

Summary
Central to cellular proliferative, survival, and metabolic responses is the serine/threonine kinase mTOR, which is activated in many human cancers. mTOR is present in distinct complexes that are either modulated by AKT (mTORC1) or are upstream and regulatory of it (mTORC2). Governance of mTORC2 activity is poorly understood. Here, we report a transmembrane molecule in hematopoietic progenitor cells that physically interacts with and inhibits RICTOR, an essential component of mTORC2. Upstream of mTORC2 (UT2) negatively regulates mTORC2 enzymatic activity, reducing AKTS473, PKCα, and NDRG1 phosphorylation and increasing FOXO transcriptional activity in an mTORC2-dependent manner. Modulating UT2 levels altered animal survival in a T cell acute lymphoid leukemia (T-ALL) model that is known to be mTORC2 sensitive. These studies identify an inhibitory component upstream of mTORC2 in hematopoietic cells that can reduce mortality from NOTCH-induced T-ALL. A transmembrane inhibitor of mTORC2 may provide an attractive target to affect this critical cell regulatory pathway.
Graphical Abstract
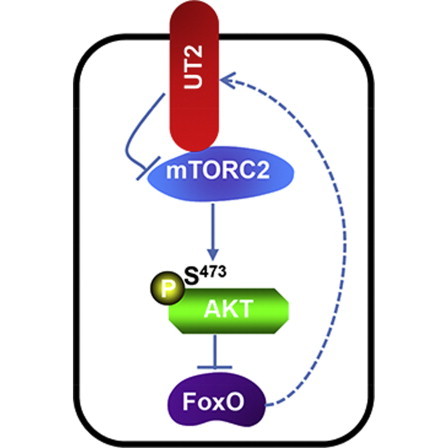
Highlights
-
•
UT2 is a transmembrane inhibitor of mTORC2 in hematopoietic progenitors
-
•
UT2 directly binds RICTOR, limiting the kinase activity of mTORC2 on pAKTS473
-
•
UT2 modulates the outcome of an mTORC2-dependent hematopoietic cancer
In this article, Scadden and colleagues show that an undefined transmembrane molecule upstream of mTORC2 (UT2) negatively regulates AKT signaling via modulation of mTORC2. Their work points to the value of models that can be used to examine niche contributions to oncogenesis and reveals a previously unrecognized transmembrane modulator of a critical pathway with therapeutic implications for cancers such as T-ALL.
Introduction
We recently described an animal model in which primary genetic lesions in bone marrow (BM) stromal cells resulted in abnormal hematopoiesis in mice and the rare development of acute leukemia (Raaijmakers et al., 2010). While evaluating three leukemias that emerged in the setting of this aberrant stroma, we noted that two of them had a shared chromosomal abnormality. Reasoning that this represented a highly nonstochastic event, we assessed the open reading frames in the altered region and found that two of them putatively encoded transmembrane molecules. Since transmembrane molecules might represent a means by which an altered BM stroma could select for abnormal hematopoietic stem/progenitors, we focused on these molecules. One of these, C14ORF37, is the subject of this report. Using overexpression and knockdown constructs, we found that C14ORF37 encodes a protein with unique characteristics in modulating a specific aspect of the AKT/mTOR growth and differentiation pathway in hematopoietic cells.
Activation of AKT in hematopoiesis as induced experimentally by deletion of Pten leads to a myeloproliferative syndrome and eventual loss of hematopoietic stem cells (Kharas et al., 2010, Yilmaz et al., 2006, Zhang et al., 2006). This is mTOR dependent, as loss of hematopoietic stem cells can be rescued by rapamycin. The mTOR complex implicated in this process appears to be mTORC1, since experimental deletion of Raptor (a canonical component of mTORC1) similarly resulted in hematopoietic failure as evidenced by a lack of reconstituting ability upon transplantation (Kalaitzidis et al., 2012, Magee et al., 2012). In addition, pharmacological inhibition of mTOR resulted in antileukemia effects in a mouse model of acute myeloid leukemia (AML) (Zeng et al., 2007, Zeng et al., 2012). In some leukemia-initiating cells, however, the AKT pathway appears to play a different role. AKT activation at position S473 is associated with subsequent phosphorylation of FOXOs, which reduces the ability of the FOXOs to enter the nucleus and serve as transcriptional regulators. However, 40% of human AML shows a gene-expression signature consistent with Foxo transcriptional activity, and FOXO inhibition in human and mouse AML cells results in terminal differentiation (Sykes et al., 2011). Deletion of Foxo1, Foxo3, and Foxo4 in a mouse model of AML resulted in differentiation and loss of leukemia-initiating cells. Therefore, the AKT pathway may have complex roles in the proliferation and differentiation abnormalities associated with myeloid malignancies.
Acute lymphocytic leukemia (ALL) is more clearly driven by activation of the AKT/mTOR pathway. Rapamycin was shown to reduce the leukemia burden in xenograft models of B cell ALL (B-ALL), and reducing AKT activity by anti-connective tissue growth factor (anti-CTGF) therapy resulted in decreased B-ALL (Lu et al., 2014). In T cell ALL (T-ALL), clonal dominance was shown in cells where AKT activation increased and was associated with resistance to antitumor glucocorticoid therapy (Blackburn et al., 2014). Another study showed that inhibition of AKT could overcome glucocorticoid resistance (Piovan et al., 2013). The T-ALL that emerged with either Pten deficiency or with NOTCH activation was dependent upon very specific elements of the AKT pathway, in particular, the mTORC2 complex. This complex includes RICTOR, among other proteins, and is upstream of AKT, phosphorylating AKT at S473. Deletion of Rictor is capable of inhibiting T-ALL associated with either Pten deletion (Kalaitzidis et al., 2012) or NOTCH activation (Lee et al., 2012). Therefore, mTORC2 is critical for some lymphoid leukemias. Here, we report that C14ORF37 encodes a protein that is capable of interacting with and inhibiting the function of mTORC2. This distinctive transmembrane molecule may be a modulator of growth-regulator signals conveyed from the BM microenvironment.
Results
Impact of UT2 on the mTORC2/AKT/FOXO Axis
C14ORF37 encodes a putative type I transmembrane protein with an intracytoplasmic tail that has no clear functional homologies. C14ORF37 is expressed by BM hematopoietic cells, with the most abundant message being found in the hematopoietic stem and progenitor cell (HSPC)-enriched Lin−SCA1+c-KIT+ (LSK) fraction and dominantly present in BM (Figures S1A–S1C available online). To determine its function, we transduced multiple small hairpin RNA (shRNA) constructs targeting C14orf37 and overexpressed cDNA in primary mouse hematopoietic cells and, for some assays, FLAG-tagged cDNA expressed in HEK293T cells. Three shRNAs targeting distinctive regions within the C14orf37 transcript provided effective knockdown (Figures S1D and S1E) and comparable effects (data are presented using one shRNA displaying >90% inhibition). Because the molecule was studied as a potential mediator of altered hematopoietic cell growth, we examined growth-regulatory pathways to determine whether any were altered in the context of changing the levels of C14ORF37 (called UT2 for reasons given below). Although there was no effect on ERK phosphorylation or PP2A, PTEN, PDK1, or PI3K p85 levels (Figure 1A), there was evidence that UT2 shRNA increased the level of pAKTS473 (Figures 1A–1C and S1F–S1H).
Figure 1.
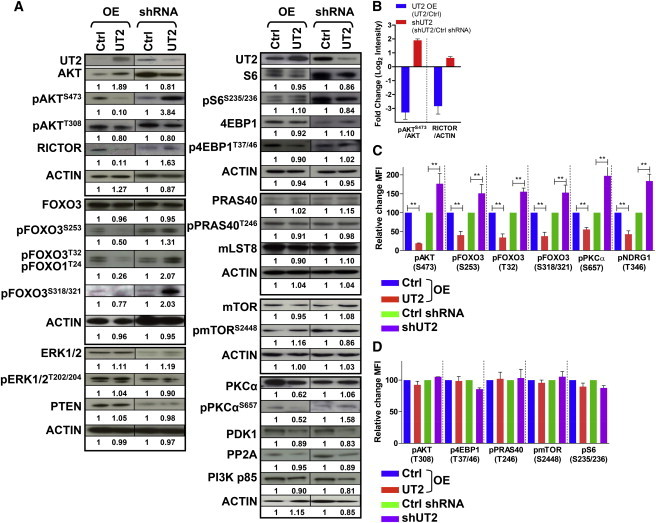
UT2 Regulates the mTORC2/AKT/FOXO Axis in Primary Hematopoietic Cells
(A) Primary hematopoietic cells from cells overexpressing or depleted for UT2 were analyzed by western blotting using the indicated antibodies. ACTIN was used as a loading control (n = 2–3 experiments).
(B) Graphs show the fold change of the indicated normalized protein ratios (pAKTS473/AKT and RICTOR/ACTIN) in (A). Data are means ± SEM (n = 3 experiments).
(C and D) Flow cytometry was performed on hematopoietic cells overexpressing or shRNA-depleted for UT2. Quantification of the fold change in mean fluorescence intensity for the indicated phosphoprotein in these cells is shown. Data are means ± SEM (n = 2–3 experiments; two-tailed, unpaired t test). ∗p < 0.05, ∗∗p < 0.01.
See also Figure S1.
Phosphorylation of AKT at S473 occurs through the RICTOR-containing complex with mTOR, mTORC2. Notably, the PDK1 phosphorylation site on AKT, T308, was not consistently changed. To further assess the impact of UT2 on AKT, we evaluated the downstream targets of pAKTT308, including pmTORS2448 and pPRAS40T246, which were not altered (Figures 1A, 1D, and S1F). Also, pAKTT308 results in activation of the RAPTOR-containing mTOR complex, mTORC1, so we evaluated the targets of mTORC1 activation: pS6S235/236 and p4EBP1T37/46. Neither of these was altered. Together, these data indicate that UT2 knockdown does not influence mTORC1. These results were further validated by overexpression of UT2: decreased pAKTS473 and RICTOR levels were noted, but no consistent changes were observed in mTORC1 components or targets. These data suggest that UT2 (C14ORF37) modulates RICTOR and alters mTORC2-dependent AKT phosphorylation without affecting mTORC1. Therefore, we have named C14ORF37 upstream of mTORC2 (UT2) in reference to its distinctive relationship with the mTOR complex containing RICTOR.
With a change in mTORC2 activity, one would expect known substrates such as PKCα (Guertin et al., 2006) and SGK (assayed by NDRG1 [García-Martínez and Alessi, 2008] phosphorylation), as well as the effects of pAKTS473, to be perturbed. We found that UT2 shRNA increased pPKCαS657 and pNDRG1T346, whereas overexpression of UT2 reduced it (Figures 1A, 1C, and S1F). Similarly, pAKTS473 is known to phosphorylate FOXOs, resulting in their retention in the cytoplasm and reduced transcriptional activity. Inhibiting UT2 by shRNA led to increased pAKTS473 and a consequent increase in the levels of pFOXO3s (S253, T32, and S318/321). The converse was seen when UT2 was overexpressed in these cells (Figures 1A, 1C, and S1F). Together, these results argue that levels of UT2 modulate the activity of mTORC2 and two of its primary known targets with expected biochemical results downstream. UT2 decreases mTORC2 functional activity.
UT2 Negatively Regulates AKT Signaling via Interaction with mTORC2
Given the impact of UT2 on the mTORC2/AKT/FOXOs axis, we postulated that UT2 may physically interact with RICTOR. To address this, we first assessed the cellular distribution of endogenous UT2 and RICTOR by immunostaining (Figure S2A). Primary hematopoietic cells from wild-type (WT) and Rictor-deficient animals were stained with antibodies against UT2 and RICTOR, and this revealed that a substantial proportion of UT2 colocalized with RICTOR. To examine whether the effects of UT2 reflected a physical interaction with candidate target proteins, we conducted immunoprecipitation (IP) experiments on HEK293T cells expressing a FLAG-UT2 fusion or FLAG-UT2 fusion with the cytoplasmic region of UT2 deleted (FLAG-UT2-ΔC) (Figure 2A). Anti-FLAG monoclonal IP demonstrated coprecipitation with UT2 and RICTOR by western blot. Furthermore, the RICTOR interaction required the cytoplasmic portion of UT2, as demonstrated by the FLAG-UT2-ΔC construct. We therefore pursued the interaction further in primary hematopoietic cells from animals with conditional homozygous deletion of Rictor (Figure 2B) and again demonstrated coIP of UT2 and RICTOR in control cells that was lost in Rictor-deleted cells (the faint band represents incomplete deletion). To assess other possible molecular interactions of UT2, we used a panel of antibodies for other known components of mTOR complexes and performed IP and western blot (Figure S2B). UT2 IP brought down RICTOR, but did not bring down RAPTOR, a canonical mTORC1 member. Cumulatively, these data indicate that UT2 interacts with mTORC2, but not mTORC1.
Figure 2.
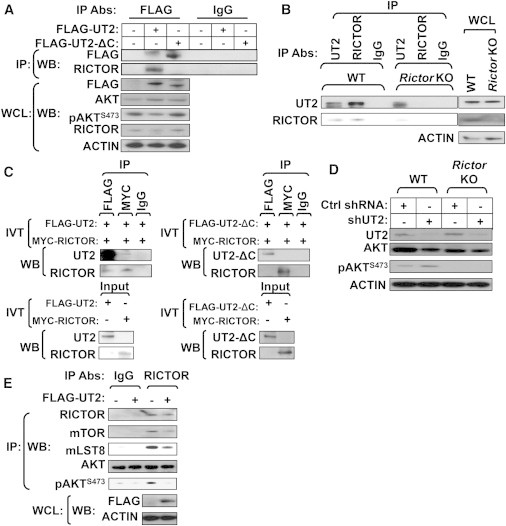
UT2 Interacts with mTORC2/RICTOR and Regulates Their Activities in Primary Hematopoietic Cells
(A) HEK293T cells transfected with vectors encoding either Flag-UT2 full length or Flag-UT2 ΔC mutant were lysed and subjected to IP using an antibody directed against FLAG and IgG, respectively. The resulting precipitates and corresponding whole-cell lysates (WCLs) were subjected to western blot analysis using the indicated antibodies (n = 3 experiments).
(B) RICTOR or UT2 was immunoprecipitated from WT and conditional homozygous deletion of Rictor BM cells and subjected to western blot analysis using the indicated antibodies (n = 6 mice pool per each genotype).
(C) In vitro-translated FLAG-UT2 full length (left) or FLAG-UT2 ΔC mutant (right) proteins were subjected to IP using the indicated antibodies in the presence of in vitro-translated myc-RICTOR protein (n = 2–3 experiments).
(D) WT and conditional homozygous deletion of Rictor BM cells expressing shRNA constructs against control or UT2 were subjected to western blot analysis using the indicated antibodies (n = 3 mice pool per each genotype).
(E) Endogenous RICTOR was immunoprecipitated from control and HEK293T cells transfected with vectors encoding FLAG-UT2 full length and subjected to in vitro kinase assays using recombinant inactive AKT as the substrate and pAKTS473 as the readout. An immunoblot for the levels of RICTOR, mTOR, and mLST8 in each immunoprecipitate is shown (n = 3 experiments).
See also Figure S2.
To further verify a direct interaction between UT2 and RICTOR, we generated in vitro-translated proteins and conducted IP experiments. IP of UT2 and RICTOR demonstrated coIP indicative of a direct interaction between the proteins in vitro (Figure 2C, left). When the same experiment was conducted with UT2 in which the C-terminal cytoplasmic domain had been deleted, the results failed to demonstrate any coIP of UT2ΔC and RICTOR (Figure 2C, right). Therefore, UT2 directly interacts with RICTOR and does so in a manner that depends on its cytoplasmic domain. Next, we examined whether UT2 altered the phosphorylation levels of pAKTS473 in primary hematopoietic cells from animals conditionally deficient in Rictor (Figure 2D). The effects of shUT2 on pAKTS473 were not observed in Rictor-deleted cells, indicating that UT2 modulates pAKTS473 in primary hematopoietic cells through RICTOR. We then performed mTORC2 in vitro kinase assays to define whether UT2 had a meaningful impact on mTORC2 function. mTORC2 was isolated from lysates of HEK293T cells by coIP with RICTOR and proportionately included other components of the complex, including mTOR and mLST8 (Figure 2E). The HEK293T cells were expressing FLAG-UT2 or controls. AKT was exogenously added as a substrate (note: it was not immunoprecipitated with RICTOR). Phosphorylation of AKT at S473 was diminished in the presence of FLAG-UT2 (Figure 2E), indicating that UT2 inhibits mTORC2 phosphorylation of AKT. Furthermore, shRNA against UT2 in primary hematopoietic cells demonstrated increased pAKTS473 levels that were accentuated by insulin or serum stimulation of starved cells (Figure S2C). Together, these data indicate that UT2 inhibits mTORC2 kinase activity as evidenced by phosphorylation of AKT.
FOXO3 Directly Regulates Ut2 Expression In Vivo
Given the impact of UT2 on the mTORC2/AKT/FOXO axis and the importance of FOXOs in regulating hematopoietic function (Paik et al., 2007, Tothova et al., 2007), we assessed whether FOXOs influenced UT2 expression. Mouse primary hematopoietic cells were transduced with recombinant retroviruses expressing FOXO3 WT, FOXO3 triple mutant (AAA, FOXO3 AKT-resistant and active form) (Brunet et al., 1999), or myristoylated AKT (myr-AKT, which leads to diminished FOXO activity) (Kharas et al., 2010, Kohn et al., 1996) (Figure 3A). Enforced expression of either the WT or AKT-resistant mutant version of FOXO3 activated Ut2 expression (Figure 3B). Primary hematopoietic cells derived from mice lacking functional purified Foxo1, Foxo3, and Foxo4 alleles (hereafter termed Foxo1/3/4 KO) displayed significantly lower levels of Ut2 transcripts compared with the WT (Figure 3C), suggesting that members of the FOXO family regulate Ut2 expression in vivo.
Figure 3.
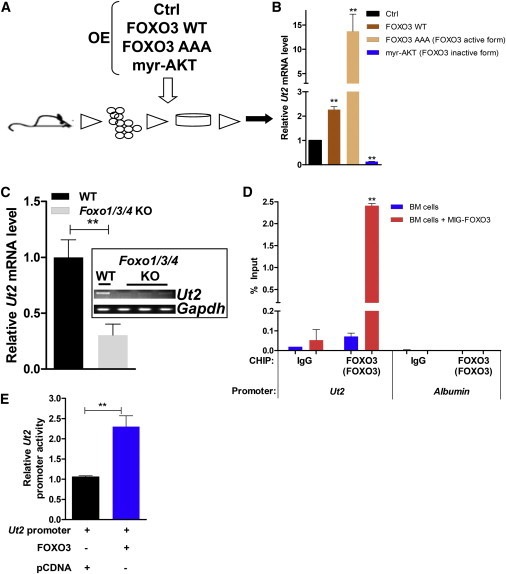
FOXO3 Activates Ut2 Expression in Primary Hematopoietic Cells
(A) Schematic representation of a gene-expression assay in primary hematopoietic cells.
(B) Quantitative RT-PCR for Ut2 expression. Data are means ± SEM (n = 3 experiments; two-tailed, unpaired t test). ∗∗p < 0.01.
(C) Ut2 expression in primary hematopoietic cells from WT and Foxo1/3/4 KO mice. Data are means ± SEM (n = 3 mice per each genotype; two-tailed, unpaired t test). ∗∗p < 0.01.
(D) ChIP analysis for MIG-FOXO3 or mock expressing BM cells, using the indicated antibodies. Data are means ± SEM (n = 3 experiments; two-tailed, unpaired t test). ∗∗p < 0.01.
(E) Relative luciferase activity of FOXO3 on the Ut2 promoter. Data are means ± SEM (n = 4 experiments; two-tailed, unpaired t test). ∗∗p < 0.01.
See also Figure S3.
Sequence analysis of the Ut2 promoter showed that there is a putative FOXO3-binding site embedded approximately 7 kb upstream of the transcriptional start site (Figure S3A). Chromatin IP (ChIP) with FOXO3-specific antibodies in primary hematopoietic cells and fibroblast cells showed that FOXO3 binds to this conserved site within the Ut2 promoter in vivo (Figures 3D and S3B). Furthermore, FOXO3 can activate luciferase expression driven by a minimal promoter containing the putative FOXO3 consensus sequence found in the Ut2 promoter (Figure 3E). Collectively, these data show that FOXO3 directly regulates the transcription of Ut2. Moreover, UT2 regulation of Rictor levels was evaluated and found to occur at the level of transcription (Figure S3C). The evidence therefore suggests that FOXO3 is a regulator of UT2 that can feed forward to further increase FOXO3 activity by inhibiting AKTS473 phosphorylation (Figure 4D). However, this is recognized to be only a partial characterization of the mechanisms that regulate FOXO3 and expression of UT2.
Figure 4.
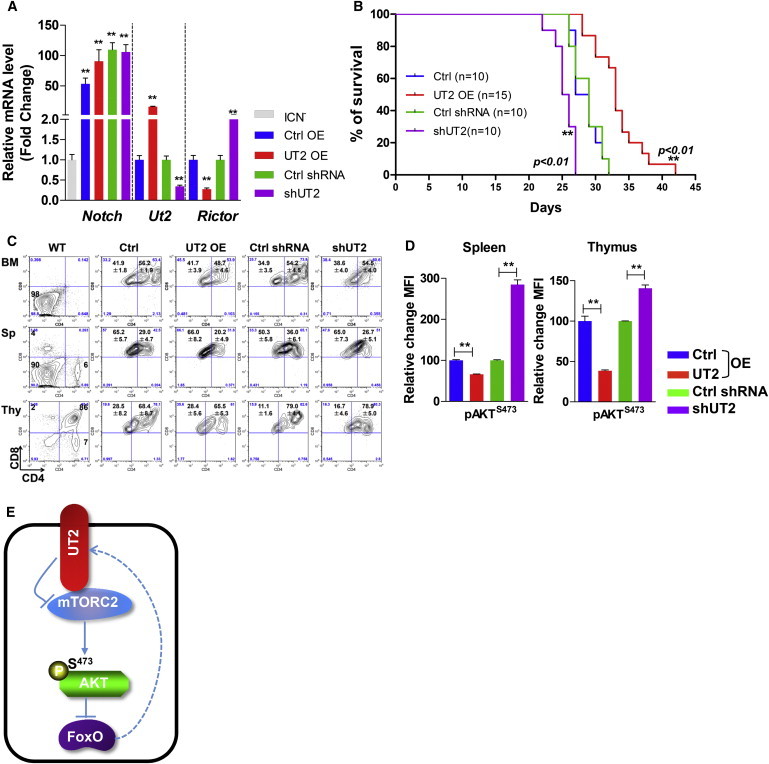
UT2 Extends the Disease Latency of T-ALL in Mice
(A) Expression profile of the indicated genes before BM transplantation (BMT) assays. Primary hematopoietic cells from C57BL/6 5-fluorouracil (5-FU)-injected donors with cells overexpressing or shRNA-depleted for UT2 were transduced with overexpressing intracellular NOTCH1 (ICN), and then BMT assays were performed. Data are means ± SEM (n = 3 experiments; two-tailed, unpaired t test). ∗p < 0.05. ∗∗p < 0.01. “Control” represents ICN-transduced cells.
(B) Kaplan-Meier survival curves for recipient mice after transplantation of ICN1-transduced BM cells from cells overexpressing or shRNA-depleted for UT2 as described in (A) (n = 10–11 biological replicated experiments, log rank test). ∗∗p < 0.01.
(C) Flow-cytometry analysis of various hematopoietic tissues from T-ALL recipient mice in (A) (n = 8–9 biological replicated experiments).
(D) Expression of phosphorylated AKT on S473 in spleen and thymus from recipient mice in (A). Animals were examined when they were moribund. Data are means ± SEM (n = 8–9 biological replicated experiments, two-tailed, unpaired t test). ∗∗p < 0.01.
(E) UT2 negatively regulates AKT signaling by modulating the activity of RICTOR/mTORC2.
See also Figure S4.
Impact of UT2 on NOTCH-Associated T-ALL
To determine whether UT2 altered the function of hematopoietic cells, we chose to use a model of malignancy because it was previously shown to be a striking phenotype of mTORC2 deficiency. More than 50% of human T-ALL cells depend on NOTCH mutations for their growth or viability (Weng et al., 2004), and a previous study showed that mTORC2 facilitates NOTCH-driven T-ALL development, whereas Rictor depletion substantially prolongs survival (Lee et al., 2012). Furthermore, in settings of Pten deletion, T-ALL develops rapidly except when the mTORC2 component, Rictor, is deleted (Kalaitzidis et al., 2012, Magee et al., 2012). To examine whether UT2 affects T-ALL, hematopoietic BM precursors were transduced with a constitutively active form of NOTCH1 (intracellular NOTCH1 [ICN]) and transplanted into recipient mice (Chiang et al., 2008, Lee et al., 2012, Piovan et al., 2013) (Figure 4A). Mice transplanted with UT2 overexpressing ICN+ cells displayed significantly prolonged disease survival, whereas the animals with knocked-down UT2 died significantly more rapidly (Figure 4B). An examination of hematopoietic tissues by flow cytometry revealed aberrant T cells in the thymus, spleen, and BM consistent with leukemic disease, although survival was prolonged in UT2-overexpressing cells (Figure 4C). The changes in mortality correlated with different levels of pAKTS473 as anticipated from UT2 modulation (Figure 4D). Together, these results demonstrate that UT2 is capable of affecting the outcome of NOTCH-associated T-ALL in association with changes in mTORC2 activity.
Discussion
These data identify a transmembrane molecule that negatively regulates AKT signaling via modulation of mTORC2 (Figure 4E). It joins the small number of mTORC2 upstream interactors, such as ribosome components (Zinzalla et al., 2011). Its transmembrane, putative cell-surface localization suggests that it may be a means by which exogenous cues can dampen mTORC2 activation. We identified this molecule by virtue of its amplification in the context of a microenvironmentally induced AML (Raaijmakers et al., 2010), in line with AKT playing a tumor-suppressive role in a substantial proportion of patients with this disease and in maintenance of leukemia stem cells in an animal model (Sykes et al., 2011). However, we cannot draw a direct link between UT2 amplification and aberrant signals from the modified stroma in that model. Of note, UT2 had modest effects on normal hematopoietic function (Figure S4). We did observe that enforced UT2-expressing donor cells contributed to lower levels of blood cells (Figure S4B), the frequency of Lin−SCA1+c-KIT+ (L−S+K+) cells (Figure S4C), and colony formation as compared with control cells (Figure S4D). Moreover, hematopoietic cells from the BM of animals with the altered microenvironment showed evidence of decreased pAKTS473 (Figure S1I) consistent with a role for UT2, but whether that was then selected for in the emergence of the leukemia cannot be discerned by our studies to date. In T-ALL, where mTORC2/AKT plays a clearer oncogenic role, we were able to demonstrate that UT2 could be a therapeutic target to modulate that disease. Collectively, our work points to the value of models that can be used to examine niche contributions to oncogenesis and reveals a previously unrecognized transmembrane modulator of a critical pathway with therapeutic implications for cancer.
Experimental Procedures
Mice and Animal Procedures
All mice were kept in a specific pathogen-free facility at Massachusetts General Hospital. All mice studies and breeding were carried out under the approval of Institutional Animal Care and Use Committee of Massachusetts General Hospital. Foxo1/3/4floxed;Mx1-Cre (Paik et al., 2007, Sykes et al., 2011, Tothova et al., 2007) and Rictorfloxed;Mx1-Cre (Shiota et al., 2006) mice were generated previously. To examine T-ALL model in mice, hematopoietic BM precursors were transduced with a constitutively active form of NOTCH (ICN) and transplanted into recipient mice (Chiang et al., 2008, Lee et al., 2012, Piovan et al., 2013). See the Supplemental Experimental Procedures for further details.
Protein Analyses
Western blot analysis was carried out according to standard methods as previously described (Sykes et al., 2011). For standard IP experiments, all cells, with the exception of those used to isolate mTORC, were lysed with Triton X-100-containing lysis buffer as previously described (Peterson et al., 2009). For IPs of mTORC, cells were lysed in mTORC lysis buffer (ice-cold CHAPS-containing lysis buffer) as previously described (Guertin et al., 2006, Huang et al., 2008, Kim et al., 2002, Peterson et al., 2009, Sarbassov et al., 2004, Sarbassov et al., 2005). The mTORC2 in vitro kinase assay was performed as described previously (Guertin et al., 2006, Huang et al., 2008, Jacinto et al., 2006, Kim et al., 2002, Peterson et al., 2009, Sarbassov et al., 2004, Sarbassov et al., 2005). ChIP experiments were performed with the use of a ChIP Assay Kit (Millipore) according to the manufacturer’s instructions (Lee et al., 2008). For serum and insulin stimulation experiments, BM cells expressing UT2 or shRNA-mediated depletion of UT2 BM cells were deprived of serum for 3 hr and then fetal bovine serum or insulin was add back at 10% serum or 1 μg/mL insulin concentration, respectively, for 30 min prior to lysis (Guertin et al., 2006, Jacinto et al., 2006). See the Supplemental Experimental Procedures for further details.
Real-Time Quantitative RT-PCR
Real-time quantitative RT-PCR was performed as previously described (Lee et al., 2008, Lee et al., 2011).
Flow Cytometry and Antibodies
BM cells and leukocytes were harvested and subjected to red cell lysis (Lee et al., 2011, Sykes et al., 2011). Phosphoflow experiments were performed as previously described (Kalaitzidis et al., 2012, Sykes et al., 2011). See the Supplemental Experimental Procedures for further details.
Statistical Analysis
The sample size required for experiments was estimated based on preliminary data. In vitro and in vivo data were analyzed with a two-tailed, unpaired Student’s t test (GraphPad Prism; GraphPad Software) and SigmaPlot 10.0 software (SPSS). Values of p < 0.05 were considered statistically significant (∗p < 0.05; ∗∗p < 0.01). The Kaplan-Meier log rank test was used to analyze mouse survival data using GraphPad Prism. No blinding or randomization was performed for any of the experiments.
Author Contributions
D.L., S.S., D.K., and D.S. designed the research, analyzed data, and wrote the paper. D.L. carried out most of the experimental work with the help of S.S., D.K., A.A.L., Y.K., M.H.G.P.R., Y.W., and S.A.A. D.S. directed the research.
Acknowledgments
We thank Joseph Avruch, David Sabatini, Sean Morrison, and Alex Soukas for advice and helpful comments. This work was supported by NIH NHLBI U01HL100402, HL97794, and HLO44851; the Ellison Medical Foundation (D.L., M.R., and D.S.), NIH NIDDK K01DK092300 (D.K.), an MGH Fund for Medical Discovery fellowship (D.L.); the ISEH (S.S.); the Dubai Foundation (Y.K.); and the MGH/HSCI CRM flow-cytometry core (L. Prickett, K. Folz-Donahue, and M. Weglarz).
Published: September 25, 2014
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2014.08.011.
Supplemental Information
References
- Blackburn J.S., Liu S., Wilder J.L., Dobrinski K.P., Lobbardi R., Moore F.E., Martinez S.A., Chen E.Y., Lee C., Langenau D.M. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 2014;25:366–378. doi: 10.1016/j.ccr.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Chiang M.Y., Xu L., Shestova O., Histen G., L’heureux S., Romany C., Childs M.E., Gimotty P.A., Aster J.C., Pear W.S. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J. Clin. Invest. 2008;118:3181–3194. doi: 10.1172/JCI35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez J.M., Alessi D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem. J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Guertin D.A., Stevens D.M., Thoreen C.C., Burds A.A., Kalaany N.Y., Moffat J., Brown M., Fitzgerald K.J., Sabatini D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Huang J., Dibble C.C., Matsuzaki M., Manning B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell. Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S.Y., Huang Q., Qin J., Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kalaitzidis D., Sykes S.M., Wang Z., Punt N., Tang Y., Ragu C., Sinha A.U., Lane S.W., Souza A.L., Clish C.B. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell. 2012;11:429–439. doi: 10.1016/j.stem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharas M.G., Okabe R., Ganis J.J., Gozo M., Khandan T., Paktinat M., Gilliland D.G., Gritsman K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.H., Sarbassov D.D., Ali S.M., King J.E., Latek R.R., Erdjument-Bromage H., Tempst P., Sabatini D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kohn A.D., Summers S.A., Birnbaum M.J., Roth R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- Lee D., Park C., Lee H., Lugus J.J., Kim S.H., Arentson E., Chung Y.S., Gomez G., Kyba M., Lin S. ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell. 2008;2:497–507. doi: 10.1016/j.stem.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D., Kim T., Lim D.S. The Er71 is an important regulator of hematopoietic stem cells in adult mice. Stem Cells. 2011;29:539–548. doi: 10.1002/stem.597. [DOI] [PubMed] [Google Scholar]
- Lee K., Nam K.T., Cho S.H., Gudapati P., Hwang Y., Park D.S., Potter R., Chen J., Volanakis E., Boothby M. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J. Exp. Med. 2012;209:713–728. doi: 10.1084/jem.20111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Kojima K., Battula V.L., Korchin B., Shi Y., Chen Y., Spong S., Thomas D.A., Kantarjian H., Lock R.B. Targeting connective tissue growth factor (CTGF) in acute lymphoblastic leukemia preclinical models: anti-CTGF monoclonal antibody attenuates leukemia growth. Ann. Hematol. 2014;93:485–492. doi: 10.1007/s00277-013-1939-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee J.A., Ikenoue T., Nakada D., Lee J.Y., Guan K.L., Morrison S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell. 2012;11:415–428. doi: 10.1016/j.stem.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J.H., Kollipara R., Chu G., Ji H., Xiao Y., Ding Z., Miao L., Tothova Z., Horner J.W., Carrasco D.R. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson T.R., Laplante M., Thoreen C.C., Sancak Y., Kang S.A., Kuehl W.M., Gray N.S., Sabatini D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovan E., Yu J., Tosello V., Herranz D., Ambesi-Impiombato A., Da Silva A.C., Sanchez-Martin M., Perez-Garcia A., Rigo I., Castillo M. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24:766–776. doi: 10.1016/j.ccr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers M.H., Mukherjee S., Guo S., Zhang S., Kobayashi T., Schoonmaker J.A., Ebert B.L., Al-Shahrour F., Hasserjian R.P., Scadden E.O. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D.D., Ali S.M., Kim D.H., Guertin D.A., Latek R.R., Erdjument-Bromage H., Tempst P., Sabatini D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shiota C., Woo J.T., Lindner J., Shelton K.D., Magnuson M.A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Sykes S.M., Lane S.W., Bullinger L., Kalaitzidis D., Yusuf R., Saez B., Ferraro F., Mercier F., Singh H., Brumme K.M. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova Z., Kollipara R., Huntly B.J., Lee B.H., Castrillon D.H., Cullen D.E., McDowell E.P., Lazo-Kallanian S., Williams I.R., Sears C. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Weng A.P., Ferrando A.A., Lee W., Morris J.P., 4th, Silverman L.B., Sanchez-Irizarry C., Blacklow S.C., Look A.T., Aster J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Yilmaz O.H., Valdez R., Theisen B.K., Guo W., Ferguson D.O., Wu H., Morrison S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Zeng Z., Sarbassov D., Samudio I.J., Yee K.W., Munsell M.F., Ellen Jackson C., Giles F.J., Sabatini D.M., Andreeff M., Konopleva M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z., Shi Y.X., Tsao T., Qiu Y., Kornblau S.M., Baggerly K.A., Liu W., Jessen K., Liu Y., Kantarjian H. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood. 2012;120:2679–2689. doi: 10.1182/blood-2011-11-393934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Grindley J.C., Yin T., Jayasinghe S., He X.C., Ross J.T., Haug J.S., Rupp D., Porter-Westpfahl K.S., Wiedemann L.M. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zinzalla V., Stracka D., Oppliger W., Hall M.N. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.