

Abstract
Glycoengineering aimed at addition of carbohydrates to proteins is an attractive approach to alter pharmacokinetic properties of proteins such as enhancing stability and prolonging the duration of action. We report a novel protein glyco-modification of BSA and recombinant thrombomodulin with O-cyanate chain-end functionalized glycopolymer via isourea bond formation. The protein glycoconjugates were confirmed by SDS-PAGE, western blot, and MALDI-TOF Mass Spectrometry. Protein C activation activity of the glyco-modified recombinant thrombomodulin was confirmed, proving no interference to activity from the glycopolymer modification. The isourea bond formation under mild conditions was demonstrated as an alternative method for protein modification with polymers.
Keywords: Carbohydrate, glycopolymer, protein, thrombomodulin, cyanate, isourea
1. Introduction
Protein-based drugs have revolutionized the treatment of many diseases and could be the major clinically used drugs in the future. However, many limitations, such as rapid clearance and instability, still prevent their practical utility. In particular, these limitations often lead to high frequency of protein administration required to maintain therapeutic dosages, which places a substantial burden on both patients and caregivers from both convenience and cost standpoints. Many efforts have been made towards enhancing protein half-life so as to reduce administration frequency and thus increase convenience and improve patient compliance. Modification of protein with water-soluble polymers, such as polyethylene glycol (PEG), has proven effective in increasing the circulation time of the drug in vivo by attenuating renal filtration, thus decreasing clearance and has been approved in clinical usage.[1] Glycoengineering aimed at addition of carbohydrates to proteins is an attractive approach to alter pharmacokinetic properties of proteins such as increasing stability and prolonging the duration of action.[2] A successful example is the discovery of darbepoetin alfa, an erythropoietin analogue that contains additional carbohydrates resulting in a threefold increase in serum half-life and increased in vivo activity compared to recombinant human erythropoietin.[3] Synthetic polymers have been widely investigated for protein modification for profound biological applications.[4] Glycopolymers that contain multiple copies of sugar moieties have been employed as natural oligosaccharide mimics and found important biological applications in biosensor, microarray and protein modification.[5] For example, a maleimide functionalized glycopolymer was developed for synthesizing glycoprotein mimics by modifying thiol-containing residues of proteins.[6] Therefore, glycoengineering of therapeutic proteins with covalently attached glycopolymers is expected to provide an effective route to improve proteins’ stability and extend their plasma half-life and also to reduce their immunogenicity as well.
Amine-targeted bioconjugations have been the major strategy for protein modification. Amide bond formation and reductive amination are the most used methods; however, low reaction efficiency and pH-dependent reaction conditions often limit their wide applications.[7] Alternatively, O-cyanate-based isourea bond formation has been proven useful as an amine-targeted bioconjugation technique for protein immobilization.[8,9] Cyanoxyl-mediated free radical polymerization (CMFRP) has been developed as a straightforward approach for synthesizing glycopolymers.[10] Recently, we demonstrated that CMFRP is a facile method for the synthesis of O-cyanate chain-end functionalized glycopolymers, which can be immobilized onto amine surface for oriented glyco-macroligand formation via isourea bond.[11,12] We found that CMFRP possesses several advantages such as direct polymerization, reaction in aqueous solution, exclusion of tedious protection and deprotection steps, compatibility with a broad range of functional groups, and a O-cyanate chain-end group. The presence of chain-terminal O-cyanate group allows for conjugation to amine group through isourea bond formation, a reaction that has been well understood, characterized, and used for affinity chromatography. Therefore, we envisioned that the chain-end O-cyanate group of the glycopolymer provides an anchor for site-specific and covalent conjugation of the glycopolymer with protein amine groups via isourea bond formation and thereby facilitates a site-specific glycopolymer-protein conjugate formation (Figure 1).
Figure 1.

Protein glyco-modification with O-cyanate chain-end functionalized glycopolymer via isourea bond formation.
Thrombomodulin (TM) is an endothelial membrane protein and acts as a physiological anticoagulant by binding thrombin and subsequently converting protein C to its active form (APC), which is an anticoagulant protease that selectively inactivates coagulation factors Va and VIIIa.[13] TM epidermal growth factor-like domains 4-6 (TM456) are the minimum required domains for TM anticoagulant activity.[14] Therefore, TM456 serves as a potential candidate for an antithrombotic agent. However, the half-life of the TM456 is extremely short compared to recombinant human soluble thrombomodulin (rhsTM), and thus limits its clinical application.[15] Herein, we proposed that the modification of TM456 with a glycopolymer could enhance its pharmacokinetic properties. In this study, we investigated protein glyco-modification with O-cyanate chain-end functionalized glycopolymer with bovine serum albumin (BSA) as a model protein and then with recombinant thrombomodulin (rTM456) as an anticoagulant agent. Both polymer-protein conjugates were generated via isourea bond formation between the amino and O-cyanate functional group.
2. Results and discussion
CMFRP is a versatile technique to synthesize polymers with multivalent carbohydrates pendants on the polymer backbone and an O-cyanate at the polymer chain end. In this study, lactose-based O-cyanate chain-end functionalized glycopolymer was synthesized from acrylaminoethyl lactoside via CMFRP as in our previous report.[16] As shown in Scheme 1, acrylamide was used in the polymerization with acrylaminoethyl lactoside so as to control the carbohydrate density as well as the solubility of the polyacrylamide polymer. Polyacrylamide also provides stability to chemical and proteolytic cleavage. Another feature is that the presence of a terminal phenyl group in the polymer allows for easy determination of lactose and acrylamide content as well as average molecular weight of the glycopolymer by 1H NMR spectrum.[17] In addition, in our previous work we were able to show that low polydispersity (Mw/Mn<1.6) glycopolymers could be produced using this approach.[18]
First, BSA was used as a model protein to test the feasibility of O-cyanate chain-end functionalized glycopolymer modification via isourea bond formation. BSA contains 57 lysine residues, which allow for multiple polymer modifications with O-cyanate functional groups. The conjugation reaction between BSA and glycopolymer was run in bicarbonate buffer (pH 8.3) for 24 hours at 4 oC. The successful conjugation of BSA and glycopolymer was confirmed by SDS-PAGE with an observed increased in molecular weight of BSA conjugates. As shown in Figure 2, the presence of protein was verified by typical Coomassie blue staining (Figure 2A), while the presence of carbohydrates was confirmed by carbohydrate-specific staining (Shiff's base formation, Figure 2B) and whole protein staining (Figure 2C). The carbohydrate-specific staining method is based on periodic acid-Schiff base formation, where carbohydrate cis-diol groups are oxidized to aldehydes by periodic acid. Subsequent reaction with fuchsine reagent forms a Schiff base between the aldehydes and the amino groups of the dye allowing for carbohydrate visualization. As a result, BSA-glycopolymer conjugate was visualized (Figure 3B, Lane 4), while BSA was not stained by the carbohydrate staining (Figure 2B, Lanes 3). The glycopolymer was not observed on the gel with carbohydrate staining (Figure 3B, Lane 2), possibly due to its neutral and hydrophilic property. The hydrophilic nature of the molecule prevented it from interacting with SDS thus leading to a lower negative charge density, resulting in reduced motility during electrophoresis. On the SDS-PAGE, the observed molecular weight of the glycopolymer-protein conjugate was higher than anticipated. This could be due to the fact that the attached glycopolymer contains long hydrophilic backbone with side chains consisting of lactose and amide functionalities, which prevent SDS from interacting with the protein and thus hamper the conjugate to move through the gel during electrophoresis. This phenomenon has been observed in a previous report when using glycopolymer to modify streptavidin via biotin binding.[17] These results indicated that the O-cyanate chain-end functionalized glycopolymer could be used for protein modification via isourea bond formation with site-specificity. In addition, the BSA-glycopolymer conjugate was also confirmed by using Matrix-Assisted Laser Desorption Ionization Time-of-Flight (MALDI-TOF) mass spectrometry (Figure 3). The BSA-glycopolymer conjugate displayed an approximately 71 kDa molecular weight, which is about 5 kDa higher than BSA (66.6 kDa). All these results indicated a successful BSA-glycopolymer conjugation via isourea bond formation.
Figure 2.

12.5 % SDS-PAGE of BSA-glycopolymer conjugate: (A). Coomassie blue staining, (B). Carbohydrate staining, (C). Total protein staining. (Lane 1: glycoprotein molecular marker, Lane 2: glycopolymer, Lane 3: BSA, Lane 4: BSA-glycopolymer conjugate). Molecular weights are indicated in kDa.
Figure 3.

MALDI TOF spectrum of BSA (red) showing a single peak at 66.6 kDa and BSA-glycopolymer (pink) showing two peaks at approximately 66.6 kDa (unreacted BSA) and 71.7 kDa (reacted) (Matrix:Water:Acetonitrile:Trifluoroacetic Acid, 50:50:0.1, v/v ratio).
Next, the O-cyanate chain-end functionalized glycopolymer was used for glyco-modification of recombinant TM (rTM456). The conjugation reaction between rTM456 and glycopolymer was run in a Tris-HCl buffer since it was found that rTM456 precipitated easily when the buffer conditions were changed. Thus, the same buffer used for rTM456 purification was applied for the conjugation reaction. The conjugate formation was confirmed by SDS-PAGE, in which the glycoconjugate was visualized by carbohydrate staining (Figure 4A, Lane 3), total protein staining (Figure 4B, Lane 3) and western blot using mouse monoclonal antibody specific to human TM (Figure 4C, Lane 3), while rTM456 (without modification) was observed at around 16 kDa by total protein staining (Figure 4B, Lane 1) and western blot using mouse monoclonal antibody specific to human TM (Figure 4C, Lane 1). No conjugate was formed in the reaction mixture of rTM456 and a glycopolymer with a hydroxyl group (Figure 4A, 4B and 4C, Lane 2), which was obtained by treating the O-cyanate chain-end functionalized glycopolymer with pyridine in water, a procedure that we had reported previously.[15] The lack of reaction with the hydroxyl-terminated glycopolymer confirmed that a glycoconjugate was formed via isourea bond. The apparent high molecular weight of the glycoconjugates may be due to a similar phenomenon as discussed above, namely that glycopolymer attachment to rTM456 prevented SDS homogenous coverage of the molecule thus preventing the formation of homogenous negative charge throughout the protein's surface, which affected the rate at which the glycoconjugates moved through the acrylamide gel. Finally, MALDI TOF analysis of the rTM456-glycopolymer conjugate revealed an increase of molecular weight of the conjugate to 21 kDa (Figure 4B), approximately 5 kDa higher than the unreacted TM (Figure 4A). All these results indicated the successful rTM456-glycopolymer conjugation via isourea bond formation.
Figure 4.

12.5 % SDS-PAGE of rTM4,5,6-glycopolymer conjugate: (A). carbohydrate staining, (B). total protein staining and (C). Western blot with mouse anti-TM antibody (Lane 1: rTM456, Lane 2: rTM456 reacted with OH-terminated glycopolymer, Lane 3: rTM456 reacted with OCN-terminated glycopolymer).
Based on the MALDI-TOF MS results, a 5 kDa molecular weight increase was observed for both BSA-glycopolymer and rTM-glycopolymer, suggesting the formation of 1:1 conjugates. BSA contains 57 lysine residues and a terminal amine and rTM456 contains one lysine residue and a terminal amine. Since the pKa of a terminal amine is close to 7-8 and that of lysine amino groups is near 9, we suspect that the reaction at pH 8.0-8.3 resulted in the specific modification of the terminal amine of both proteins. In addition, steric hindrance might have blocked polymer modification on the other sites of proteins. A more detailed investigation of the specificity of modification is needed in our continued study.
TM is an endothelial membrane protein and acts as an anticoagulant by binding to thrombin and subsequently converting protein C to its active form (APC), which selectively inactivates coagulation factors Va and VIIIa.[13] In this study, the protein C activation activities of rTM456 (control) and rTM456-glycopolymer were assessed using a previously established method.[19] As a result, rTM456-glycopolymer showed the similar protein C activation activity to rTM456 for both Michaelis constant (Km) and turnover number (kcat) (Table 1), meaning that the glycopolymer modification did not negatively affect the rTM456 activity.
Table 1.
Protein C activation activities of rTM456 and rTM456-glycopolymer conjugate
| rTM456 | rTM456-glycopolymer | |
|---|---|---|
| Km (μM) | 0.75±0.2 | 0.81±0.12 |
| kcat (Min−1) | 0.25±0.08 | 0.19±0.20 |
| kcat/Km (Min−1 • μM−1) | 0.29±0.11 | 0.20±0.15 |
Standard deviations among the three measurements are shown after the ‘±’ sign.
3. Conclusions
In this study, we have demonstrated a novel strategy for protein modification with O-cyanate chain-end functionalized glycopolymer via isourea bond formation. This modification provides beneficial properties for possible therapeutic applications of proteins. In particular, single chain-end O-cyanate group provides an anchor point by its specific conjugation with amine groups of proteins and facilitates highly oriented assembly of the glycopolymer. In addition, the ease of synthesis of O-cyanate chain-end functionalized glycopolymers via CMFRP and the mild conditions for isourea bond formation in different buffers make this approach versatile and general for primary amine-oriented protein modification.
4. Experimental
4.1. Materials and methods
All solvents and reagents were purchased from commercial sources and were used as received, unless otherwise noted. Deionized water was used for reactions, dialyses, and measurements. Dialyses were performed using cellulose membranes. 12.5% Polyacrylamide gels were cast using Mini-PROTEAN® Tetra Handcast System from Biorad (Hercules, CA). Electrophoresis was performed at 130 V for 120 min. at room temperature. Glycoprotein and RAPID stain total protein stains were obtained as a kit from G-Biosciences (St. Louis, MO). Bovine serum albumin (BSA) and sinapic acid (SA for MALDI matrix) were obtained from Sigma-Aldrich (St. Louis, MO). Purified recombinant human PC and human thrombin were from Haematologic Technologies Inc. (Essex Junction, VT). Human antithrombin III and chromogenic substrate Spectrozyme PCa were purchased from American Diagnostica Inc. (Stamford, CT). 1H NMR spectra were obtained from Bruker HD 400 NMR spectrometer at room temperature. All sample concentrations were 10 mg mL-1, and appropriate deuterated solvents were used. UV-vis absorbance measurements were performed on a Varian Bio50 UV-vis spectrometer. MALDI TOF spectra were obtained using Bruker Autoflex III smartbeam spectrometer.
4.2. Synthesis of O-cyanate chain-end functionalized glycopolymer
4.2.1. Synthesis of lactosyl acrylamide (2-N-acryloyl-aminoethoxyl-4-O-(β-D-galactopyranosyl)-β-D-glycopyranoside)
The lactosyl acrylamide was synthesized by following our previous report.[16]
4.2.2. Polymerization
Sodium nitrite (7 mg, 1.0 mmol) and 4-chloroaniline (11 mg, 0.09 mmol) were dissolved in 3 mL mixture of H2O/THF (1:1, V/V), followed by adding HBF4 (122 mg, 1.5 mmol) then incubating for 30 mins. Next, lactosyl acrylamide monomer (90 mg, 13 μmol), NaOCN (27 mg, 0.42 mmol) and acrylamide (105 mg, 13 μmol) were added. The reaction solution was incubated for 16 h at 60 °C, followed by dialysis (3500 Da MW cutoff) to afford lactose-based O-cyanate chain-end functionalized glycopolymer (275 mg, 65% yield). The O-cyanate chain-end functionalized glycopolymer was characterized by 1H and 13C NMR spectroscopy (Supporting Information Figures S1 and S2).
4.2.3. Conversion of O-cyanate group to hydroxyl group of the glycopolymer
The chain-end O-cyanate group of glycopolymer was converted to a hydroxyl group by the reaction with pyridine in water as in our previous report.[18]
4.3. Synthesis of BSA-glycopolymer conjugate
BSA (0.2 mg) was reacted with O-cyanate chain-end functionalized glycopolymer (4 mg) in 16 μL of 0.1 M NaHCO3 (pH 8.3) containing 0.5 M NaCl at 4 °C for 24 hours. The reaction mixture was dialyzed (13 kDa cutoff) against DI water to remove the unreacted glycopolymer and buffer solution and then was lyophilized to afford the BSA-glycopolymer conjugate (0.4 mg).
4.4. Characterization of BSA-glycopolymer conjugate
BSA-glycopolymer conjugate was analyzed by SDS PAGE, followed by Coomassie blue and carbohydrate staining, respectively. Coomassie blue staining was performed by following the Laemmli method[20] under reducing and non-reducing condition. Carbohydrate staining was conducted by following the protocol provided with the glyco-staining kit (G-Biosciences, St. Louis, MO). MALDI TOF experiments were carried out by first dissolving the material in water (1 mg/mL), then mixing with a saturated sinapic acid matrix solution (Water:Acetonitrile:Trifluoroacetic Acid, 50:50:0.1, v/v ratio). Then, 1 μL of the sample was deposited on MALDI MS steel plate, dried and analyzed by Bruker Autoflex III smartbeam spectrometer in linear positive mode.
4.5. Expression and purification of recombinant thrombomodulin EGF4,5,6 domains (rTM456
The rTM456 was prepared by transformation of expression plasmid encoding rTM456 into E. coli BL21 (DE3) as previously reported.[19] Briefly, bacteria were grown in LB medium with 35 mg/L kanamycin at 37 °C. Upon obtaining a value of OD600nm between 0.8 and 1.0, protein expression was induced with the addition of IPTG (isopropyl-β-D-1-thiogalactopyranoside, 0.5 mM), and the culture was incubated overnight at 25°C. Cells were harvested by centrifugation for 30 min. at 8000 × g at 4 °C. The pellets were collected and dissolved in a cell lysis buffer (20 mM Tris-HCl, 300 mM NaCl, pH 8.0) to induce cell membrane rupturing and release of cytosolic proteins. In order to prevent cytosolic protein breakdown by cellular proteases, a serine protease inhibitor, PMSF (10 μg/mL), was added. The cells were then sonicated with a probe sonicator (Sonifier 450, Branson Ultrasonics, CT, USA) 10 times for 30 sec., followed by 2 min. storage on ice after every sonication. Cell disruption was evident after partial clearing of the suspension. The broken cells were then centrifuged at 15000 × g for 30 min. and the supernatant was collected for purification. The supernatant was directly applied to an immobilized metal affinity chromatography column (HisTrap FF, GE), which was previously equilibrated with a washing buffer (20 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, pH 8.0). The column was washed with 10 column volumes of washing buffer then flushed with 3 column volumes of elution buffer (20 mM Tris-HCl, 300 mM NaCl, 250 mM imidazole, pH 8.0) to afford ‘His’ and ‘S’ tagged rTM456. Finally, recombinant enterokinase was used for site-specific cleavage to remove the fusion tags and generate the target protein rTM456. Recombinant enterokinase was removed by following instruction from kit, and digested solution was loaded into Ni2+ affinity chromatography to remove cleaved tag. The target protein, rTM456 was collected after passing through chromatography and dialyzed against Tris-HCl buffer (pH 8.0) and characterized by SDS-PAGE (17 kDa MW).
4.6. Synthesis and characterization of rTM456-glycopolymer conjugate
20 μL of 20 mM Tris-HCl buffer solution (pH 8.0)containing 2 mg of O-cyanate chain-end functionalized glycopolymer was added to 100 μL of the same buffer containing a solution containing 10 μg (0.6 nmol) of rTM456. The mixture was stirred at 4 °C for 24 h. Then the reaction mixture was dialyzed (13 kDa cutoff) against DI water for 48 h to remove the unreacted glycopolymer and then lyophilized to afford the rTM456-glycopolymer conjugate (20 μg). The control reaction of rTM456 with OH-terminated glycopolymer was conducted under the same conditions as above. SDS-PAGE gel (12.5%) was run, followed by glyco-staining and total protein staining to confirm the reaction product by following the manufacturer procedure (GBiosciences). Western blot of rTM456, rTM456 reacted with O-cyanate-terminated glycopolymer and rTM456 reacted with OH-terminated glycopolymer were performed using mouse monoclonal antibody specific to human thrombomodulin, respectively. MALDI TOF experiments were carried out by first dissolving the rTM456-glycopolymer conjugate in water (1 mg/mL) and then mixing with a saturated sinapic acid matrix solution (Water:Acetonitrile:Trifluoroacetic Acid, 50:50:0.1, v/v ratio). 1 μL of sample was then deposited on polished steel plate, dried and sampled.
4.7. Protein C activation activity of rTM456-glycopolymer conjugate
Activation experiments were conducted at 37 °C in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 0.1% BSA, and 5 mM Ca2+. Protein activation solution contained 0.5 nM of rTM456, 5 nM of thrombin, and 0.1 μM of protein C. Activation was stopped by adding antithrombin III (300 μg/mL final concentration). The activated protein C present was measured by hydrolysis of Spectrozyme PCa at 25 °C in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.4, containing 1 mg mL-1 of BSA. The produced active PC was measured through hydrolysis of Spectrozyme PCa by comparing a standard curve, in which the concentration of active PC to the rate of pnitroanilide (pNA) formation was measured. The hydrolysis of Spectrozyme PCa was performed for 10 min in the assay buffer at 37 °C, in which pNA was produced and its concentration measured by monitoring at λ = 405 nm with a UV spectrophotometer.
Supplementary Material
Figure 5.

MALDI TOF spectra of (A) rTM456 (B) and rTM456-glycopolymer (Matrix:Water:Acetonitrile:Trifluoroacetic Acid, 50:50:0.1, v/v ratio).
Scheme 1.
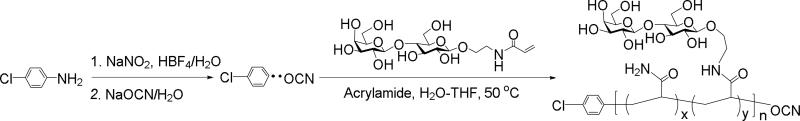
Synthesis of O-cyanate chain-end functionalized glycopolymer with lactose pendant units via CMFRP.
Acknowledgements
This work was partly supported by grants from the NIH (1R01HL102604-04, X.-L. Sun), National Science Foundation MRI Grant (CHE-1126384, X.-L. Sun), Faculty Research Development Fund and the research fund at the Center for Gene Regulation in Health and Disease (GRHD) at Cleveland State University supported by Ohio Department of Development (ODOD). MALDI TOF data was supported by the National Science Foundation under Grant No. MRI-0821515 at Case Western Reserve University.
References
- 1.Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2008;22:315–29. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- 2.Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. Journal of pharmaceutical sciences. 2005;94:1626–35. doi: 10.1002/jps.20319. [DOI] [PubMed] [Google Scholar]
- 3.Elliott S, Lorenzini T, Asher S, Aoki K, Brankow D, Buck L, Busse L, Chang D, Fuller J, Grant J, Hernday N, Hokum M, Hu S, Knudten A, Levin N, Komorowski R, Martin F, Navarro R, Osslund T, Rogers G, Rogers N, Trail G, Egrie J. Enhancement of therapeutic protein in vivo activities through glycoengineering. Nature biotechnology. 2003;21:414–21. doi: 10.1038/nbt799. [DOI] [PubMed] [Google Scholar]
- 4.Grover GN, Maynard HD. Protein-polymer conjugates: synthetic approaches by controlled radical polymerizations and interesting applications. Current opinion in chemical biology. 2010;14:818–27. doi: 10.1016/j.cbpa.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Q, Dordick JS, Linhardt RJ. Synthesis and application of carbohydrate-containing polymers. Chemistry of Materials. 2002;14:3232–3244. [Google Scholar]
- 6.Geng J, Mantovani G, Tao L, Nicolas J, Chen G, Wallis R, Mitchell DA, Johnson BR, Evans SD, Haddleton DM. Site-directed conjugation of “clicked” glycopolymers to form glycoprotein mimics: binding to mammalian lectin and induction of immunological function. Journal of the American Chemical Society. 2007;129:15156–63. doi: 10.1021/ja072999x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baslé E, Joubert N, Pucheault M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010;17:213–227. doi: 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Porath J, Axen R, Ernback S. Chemical coupling of proteins to agarose. Nature. 1967;215:1491–1492. doi: 10.1038/2151491a0. [DOI] [PubMed] [Google Scholar]
- 9.Tugulu S, Silacci P, Stergiopulos N, Klok HA. RGD-Functionalized polymer brushes as substrates for the integrin specific adhesion of human umbilical vein endothelial cells. Biomaterials. 2007;28:2536–46. doi: 10.1016/j.biomaterials.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Grande D, Baskaran S, Baskaran C, Gnanou Y, Chaikof EL. Glycosaminoglycan-mimetic biomaterials. 1. Nonsulfated and sulfated glycopolymers by cyanoxyl-mediated free-radical polymerization. Macromolecules. 2000;33:1123–1125. [Google Scholar]
- 11.Narla SN, Sun XL. Orientated glyco-macroligand formation based on site-specific immobilization of O-cyanate chain-end functionalized glycopolymer. Organic & biomolecular chemistry. 2011;9:845–50. doi: 10.1039/c0ob00556h. [DOI] [PubMed] [Google Scholar]
- 12.Narla SN, Sun XL. Glyco-macroligand microarray with controlled orientation and glycan density. Lab on a chip. 2012;12:1656–63. doi: 10.1039/c2lc21224b. [DOI] [PubMed] [Google Scholar]
- 13.Esmon CT, Gu JM, Xu J, Qu D, Stearns-Kurosawa DJ, Kurosawa S. Regulation and functions of the protein C anticoagulant pathway. Haematologica. 1999;84:363–8. [PubMed] [Google Scholar]
- 14.Zushi M, Gomi K, Yamamoto S, Maruyama I, Hayashi T, Suzuki K. The last three consecutive epidermal growth factor-like structures of human thrombomodulin comprise the minimum functional domain for protein C-activating cofactor activity and anticoagulant activity. The Journal of biological chemistry. 1989;264:10351–3. [PubMed] [Google Scholar]
- 15.Suzuki M, Mohri M, Yamamoto S. In vitro anticoagulant properties of a minimum functional fragment of human thrombomodulin and in vivo demonstration of its benefit as an anticoagulant in extracorporeal circulation using a monkey model. Thrombosis and haemostasis. 1998;79:417–22. [PubMed] [Google Scholar]
- 16.Sun XL, Grande D, Baskaran S, Hanson SR, Chaikof EL. Glycosaminoglycan mimetic biomaterials. 4. Synthesis of sulfated lactose-based glycopolymers that exhibit anticoagulant activity. Biomacromolecules. 2002;3:1065–1070. doi: 10.1021/bm025561s. [DOI] [PubMed] [Google Scholar]
- 17.Sun XL, Faucher KM, Houston M, Grande D, Chaikof EL. Design and synthesis of biotin chain-terminated glycopolymers for surface glycoengineering. Journal of the American Chemical Society. 2002;124:7258–9. doi: 10.1021/ja025788v. [DOI] [PubMed] [Google Scholar]
- 18.Hou S, Sun XL, Dong CM, Chaikof EL. Facile synthesis of chain-end functionalized glycopolymers for site-specific bioconjugation. Bioconjugate chemistry. 2004;15:954–9. doi: 10.1021/bc0499275. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Weingart J, Jiang R, Peng J, Wu Q, Sun XL. Bio-inspired liposomal thrombomodulin conjugate through bio-orthogonal chemistry. Bioconjugate chemistry. 2013;24:550–9. doi: 10.1021/bc300399f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.