

Abstract
Impaired nutrient delivery to the brain due to decreased blood flow contributes to cognitive decline and dementia in Alzheimer’s disease (AD). Considering this, many studies have suggested that neuroprotective agents like those used in stroke could prevent AD onset or progression by promoting cell survival. However, research in the past decade suggests that the culprit behind the cognitive loss in AD models is actually the soluble tau accumulating inside of surviving neurons. In fact, tau reductions improve cognition in mouse models of AD, even those that only deposit amyloid plaques. There is emerging evidence that neuroprotection alone in these AD models may be insufficient to restore neuron function and cognition. Only when soluble tau is reduced on a neuroprotective background could memory be rescued. Thus, once a neuron begins to accumulate tau, it may survive in a malfunctioning capacity, leading to impaired electrical signaling and memory formation in the brain. These data imply that multiple drugs may be necessary to ameliorate the different disease components. In fact, strategies to preserve neurons without affecting the soluble protein burden within neurons may accelerate the disease course.
1. Cerebral angiopathy and blood flow dysregulation in AD
Current research in AD suggests that cerebral angiopathy (CA) can occur from excess neuronal secretion of Aβ, and from preexisting vascular disease (Iadecola, 2004, Zlokovic, 2008). Aβ can be cleared from the brain by vascular transport across the blood-brain-barrier (BBB). This free Aβ, can bind to different transport binding proteins like apolipoprotein J (Ghiso, 1993) and E (Yang, 1997), transthyretin (Schwarzman, 1994), lipoprotein receptors (Matsubara, 1999) and several others. Vascular smooth muscle cells (VSMC) have recently been an intense area of focus due to their ability to internalize and clear Aβ and contract the capillary (Urmoneit, 1997). Thus, modulation of VSMC could provide a cellular and molecular link between vascular disorders and AD. The VSMC can clear Aβ by using lipoprotein receptor-related protein-1 (LRP-1) to sequester it (Shibata, 2000, Urmoneit, 1997). It is speculated that when the neuron begins secreting excess amyloid, the VSMC becomes saturated, allowing Aβ to accumulate. Aβ is also capable of potentiating the cell’s constriction capability (Niwa, 2001, Paris, 2002). In fact, when Aβ 1–40 peptide is administered to wildtype mice by topical superfusion the subjects display reduced resting cerebral blood flow (Niwa, 2001). Also, it has been found that serum response factor and myocardin are upregulated in AD tissue (Chow, 2007). They are transcription factor proteins that facilitate the VSMC-differentiated phenotype. Hence, AD patients may have a hypercontractile phenotype that can be further potentiated by Aβ. The amyloid cascade hypothesis is further supported by studies showing that the overexpression of APP in mice is capable of causing pathologic changes before the detectable appearance of amyloid plaques. In the PDAPP mice, dentate gyrus volume is reduced before plaque formation (Redwine, 2003). Mice overexpressing the Swedish APP mutation exhibit a reduction in hyperemia (Niwa, 2000). On the other hand, vascular disease on its own is also capable of reducing blood flow across the blood-brain-barrier, creating a hypoxic state. Hypoxia has been shown to decrease the ability of VSMC to clear Aβ (Bell, 2009). Anothr complication from arising from CA is that reduced blood flow leads to energy deprivation, which in turn potentiates BACE1 levels (O'Connor, 2008). Since BACE1 is the rate-limiting step of Aβ production, more BACE1 protein creates more Aβ peptide. Ultimately, a cascade can arise from CA that becomes self-perpetuating, manifesting in dementia.
2. Neuroprotective agents for AD
Because of the strong vascular component in AD, there has been a search for compounds that can elongate the life of neurons. Many drugs have been proposed to have possible neuroprotective effects in AD based on their ability to scavenge antioxidants and free radicals. Examples of these drugs are, indole-3-propionic acid (IPA), vitamin E and resveratrol.
IPA is an inhibitor of Aβ fibril formation, an antioxidant, and neuroprotectant. To assess its ability to protect neurons from ischemic damage it was administered for 15 days at a 10 mg/kg dose to mice. Tissue was collected and IPA was shown to spare neurons from ischemic damage ~300% (Hwang, 2009). IPA has also been shown to inhibit Aβ fibril formation but other indole derivatives like indole 3-acetic acid, indole 3-carbinol and tryptophol were more effective (Morshedi, 2007).
Vitamin E is currently in phase III of clinical trials to treat AD. However, beneficial effects of vitamin E in patients with moderate to severe AD have been modest (Brewer, 2010). Vitamin E treatment of 2000 IU slowed the functional deterioration of patients, improving their daily living, but failing to improve the mini-mental exam score (Grundman, 2000, Petersen, 2005).
Resveratrol can promote antioxidant activity, neuroprotective effects, and activation of sirtuins and their positive effect on aging (Albani, 2010). This compound, derived from grapes, improves cognition and reduces plaque pathology in animal models of AD (Karuppagounder, 2009, Kim, 2007, Wang, 2006). Resveratrol is one of the more promising compounds that has entered clinical trials.
Pharmacological agents like resveratrol and IPA may indeed be neuroprotective, but questions about a strategy designed to spare sick neurons are beginning to emerge. In some sense, this is in direct violation of Darwinian principles: Culling of the weak or sick from the herd is better for the species population as a whole. This same principle may be in play in the brain. Perhaps sparing neurons that harbor aberrantly accumulated proteins could worsen brain function. Thus, extending the life of neurons that have already begun to accumulate tau may not be an effective strategy. Perhaps the brain is able to adapt when a neuron dies, as has been shown in stroke and occlusion diseases; the plasticity in the brain can reroute processes to perform essential functions. However, when a neuron is chronically sick, but surviving, the brain may continue to route information through it, but that neuron may simply be unable to propagate the signal (Figure 1).
Figure 1.
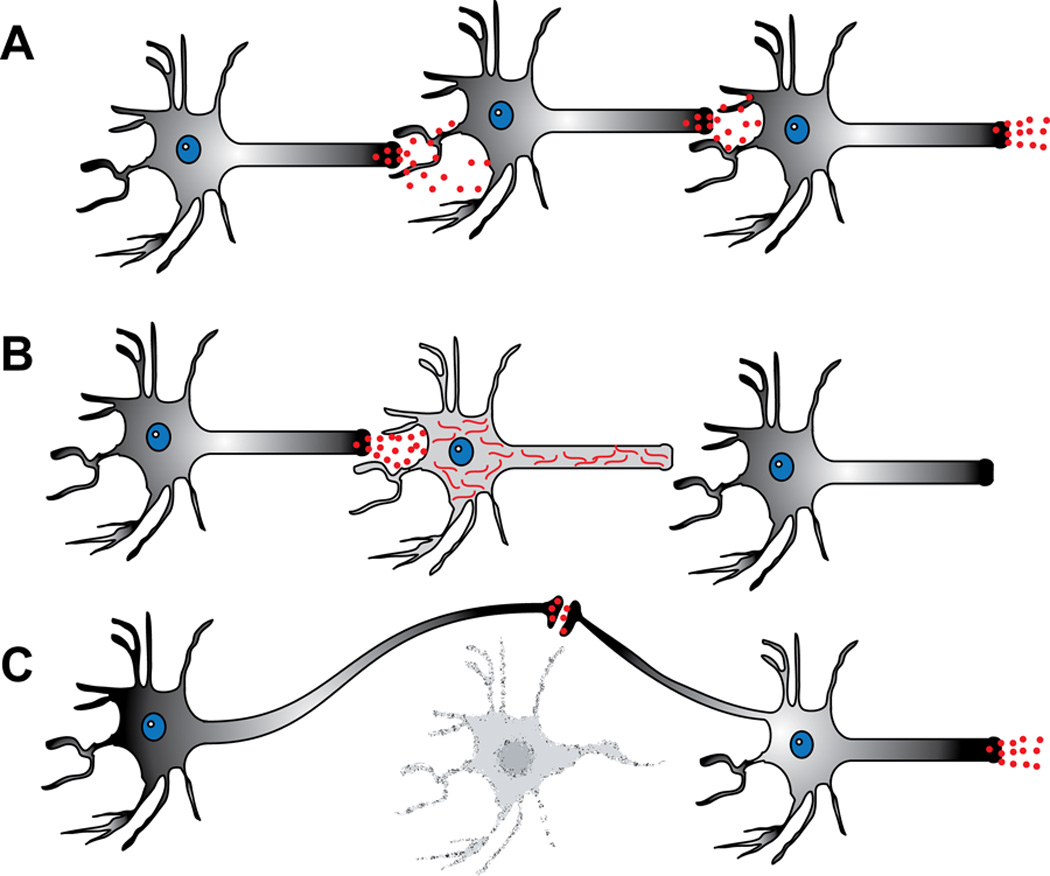
Sub-optimally functioning neurons surviving with proteotoxic tau accumulation subvert brain plasticity that normally occurs in response to neuronal death. (A) Neuronal transmission in normal brain. (B) Sub-optimally performing neurons due to tau accumulation cannot transmit a signal to the post-synaptic neuron, and the brain fails to re-route connectivity since the neuron is not dead. (C) Neuronal death facilitates network re-routing, allowing the plastic brain to adapt and re-establish downstream connectivity.
3. Pathologically visible tau or invisible soluble intermediates; what should we be targeting?
Although there is strong data demonstrating the adverse effects of vascular dysfunction and amyloid accumulation in AD, research in the last decade suggests that tau may be a better therapeutic target. Recent work has demonstrated a critical role for tau in amyloid-induced deficits of the hAPP mice (Roberson, 2007). Since these mice do not develop tau pathology it was long assumed that tau did not have a role in their cognitive deficits. However, when the hAPP transgenic mice were crossed onto a tau null background, the cognitive deficits induced by amyloid accumulation were ameliorated. Moreover, tau depletion protected against both kainate and GABAA receptor antagonist (pentylenetetrazol) induced seizures. Furthermore, tau reduction did not change Aβ deposition, neuritic distrophy, or aberrant sprouting. This study suggests that tau may be a better therapeutic target because it is necessary for Aβ-mediated cognitive deficits and excitotoxicity. Also, it suggests that cognition can be improved despite amyloid pathology.
Another piece of evidence that supports tau-induced cognitive decline is the creation of the rTg4510 mouse model (Santacruz, 2005), which is doubly transgenic for the tetracycline operator driven by the CamKII promoter and human P301L tau driven by the PrP promoter and regulated by the tetracycline-responsive element. These mice accumulate tau tangles and develop severe neurodegeneration and cognitive deficits. A large group of rTg4510 mice were trained to find the hidden escape platform in the Morris water maze (MWM) at 2.5 months of age. It was found that tau was not affecting all mice equally. As a result, the mice were split into two cohorts. To test the effect of tau reduction and pathology on cognition in a sub-optimally performing cohort, tau expression was shut off at 2.5 months of age in half of the mice, while the other half received control diet. The mice were retrained and tested at 4.5 months, an age prior to onset of neurodegeneration. Suppressing tau expression for 2 months enabled mice that had previously performed poorly in the MWM to now learn and recall the location of the escape platform, while the performance of the vehicle-treated rTg4510 mice continued to decline. Next, the higher performing cohort had doxycycline treatment initiated at 5.5 months, a point when neurodegeneration and frank tangle pathology begins in this model. These mice had improved cognitive function despite increasing tangle formation. These two studies showed that not only is soluble tau able to impair learning and memory, but tangles failed to correlate with memory improvement.
A role for soluble tau in altering cognitive function was re-emphasized in a subsequent study using the rTg4510 model (O'Leary, 2010). Seven-month old rTg4510 mice were treated with the pleiotropic compound, methylene blue (MB), which is currently in clinical trials for AD. Direct hippocampal infusion of MB improved learning and memory in the rTg4510 mice, but neither pathology nor neuronal morphology were altered: Only soluble tau was extensively reduced.
In a more recent study, the role of caspase cleavage of tau was tested in the rTg4510 model. Caspase activation was shown to occur before tangle formation, tangle-bearing neurons were long-lived, and aggregated tau was able to suppress caspase activity (de Calignon, 2010). Conversely, injection with a virus expressing wildtype human tau induced caspase cleavage of tau, suggesting that soluble cytosolic tau could activate caspase. This work suggests that tau tangles may protect the neuron from apoptosis, while soluble tau can induce caspase activation and possibly neurotoxicity.
There is still more evidence for the importance of soluble tau in disrupting neuronal function. Mice over-expressing wildtype human tau show no tau insolubility or neuronal loss, yet these mice have memory deficits and synaptic dysfunction. Conversely mice over-expressing the P301L variant of human tau did develop neuronal loss and tau insolubility, but these mice were cognitively intact and synapses functional (Kimura, 2010). This concept provides further support to the notion that accumulation of soluble tau essentially clogs the neuron, but does not kill it, leading to reduced overall brain function. In fact, neuronal death and insolubility of tau is less deleterious compared to soluble tau accumulation and survival of sub-optimally performing neurons.
There are several mechanisms through which soluble tau might accumulate and take on a deleterious function. Pre-fibrillar Aβ can cause microtubule disassembly, resulting in a loss of function for tau and an increased cytosolic burden of free tau (King, 2006, Rapoport, 2002). Aberrant phosphorylation and mutations associated with some tauopathies also reduce tau’s affinity for the microtubules (Wagner, 1996), possibly enlarging the pool of tau that is not microtubule bound (Dayanandan, 1999). Another possible way that soluble tau becomes enriched in the cytosol would be by over-expression. Indeed this occurs naturally in sporadic Parkinson’s disease (Simon-Sanchez, 2009). It has been shown that over-expression of widltype human tau leads to excess free tau in the cytosol (Andorfer, 2003), providing a greater opportunity for tau to interact with itself and aggregate into a soluble non-functioning intermediate. Thus, perhaps when tau loses its microtubule function, it can gain a toxic function. For example, tau can mediate Aβ excitotoxicity by allowing fyn to phosphorylate the NMDA receptor subunit 2 (Ittner, 2010). In this study, crossing the APP23 transgenic mouse line with tau null mice disrupted the dendritic targeting of fyn. Interestingly, the same result was achieved by overexpressing a truncated form of tau that lacks the microtubule binding domains of tau. Again, this suggests that soluble, free tau without the capacity to interact with microtubules may be even more deleterious to neuronal function.
All of this evidence strengthens the rationale for developing strategies to deplete free, soluble tau to treat AD. While reducing all tau has been thought likely to be harmful, there are several findings that suggest the brain may be more tolerant of such a strategy than first imagined. In particular, tau knockout mice are functionally intact, due in part to compensation by MAP1a (Harada, 1994, Tucker, 2001). Moreover, fast axonal transport is not affected in tau knockout mice (Yuan, 2008). Considering this evidence and that many of the patients who would receive anti-tau therapies would be elderly, reducing tau may indeed be a well-tolerated strategy to ameliorate AD symptoms and modify disease course.
4. Lifespan versus healthspan for neurons: Do Darwinian principles apply in a degenerating brain?
The phenothiazine class of compounds includes the controversial compound, methylene blue (MB). The pleiotropy of MB has raised a number of concerns about its clinical application. More than a decade passed between the time that MB was first reported to prevent tau aggregation in solution and the time that the first reports emerged describing successful use of MB in the clinic (Wischik, 2009, Wischik, 1996). Since then, a number of studies have begun to dissect the mechanisms contributing to the efficacy of this drug. MB was shown to reduce Aβ levels in the 3xTg-AD mouse model and improve learning and memory (Medina, 2010). In this study MB treatment was given for 4 months, beginning at the age of 6 months. MB treatment ameliorated learning and memory deficits in this mouse model, reduced Aβ levels, but failed to alter tau.
Other studies have ascribed one mechanism of MB action to its ability to inhibit the molecular chaperone Hsp70. Indeed this activity was shown to destabilize tau in cells and mice (Jinwal, 2009). MB treatment was recently shown to improve cognitive function in rTg4510 mice, but only when sufficiently high doses were present in the brain (O'Leary, 2010). Interestingly, tau tangle pathology as assessed by histology was unchanged in these mice compared to those treated with vehicle. When stereological analyses were performed on tissues from these mice, MB facilitated neuroprotection in all treated mice; however, this neuroprotection did not correlate with cognitive function. Biochemical analyses of tissue from these mice showed that soluble tau levels were reduced by MB treatment, but only in mice with high concentrations in their brain. Surprisingly, reduced soluble tau burden correlated with improved cognitive function in this study.
An additional report recently showed that reducing soluble tau burden with a purely genetic approach could improve neuronal function in the rTg4510 mice, as measured by long term potentiation (Abisambra, 2010). Moreover, over-expression of wildtype human tau in mice does not beget tau insolubility or neuronal loss, but does cause cognitive dysfunction, while over-expression of mutant tau causes neuronal loss and insolubility of tau, but does not lead to memory deficits (Kimura, 2010). These results allow for several conclusions to be drawn. First, neuroprotection alone is insufficient to rescue memory function in tau transgenic mice, decoupling two processes that have always been intertwined. Secondly, reductions in tau tangle pathology are not necessary for neuroprotection or cognitive improvement. Lastly, reducing soluble tau levels is necessary for cognitive improvement in this model, but it is not required for neuroprotection.
This work has several important implications regarding classical paradigms about the relationship of neuronal survival, neuronal health and overall brain function. When soluble proteins accumulate in the cytosol, the endgame may be neuronal death, but there are likely many neurons that continue to survive for long periods of time in a sub-optimal, or functionally impaired, state. This is likely due to the intracellular accumulation of “sticky” proteins. Strategies aimed at only protecting these sick neurons may ultimately be deleterious to brain function. Instead, focusing on amelioration of the protein accumulation in these neurons to restore their function may prove more therapeutic. Simply prolonging the life of non-functioning neurons may prevent compensatory brain plasticity mechanisms from being triggered to overcome the loss of function of a discreet circuit (Figure 1). While neuroprotective strategies may indeed be beneficial, it might be critical to couple such agents with a treatment that can reduce the soluble protein accumulation that burdens neurons in many neurodegenerative diseases. Neuroprotection has proven successful in stroke; however these neurons do not have toxic soluble proteins accumulating within. In neurodegenerative diseases resulting from proteotoxicity, neuroprotection without reducing the toxic protein burden may be harmful.
5. Conclusions
The population with AD is expected to grow tremendously within the next 20 years as the first wave of baby-boomers reaches 65 years of age this year. In addition to AD, there are more than 15 other neurodegenerative diseases where tau is pathogenic. Research from the most recent decade suggests that ameliorating soluble tau protein accumulation may provide patients with greater cognitive improvement and behavioral health. This coupled with neuroprotective agents may be an even more effective clinical strategy. However neuroprotection without also rescuing the soluble proteotoxic burden in neurons may be unsuccessful, essentially allowing malfunctioning neurons to evade the “survival of the fittest” principle. Thus the brain may be unable to recognize a need to reconfigure damaged circuitry. Strategies aimed at only neuroprotection may ultimately accelerate the course of these disorders.
References
- 1.Abisambra JF, Blair LJ, Hill SE, et al. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci. 2010;30:15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albani D, Polito L, Signorini A, Forloni G. Neuroprotective properties of resveratrol in different neurodegenerative disorders. Biofactors. 2010;36:370–376. doi: 10.1002/biof.118. [DOI] [PubMed] [Google Scholar]
- 3.Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 4.Bell RD, Deane R, Chow N, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brewer GJ. Why vitamin E therapy fails for treatment of Alzheimer's disease. J Alzheimers Dis. 2010;19:27–30. doi: 10.3233/JAD-2010-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow N, Bell RD, Deane R, et al. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proc Natl Acad Sci U S A. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayanandan R, Van Slegtenhorst M, Mack TG, et al. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999;446:228–232. doi: 10.1016/s0014-5793(99)00222-7. [DOI] [PubMed] [Google Scholar]
- 8.de Calignon A, Fox LM, Pitstick R, et al. Caspase activation precedes and leads to tangles. Nature. 2010;26:787–789. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghiso J, Matsubara E, Koudinov A, et al. The cerebrospinal-fluid soluble form of Alzheimer's amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J. 1993;293(Pt 1):27–30. doi: 10.1042/bj2930027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundman M. Vitamin E and Alzheimer disease: the basis for additional clinical trials. Am J Clin Nutr. 2000;71:630S–636S. doi: 10.1093/ajcn/71.2.630s. [DOI] [PubMed] [Google Scholar]
- 11.Harada A, Oguchi K, Okabe S, et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- 12.Hwang IK, Yoo KY, Li H, et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J Neurosci Res. 2009;87:2126–2137. doi: 10.1002/jnr.22030. [DOI] [PubMed] [Google Scholar]
- 13.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 14.Ittner LM, Ke YD, Delerue F, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 15.Jinwal UK, Miyata Y, Koren J, 3rd, et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. Neurochem Int. 2009;54:111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura T, Fukuda T, Sahara N, et al. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem. 2010;285:38692–38699. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsubara E, Ghiso J, Frangione B, et al. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer's disease and Down's syndrome. Ann Neurol. 1999;45:537–541. [PubMed] [Google Scholar]
- 21.Medina DX, Caccamo A, Oddo S. Methylene Blue Reduces Abeta Levels and Rescues Early Cognitive Deficit by Increasing Proteasome Activity. Brain Pathol. 2010 doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morshedi D, Rezaei-Ghaleh N, Ebrahim-Habibi A, Ahmadian S, Nemat-Gorgani M. Inhibition of amyloid fibrillation of lysozyme by indole derivatives--possible mechanism of action. FEBS J. 2007;274:6415–6425. doi: 10.1111/j.1742-4658.2007.06158.x. [DOI] [PubMed] [Google Scholar]
- 23.Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol. 2001;281:H2417–H2424. doi: 10.1152/ajpheart.2001.281.6.H2417. [DOI] [PubMed] [Google Scholar]
- 24.Niwa K, Younkin L, Ebeling C, et al. Abeta 1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A. 2000;97:9735–9740. doi: 10.1073/pnas.97.17.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Connor T, Sadleir KR, Maus E, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Leary JC, 3rd, Li Q, Marinec P, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paris D, Townsend KP, Humphrey J, Obregon DF, Yokota K, Mullan M. Statins inhibit A beta-neurotoxicity in vitro and A beta-induced vasoconstriction and inflammation in rat aortae. Atherosclerosis. 2002;161:293–299. doi: 10.1016/s0021-9150(01)00660-8. [DOI] [PubMed] [Google Scholar]
- 28.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 29.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Redwine JM, Kosofsky B, Jacobs RE, et al. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci U S A. 2003;100:1381–1386. doi: 10.1073/pnas.242746599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 32.Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwarzman AL, Gregori L, Vitek MP, et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 37.Urmoneit B, Prikulis I, Wihl G, et al. Cerebrovascular smooth muscle cells internalize Alzheimer amyloid beta protein via a lipoprotein pathway: implications for cerebral amyloid angiopathy. Lab Invest. 1997;77:157–166. [PubMed] [Google Scholar]
- 38.Wagner U, Utton M, Gallo JM, Miller CC. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J Cell Sci. 1996;109(Pt 6):1537–1543. doi: 10.1242/jcs.109.6.1537. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Ho L, Zhao Z, et al. Moderate consumption of Cabernet Sauvignon attenuates Abeta neuropathology in a mouse model of Alzheimer's disease. FASEB J. 2006;20:2313–2320. doi: 10.1096/fj.06-6281com. [DOI] [PubMed] [Google Scholar]
- 40.Wischik C, Staff R. Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of Tau-aggregation inhibitor therapy. J Nutr Health Aging. 2009;13:367–369. doi: 10.1007/s12603-009-0046-5. [DOI] [PubMed] [Google Scholar]
- 41.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang DS, Smith JD, Zhou Z, Gandy SE, Martins RN. Characterization of the binding of amyloid-beta peptide to cell culture-derived native apolipoprotein E2, E3, and E4 isoforms and to isoforms from human plasma. J Neurochem. 1997;68:721–725. doi: 10.1046/j.1471-4159.1997.68020721.x. [DOI] [PubMed] [Google Scholar]
- 43.Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]