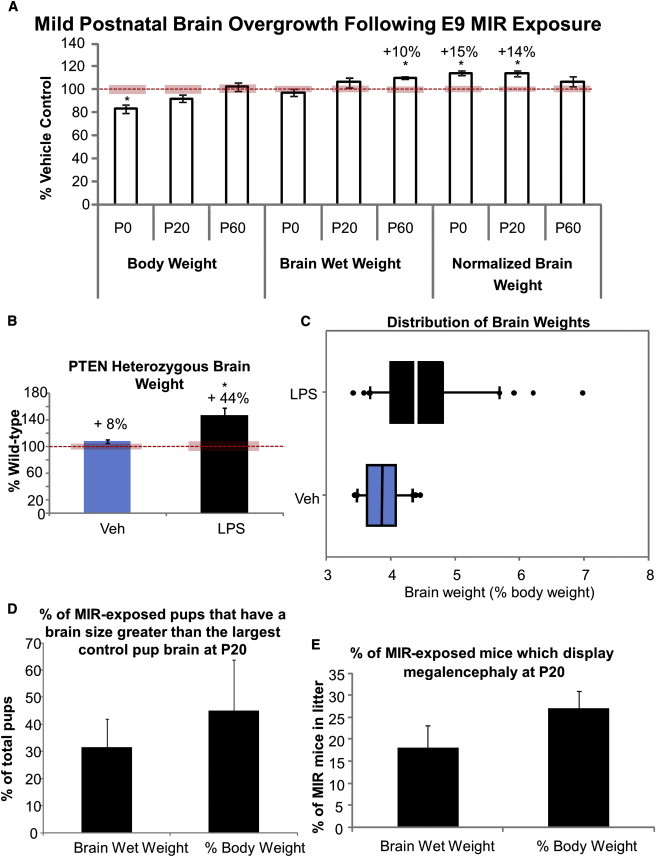
MIR Exposure Increases Brain Size Postnatally
(A) Mice body and brain weight and brain weight normalized to body weight of LPS-treated offspring at P0, P20, and P60 expressed as percent of saline-treated control.
(B) Brain wet weight after vehicle or LPS treatment in PTEN heterozygous mice (n = 5) compared with their WT littermates (n = 8).
(C) Distribution of normalized brain weights in a single litter. The bottom and top of the boxplot represent the first and third quartiles of the brain weight distribution, with the second quartile (the median) indicated by the band inside the box and outliers plotted as individual points.
(D) Percentage of pups with brain wet weights or normalized brain weights larger than the largest control brain.
(E) Percentage of mice that displayed megalencephaly (2 SD greater than the mean control brain size). Unless specified, all data are from n = 3 litters of 10–18 pups each. All data are presented as mean ± SEM. The red bars represent the normalized SEM of the controls.
Although as a group the MIR-exposed offspring displayed an average increase in brain size as compared with controls, not all of the pups in a litter experienced brain overgrowth. There was a wide distribution of brain wet weights in MIR-exposed litters. At birth, no pups from the LPS treatment group had a brain wet weight higher than the highest brain wet weight in the control pups (Figure 1C). However, by P20, about 30% of the pups had brain weights greater than the highest brain weight in the control group, and when brain size was normalized to body weight, approximately 45% of MIR-exposed pups had larger brains than the largest controls (Figure 1D). Furthermore, up to 25% of MIR-exposed mice displayed megalencephaly (2 SD above the mean for typically developing age-matched controls; Figure 1E). Therefore, the overgrowth we observed was clearly evolving postnatally, but did not universally affect all offspring. No sex differences were observed in LPS-induced brain overgrowth (Figure S1B).
Early Embryonic MIR Exposure Expands Cortical Size and Has Long-Term Stimulatory Effects on Forebrain Microglia and Proliferation in the Neurogenic SVZ Niche of Offspring
We observed an increased cortical thickness and cortical area, but no change in corpus callosum thickness in the pups from the LPS treatment group compared with controls (Figure 2A; p < 0.05). Embryonic LPS treatment resulted in a significant increase in IBA1+ microglia throughout the brain, with only rare microglia located within the SVZ itself. The increase in microglia was greater at E18 but remained significantly elevated at birth (Figures 2B and S2A; p < 0.05). It remains to be determined whether this increase in microglia plays a role in stimulating progenitor proliferation (Antony et al., 2011) or in the phagocytotic pruning of cells produced in excess following MIR exposure (Cunningham et al., 2013).
Figure 2.
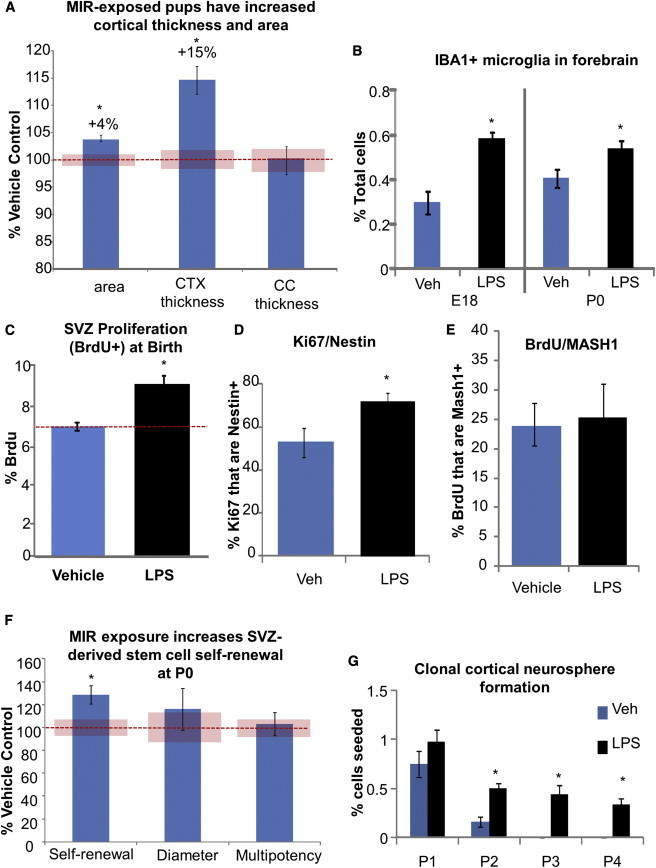
Cortical Size and SVZ Stem and Progenitor Cell Proliferation Are Increased following LPS Treatment
(A) Cortex thickness and area and corpus callosum thickness in tissue sections from P0 pup brains.
(B) Number of IBA1+ microglia in the forebrain of MIR-exposed and control offspring on E18 and P0.
(C) Stereological counts of BrdU+ cells as a percentage of total cells in the SVZ at P0 (n = 4 animals per group).
(D) Fluorescence-activated cell sorting (FACS) quantification of acutely dissociated P0 SVZ stained for Ki67 and Nestin.
(E) Stereological counts of BrdU+/MASH1+ cells in the SVZ (n = 4 per group).
(F) Sphere formation and diameter, and multipotency of clonal cultures from acutely dissociated P0 SVZ.
(G) Numbers of clonal neurospheres from postnatal cortex in control and MIR-exposed offspring at various passages. Unless specified, all data are from n = 3 litters of 10–18 pups each; ∗p < 0.05. All data are presented as mean ± SEM. The red bars represent the normalized SEM of the controls.
Our mild LPS treatment at E9 also induced a significant increase in postnatal SVZ proliferation (Figure 2C; p < 0.05) that corresponded to an increase in proliferative Nestin progenitor cells (Figures 2D, S2B, and S2C) that were also MASH1 positive (Figures 2E and S2D), indicating that a neural progenitor population underlies this MIR effect. To further clarify whether the enhanced SVZ cell division also indicated an increase in neural stem cell self-renewal, we performed a clonal density neurosphere-forming assay. Acutely dissociated SVZ cells from the MIR-exposed pups displayed significantly enhanced self-renewal (clonal sphere number) and increased proliferation (sphere diameters), and they were similarly multipotent, producing neurons, astrocytes, and oligodendrocytes (Figure 2F; p < 0.05). These effects were also not gender specific (Figure S2E). Subsequent serial clonal passages after the primary clonal culture did not maintain this increased self-renewal (data not shown), indicating that the cells were likely stimulated by their in vivo environment—an effect that was not sustained during long-term culture in the same high-growth factor environment in vitro. Early postnatal, cortical-derived progenitor cells typically have a limited self-renewal and neurogenic capacity (Seaberg et al., 2005). However, we found that cortical progenitors derived from MIR-exposed pups at birth had a significantly prolonged self-renewal capacity compared with vehicle-treated pups, suggesting the possibility that ectopic stem cells were present in the cortex or that there was delayed maturation of cells after MIR (Figure 2G; p < 0.05).
MIR Exposure Is Associated with Abnormal Behaviors in Offspring
We examined the MIR-exposed mice for several autism-associated behaviors. Neonatal (P7) pups from the LPS treatment group displayed a deficit in ultrasonic vocalizations when separated from their dam (Figure 3A; p < 0.05). In a three-chamber social interaction test, the MIR-exposed mice showed lower levels of interaction with the mouse in the social chamber and more time sniffing an empty cup compared with controls. MIR-exposed mice also preferred to spend most of their time in the chamber with the empty cup and less time in the chamber with the mouse (Figure 3B; p < 0.01). Additionally, unlike controls, MIR-exposed offspring showed less preference for social novelty, spending less time interacting with a new, unknown mouse (Figure S3; p < 0.01). Similarly, the elevated plus maze indicated that MIR mice had a higher level of anxiety due to a decrease in the amount of time spent in the open arms of the maze and an increase in the latency to enter the open arms during the testing period (Figure 3C; p < 0.05). At P60 the MIR-exposed mice displayed excessive repetitive grooming behavior, which was highly correlated with brain size (Figure 3D; p < 0.01, r = 0.918). Behavioral deficits were not gender dependent (data not shown). Finally, the MIR mice showed normal postnatal weight gain (Figure 3E) and normal mobility during behavioral testing (Figure 3F), demonstrating that their behavioral deficits were not due to gross motor problems.
Figure 3.
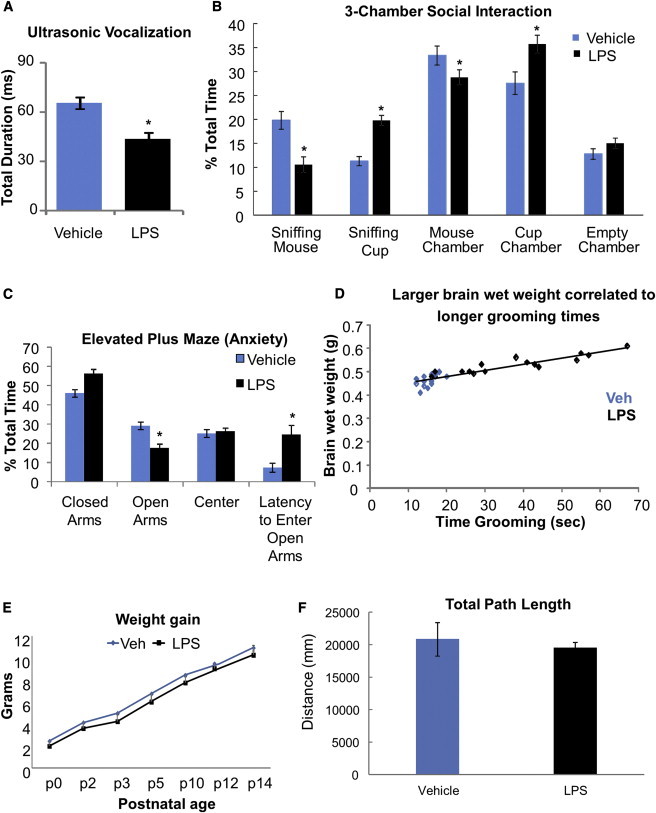
MIR-Exposed Mice Display Autism-Associated Deficits in Behaviors
(A) Mean duration of ultrasonic vocalizations by P7 pups when separated from the dam.
(B) Mean percent total time spent in each condition in the three-chamber social interaction test.
(C) Mean percent total time spent in each maze location and the latency to enter open arms in the elevated plus maze test.
(D) Correlation between brain size and repetitive grooming behavior. Data shown were obtained from each pup from two litters.
(E) Average weight gain in control and treated animals over 14 postnatal days.
(F) Average total path length traveled during the three-chamber social interaction test. n = 16 mice per group; ∗p < 0.05. All data are presented as mean ± SEM.
MIR Induces Dysregulation of the Redox Balance in the SVZ
Endogenous cellular ROS levels were elevated in the postnatal SVZ of MIR-exposed pups in both males and females (Figure 4A). In vivo inhibition of the ROS-generating enzyme NOX following LPS treatment reduced brain overgrowth in WT and PTEN heterozygous offspring but produced brain undergrowth in some pups (Figures 4B and S4A; p < 0.01). Inhibition of NOX from the time of LPS injection on E9 until birth also prevented the increase in forebrain microglia (Figures 4C and S4B; p < 0.01) and SVZ stem cell self-renewal (Figure 4D; p < 0.01). We observed an increase in AKT and pS6 activation in acutely dissected SVZ from MIR-exposed pups, which could be rescued by prenatal inhibition of NOX with apocynin (Figures 4E and S4C). Finally, we tested the ability of NOX inhibition to rescue the two most significantly abnormal behaviors caused by MIR exposure: vocalization and grooming. We found that it could rescue the excessive grooming behavior at P20, but not the early vocalization deficit (Figures 4F and 4G; p < 0.05). It is possible the daily, prenatal apocynin treatment paradigm we utilized was overly aggressive, resulting in smaller brains in some mice at birth. If mild brain undergrowth also affects early vocalization behavior, this could explain why we failed to see rescue of abnormal behavior until older ages, at which time brain size was normalized in treated offspring. This highlights the difficulty of developing effective therapeutic interventions that aim to restore a normal redox balance in the developing brain, since both too much and too little cellular ROS can have deleterious effects (Le Belle et al., 2011).
Figure 4.
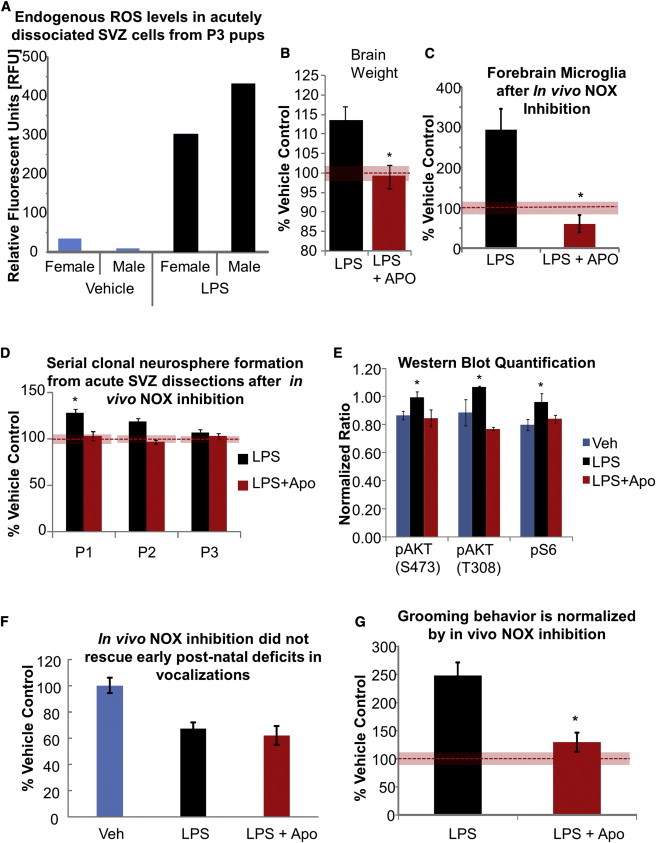
MIR-Exposed Pups Have Dysregulated SVZ ROS Levels, which Contributes to Brain Overgrowth
(A) Endogenous ROS levels in SVZ cells measured by DCFDA dye on P3 (results are pooled samples of three or four mice per group).
(B) Brain weights (corrected for body weight) of MIR-exposed pups with and without NOX inhibition (APO) expressed as a percentage of vehicle controls.
(C) IBA1+ microglia present at birth in the forebrain of MIR-exposed pups with and without NOX inhibition, determined by FACS acquisition and expressed as a percentage of vehicle controls.
(D) Mean number of clonal neurospheres from the SVZ of MIR-exposed pups as a percentage of vehicle control at each passage indicated.
(E) AKT and S6 activation (mean intensity normalized to beta-actin signal) in SVZ cells from MIR-exposed pups with and without in vivo NOX inhibition measured at birth by western blot (n = 3 per group).
(F) Duration of ultrasonic vocalizations of treated and control pups (n = 6 per group). Results are the mean percent vehicle controls.
(G) Grooming duration in MIR-exposed offspring with and without NOX inhibition. Results are the mean percent vehicle controls, n = 16 per group. Unless specified, all data are from n = 3 litters of 10–18 pups each; ∗p < 0.05. All data are presented as mean ± SEM. The red bars indicate the SEM of the normalized control data.
Discussion
The fact that mild brain overgrowth is the most common endophenotype in ASD suggests that the mechanism underlying this pervasive accelerated growth could result from the convergence of different genetic susceptibilities in combination with environmental stimulation onto common pathways. It is our theory that a large subset of individuals with autism and mild brain overgrowth has the final common pathway of enhanced ROS-signaling with resultant PI3K/AKT pathway stimulation, even though different genetic mutations and immune-activating environmental factors may contribute to this activation. The types of genetic mutations that could impact the cellular redox balance in response to MIR include alterations in antioxidant genes, immune-response genes, and ROS-generating genes (e.g., NADPH oxidase), or in genes encoding proteins that can be inactivated and regulated by ROS, such as PTEN. Many of these mutations have already been identified in ASD populations (Luo et al., 2012; Klein et al., 2013; Rose et al., 2012; Voineagu et al., 2011).
Clearly, not every maternal immune-activating event during early pregnancy results in the development of autism, raising the question as to what influences the deleterious fetal response to MIR in those infants who do develop autism. In our MIR model, we found that not all pups in a litter experienced brain overgrowth and this was not related to gender, consistent with what others have found regarding autoimmune-related brain overgrowth in both children (Nordahl et al., 2013) and primates (Bauman et al., 2013). Furthermore, although we found that rescue of MIR-induced brain overgrowth and pathway activation was possible with prenatal NOX inhibition, it also had the negative effect of causing brain undergrowth in some pups. Thus, there is a heterogeneous response to an altered redox balance during brain development among offspring, suggesting that the effects of this dysregulation may depend on underlying individual susceptibilities. Here, we examined a specific genetic susceptibility, PTEN. Mutations in PTEN affect only 1 in 242,063 individuals in the general population, but are found in approximately 4% of ASD individuals (Hobert et al., 2013; Varga et al., 2009). As a proof of principle, we have shown that PTEN heterozygosity interacts with MIR to produce even greater brain overgrowth than can be induced in WT mice. This is in agreement with previous evidence that heterozygous PTEN deletion interacts with ROS stimulation by enhancing stem cell proliferation even more than can be achieved in WT cells (Le Belle et al., 2011). Similarly, TSC haploinsufficiency has been shown to interact with MIR to cause autism-related behaviors in pups (Ehninger et al., 2012). Although our MIR model most closely replicates key phenotypes associated with autism, there is a considerable overlap of some genetic and environmental risk factors, brain pathology, and behavioral abnormalities with other neurodevelopmental disorders, such as schizophrenia (Fatemi et al., 2002). Consequently, MIR-mediated brain overgrowth and the underlying cellular and signaling mechanisms that we have identified could represent a final common pathway for the abnormal phenotypes shared by several neuropsychiatric disorders.
Many cellular mechanisms have the potential to contribute to enlarged brain size and to faulty connections among centers in the brain that are thought to be linked to abnormal behaviors in ASD. While neurite outgrowth and pruning normally occur during the time period in which the accelerated brain growth is observed in humans, they are not the only candidates capable of contributing to this phenomenon. We hypothesize that increased cortical cell numbers resulting from enhanced SVZ proliferation and, possibly, the greater self-renewal capacity of cortical progenitors could have a cascade effect on later downstream processes in development through a direct increase in neuronal numbers, as has been reported in autism (Courchesne et al., 2011), or indirectly through altered glial cell numbers (Herbert, 2005). These processes could then trigger abnormalities in connectivity resulting from altered lamination, neurite outgrowth (Courchesne et al., 2011; Stoner et al., 2014), or other processes. Our findings indicate that environmental factors such as mild maternal inflammation and subsequent alterations in the neural stem cell redox balance can significantly alter brain development in affected offspring and could set up a cascade of cellular changes that lead to brain overgrowth and abnormal behaviors in autism.
Experimental Procedures
Animals
Unless otherwise specified, all experiments were carried out on time-mated CD1 mice from Charles River Laboratories. Female PTEN floxed mice were crossed with hemizygous Nestin-Cre male mice (The Jackson Laboratory), generating pregnant dams carrying both PTEN heterozygous and WT offspring. MIR was induced with low-dose LPS (E. coli serotype 0111:B4; 0.008 mg/kg, Sigma) administered in a single I.P. injection on E9 of gestation. In vivo administration of apocynin (5 mg/kg/day, I.P.; Sigma) was performed daily from E9 until birth. Bromodeoxyuridine (BrdU; 50 mg/kg/injection) was injected subcutaneously at birth and pups were perfused 4 hr later. Replicates of three pregnant dams with 10–18 pups per litter were used in all experiments unless specified otherwise.
Immunohistochemistry
Brain sections were immunostained with BrdU (Exalpha), IBA1 (AbCam), Nestin (BD Biosciences), or MASH1 (BD Biosciences) antibodies with the appropriate Alexa fluorescent secondary antibodies, and positive cells were quantified using the unbiased optical fractionator approach (StereoInvestigator; MicroBrightField). Hoescht counterstain was used to measure cortical thickness and area, as well as corpus callosum thickness, using image analysis software (MCID; Imaging Research).
Behavioral Testing
Ultrasonic vocalization, three-chamber social interaction, and elevated plus maze tests were performed on adult mice according to the methods of Crawley (2007). A repetitive grooming test was performed according to the methods of Peñagarikano et al. (2011).
Statistical Analysis
All data are expressed as mean ± SEM unless otherwise indicated; t tests were performed using Microsoft Excel to determine the statistical significance of treatment sets. For multiple comparisons, one- or two-way ANOVA was performed as appropriate, and Bonferroni post hoc t tests were done to determine significance. The Spearman rank order test was used for correlation analysis. Alpha values were 0.05 except when adjusted by the post hoc tests.
For more information, please refer to Supplemental Experimental Procedures.
Acknowledgments
This work was supported by the following grants and awards: Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to H.I.K.); Cure Autism Now Fellowship (to J.E.L.); Autism Speaks Basic and Clinical grant (to H.I.K.); Autism Speaks Environmental Sciences grant (to J.E.L.); Center for Autism Research and Treatment (CART) Pilot Grant Award #06LEB2008, which is supported by NIH/NICHD grant P50-HD-055784 (to J.E.L.); and NIH grant MH65756 (to H.I.K.). Flow cytometry and cell sorting was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and the Center for AIDS Research Flow Cytometry Core Facility, which is supported by NIH awards CA-16042 and AI-28697, the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, and the UCLA Chancellor’s Office.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Antony J.M., Paquin A., Nutt S.L., Kaplan D.R., Miller F.D. Endogenous microglia regulate development of embryonic cortical precursor cells. J. Neurosci. Res. 2011;89:286–298. doi: 10.1002/jnr.22533. [DOI] [PubMed] [Google Scholar]
- Bauman M.D., Iosif A.M., Ashwood P., Braunschweig D., Lee A., Schumann C.M., Van de Water J., Amaral D.G. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl. Psychiatr. 2013;3:e278. doi: 10.1038/tp.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloosesky R., Weiner Z., Ginsberg Y., Ross M.G. Maternal N-acetyl-cysteine (NAC) protects the rat fetal brain from inflammatory cytokine responses to lipopolysaccharide (LPS) J. Matern. Fetal Neonatal Med. 2012;25:1324–1328. doi: 10.3109/14767058.2011.632793. [DOI] [PubMed] [Google Scholar]
- Bill B.R., Geschwind D.H. Genetic advances in autism: heterogeneity and convergence on shared pathways. Curr. Opin. Genet. Dev. 2009;19:271–278. doi: 10.1016/j.gde.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Kim G.S., Okami N., Narasimhan P., Chan P.H. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol. Dis. 2011;42:341–348. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement H.W., Vazquez J.F., Sommer O., Heiser P., Morawietz H., Hopt U., Schulz E., von Dobschütz E. Lipopolysaccharide-induced radical formation in the striatum is abolished in Nox2 gp91phox-deficient mice. J. Neural Transm. 2010;117:13–22. doi: 10.1007/s00702-009-0327-5. [DOI] [PubMed] [Google Scholar]
- Courchesne E., Carper R., Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. JAMA. 2003;290:337–344. doi: 10.1001/jama.290.3.337. [DOI] [PubMed] [Google Scholar]
- Courchesne E., Mouton P.R., Calhoun M.E., Semendeferi K., Ahrens-Barbeau C., Hallet M.J., Barnes C.C., Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- Crawley J.N. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007;17:448–459. doi: 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C.L., Martínez-Cerdeño V., Noctor S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013;33:4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D., Sano Y., de Vries P.J., Dies K., Franz D., Geschwind D.H., Kaur M., Lee Y.S., Li W., Lowe J.K. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry. 2012;17:62–70. doi: 10.1038/mp.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi S.H., Earle J., Kanodia R., Kist D., Emamian E.S., Patterson P.H., Shi L., Sidwell R. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell. Mol. Neurobiol. 2002;22:25–33. doi: 10.1023/a:1015337611258. [DOI] [PubMed] [Google Scholar]
- Fidler D.J., Bailey J.N., Smalley S.L. Macrocephaly in autism and other pervasive developmental disorders. Dev. Med. Child Neurol. 2000;42:737–740. doi: 10.1017/s0012162200001365. [DOI] [PubMed] [Google Scholar]
- Gregorian C., Nakashima J., Le Belle J., Ohab J., Kim R., Liu A., Smith K.B., Groszer M., Garcia A.D., Sofroniew M.V. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J. Neurosci. 2009;29:1874–1886. doi: 10.1523/JNEUROSCI.3095-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M., Erickson R., Scripture-Adams D.D., Dougherty J.D., Le Belle J., Zack J.A., Geschwind D.H., Liu X., Kornblum H.I., Wu H. PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. USA. 2006;103:111–116. doi: 10.1073/pnas.0509939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J., Cleveland S., Torres A., Phillips J., Cohen B., Torigoe T., Miller J., Fedele A., Collins J., Smith K. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert M.R. Large brains in autism: the challenge of pervasive abnormality. Neuroscientist. 2005;11:417–440. doi: 10.1177/0091270005278866. [DOI] [PubMed] [Google Scholar]
- Hobert J.A., Embacher R., Mester J.L., Frazier T.W., 2nd, Eng C. Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur. J. Hum. Genet. 2013;22:273–276. doi: 10.1038/ejhg.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S., Sharifi-Hannauer P., Martinez-Agosto J.A. Macrocephaly as a clinical indicator of genetic subtypes in autism. Autism Res. 2013;6:51–56. doi: 10.1002/aur.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Belle J.E., Orozco N.M., Paucar A.A., Saxe J.P., Mottahedeh J., Pyle A.D., Wu H., Kornblum H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8:59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Sanders S.J., Tian Y., Voineagu I., Huang N., Chu S.H., Klei L., Cai C., Ou J., Lowe J.K. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am. J. Hum. Genet. 2012;91:38–55. doi: 10.1016/j.ajhg.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordahl C.W., Braunschweig D., Iosif A.M., Lee A., Rogers S., Ashwood P., Amaral D.G., Van de Water J. Maternal autoantibodies are associated with abnormal brain enlargement in a subgroup of children with autism spectrum disorder. Brain Behav. Immun. 2013;30:61–65. doi: 10.1016/j.bbi.2013.01.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson P.H. Maternal infection and immune involvement in autism. Trends Mol. Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O., Abrahams B.S., Herman E.I., Winden K.D., Gdalyahu A., Dong H., Sonnenblick L.I., Gruver R., Almajano J., Bragin A. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebello C.M., Ramos J.L. Association between maternal-fetal genetic histocompatibility and maternal undernutrition in mice: influence on intrauterine growth. Clinics (Sao Paulo) 2006;61:127–132. doi: 10.1590/s1807-59322006000200007. [DOI] [PubMed] [Google Scholar]
- Rose S., Melnyk S., Pavliv O., Bai S., Nick T.G., Frye R.E., James S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatr. 2012;2:e134. doi: 10.1038/tp.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco R., Militerni R., Frolli A., Bravaccio C., Gritti A., Elia M., Curatolo P., Manzi B., Trillo S., Lenti C. Clinical, morphological, and biochemical correlates of head circumference in autism. Biol. Psychiatry. 2007;62:1038–1047. doi: 10.1016/j.biopsych.2007.04.039. [DOI] [PubMed] [Google Scholar]
- Schumann C.M., Bloss C.S., Barnes C.C., Wideman G.M., Carper R.A., Akshoomoff N., Pierce K., Hagler D., Schork N., Lord C., Courchesne E. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J. Neurosci. 2010;30:4419–4427. doi: 10.1523/JNEUROSCI.5714-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaberg R.M., Smukler S.R., van der Kooy D. Intrinsic differences distinguish transiently neurogenic progenitors from neural stem cells in the early postnatal brain. Dev. Biol. 2005;278:71–85. doi: 10.1016/j.ydbio.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Stoner R., Chow M.L., Boyle M.P., Sunkin S.M., Mouton P.R., Roy S., Wynshaw-Boris A., Colamarino S.A., Lein E.S., Courchesne E. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014;370:1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga E.A., Pastore M., Prior T., Herman G.E., McBride K.L. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 2009;11:111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Parada L.F. PTEN signaling in autism spectrum disorders. Curr. Opin. Neurobiol. 2012;22:873–879. doi: 10.1016/j.conb.2012.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.