

Summary
High-level resistance to β-lactam antibiotics in methicillin-resistant Staphylococcus aureus (MRSA) is due to expression of penicillin-binding protein 2a (PBP2a), a transpeptidase that catalyzes cell-wall crosslinking in the face of the challenge by β-lactam antibiotics. The activity of this protein is regulated by allostery at a site 60 Å distant from the active site, where crosslinking of cell wall takes place. This review discusses the state of knowledge on this important enzyme of cell-wall biosynthesis in MRSA.
Keywords: methicillin-resistant Staphylococcus aureus, allosteric regulation, conformational change, β-lactam antibiotics, resistance mechanism
Introduction
The Gram-positive bacterium Staphylococcus aureus is a global concern in light of broad resistance to the majority of available treatment options. It is a common cause of skin and respiratory system infections. Penicillins, which inhibit cell-wall synthesis, were the first-line treatment of infections by S. aureus. But within a few years of penicillin use, 50% of S. aureus infections were resistant to the drug due to the catalytic function of β-lactamase, a resistance enzyme that hydrolytically turns over these antibiotics, rendering them inactive. A second generation of semi-synthetic penicillins was introduced in 1959 expressly to circumvent the β-lactamase activity. These antibiotics, which include methicillin and oxacillin, among others, were resistant to the action of the S. aureus β-lactamase. However, the first methicillin-resistant Staphylococcus aureus (MRSA), strain was identified in the UK in 1961 (1). It became a global problem with two years. This resistance phenotype was due to the acquisition of a gene that encodes for a protein that overcomes the challenge by a broad range of β-lactam antibiotics (including penicillins, cephalosporins and carbapenems; Figure 1), which is the subject of discussion in the ensuing sections. Currently, the drugs of choice for treatment of infections by MRSA are vancomycin (2), daptomycin (3), ceftaroline (4) and linezolid (5). Even with controlled use, strains that show reduced susceptibility, or outright resistance, to these drugs have arisen (6–9).
Figure 1.

The core structures of penicillins, cephalosporins, and carbapenems mimic the d-Ala-d-Ala of the peptide stem of the cell wall.
Cell-wall synthesis and the roles of penicillin-binding proteins
Survival of bacteria depends on the health of the cell wall. During growth and division, bacteria biosynthesize cell wall, which is a polymer made up of the peptidoglycan as the principle building unit. The peptidoglycan is composed of repeats of the disaccharide N-acetylglucosamine (NAG)-N-acetylmuramic acid (NAM) with peptide stems on the NAM unit. The peptide stems of neighboring peptidoglycan strands are the points of crosslinking to give the mature cell wall (10, 11) (Figure 2). The peptide stem varies by organism, but in S. aureus it is the pentapeptide l-Ala-γ-d-Glu-l-Lys(Gly)5-d-Ala-d-Ala; the pentaglycyl moiety being linked to the lysine side chain. Synthesis of the peptidoglycan backbone is carried out by the catalytic function of transglycosylases, with transpeptidases—also referred to as penicillin-binding proteins (PBPs)—performing the crosslinking reaction. Both activities could be present in the same protein, but not necessarily. Coordination of these reactions is an elaborate process, which ultimately results in the cell-wall structure (12, 13). As the activity of PBPs is essential for bacterial survival, they are desirable targets of antibiotics, specifically by β-lactams. In S. aureus, there exist four native PBPs: PBP1, PBP2, PBP3, and PBP4. There is a fifth PBP in MRSA, namely PBP2a. This is the aforementioned resistance determinant in MRSA, encoded by the gene mecA, which was acquired from a non-S. aureus origin. Tipper and Strominger argued that PBPs recognize and are efficiently inhibited by β-lactam antibiotics due to the similarity of the β-lactam backbone to the acyl-d-Ala-d-Ala segment of the peptide stem in the peptidoglycan, the physiological substrate of PBPs (14) (Figure 1). PBP2a is a unique transpeptidase that is not inhibited well by β-lactam antibiotics. Hence, it is able to continue peptidoglycan crosslinking in the face of the challenge by these antibiotics, when other PBPs with transpeptidase activity are inhibited.
Figure 2.

Crosslinking of peptidoglycan strands in cell-wall synthesis. Elongation of the glycan strand is carried out by PBPs with transglycosylase activity. The transpeptidation reaction, where the terminal d-Ala is displaced and the peptide stems are crosslinked is accomplished by PBPs such as PBP2a in MRSA.
Mechanism of resistance in Staphylococcus aureus
Bacteria have evolved numerous strategies to counter the effect of antibiotics, which have been reviewed elsewhere (15–18). Staphylococcus aureus produces the PC1 β-lactamase, the product of the blaZ gene, which was the original mechanism of resistance to β-lactam antibiotics in this organism. We described above that the β-lactamase hydrolyzes the β-lactam ring, rendering it inactive. Methicillin-resistant S. aureus has also acquired the mecA gene, which encodes PBP2a (19–23), enabling the bacteria to sustain cell-wall synthesis when other PBPs are inhibited by β-lactam antibiotics. Additional genes that are necessary for methicillin resistance have also been identified, among them fem (factors essential for expression of methicillin resistance) and aux (auxiliary) factors (24, 25).
As stated earlier, S. aureus acquired the mecA gene from an unknown source, which might be another staphylococcal organism, as mecA homologues have been identified in Staphylococcus sciuri, Staphylococcus vitulinus, and Staphylococcus fleurettii (26–28). Although resistance by the organism is contingent on the presence of the mecA gene, some strains that contain mecA exhibit varied phenotypic expression (29, 30). Staphylococcus aureus PBP2a is a high-molecular-weight class B PBP and is found only in resistant bacteria. The transpeptidase active site of PBP2a, as in the cases of other PBPs, contains the active-site serine (S403) at the N-terminus of the α2 helix in the sequence motif SXXK. The β-lactam antibiotic binds to the active site and forms a rapidly reversible Michaelis complex (EI) (Figure 3). This complex is converted to a stable covalent adduct by nucleophilic attack by S403 on the β-lactam ring (acyl-enzyme species, E-I). The deacylation reaction is slow, with t1/2 values between 26 and 77 hours (31), which renders acylation effectively irreversible. The rate of acylation of PBP2a is slow (t1/2 = 3–12 min) and the dissociation constants for all classes of β-lactams (~600–1600 μM) are unfavorable (31), both of which contribute to the manifestation of resistance. These observations were interpreted with X-ray structures for the apo enzyme (32) and the enzyme in complex with antibiotics such as ceftaroline (33) and ceftobiprole (34), showing that the main obstacle to β-lactam acylation of the enzyme is the inaccessibility of the active site.
Figure 3.
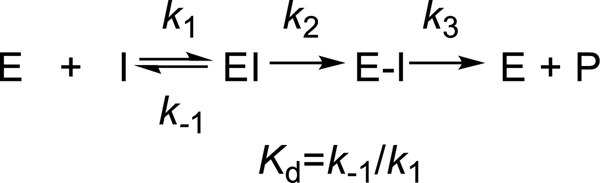
The minimal scheme for interactions of PBP2a with β-lactam antibiotics. The β-lactam interacts reversibly with the free enzyme before forming the stable acyl-enzyme species (E–I). Deacylation is slow (k3) and the reaction is essentially irreversible.
Structural basis for resistance
Lim and Strynadka published the first crystal structure of soluble PBP2a at 1.8 Å resolution in both the apo (Figure 4A) (PDB ID 1VQQ) and the antibiotic-acylated forms (32). These structures showed the enzyme as an elongated protein with a transpeptidase domain (residues 327–668) and what the authors referred to as a non-penicillin-binding domain (residues 27–326), which includes an N-terminal extension subdomain (residues 27–138). The full-length protein also contains a transmembrane anchor segment (residues 1–23), which can be removed to make the protein soluble for characterization (31, 35, 36). Structures of apo-PBP2a and nitrocefin-acylated PBP2a revealed a closed active site, such that an intact β-lactam could not gain access to the active site. This suggested the need for a conformational change at the active site in order to accommodate the antibiotic. The closed active site can explain resistance, but not the physiological reaction of PBP2a. The enzyme is able to efficiently perform the peptidoglycan-crosslinking reaction, which requires the ability of the active site to accommodate the two strands of the peptidoglycan, which entail a volume in excess of 1000 Å3. This indicates that the protein should undergo conformational changes in the course of catalysis, which has been documented to be the case, as we will elaborate.
Figure 4.

X-ray structures of PBP2a in A. the apo-enzyme form (PDB code 1VQQ) B. complex with ceftaroline bound at the active site and allosteric site (3ZG0) C. complex of PBP2a with ceftobiprole acylating the active site (4DKI). The side chains of Y446 and M641, which act as gatekeepers of the active site, are shown as black sticks (1 o’clock). D. Chemical structures of ceftobiprole and ceftaroline. The R1 and R2 groups are shown in blue and red, respectively.
Circumvention of the problem by new β-lactam antibiotics
While PBP2a has low affinity for most β-lactam antibiotics, it has higher affinity for ceftaroline, the most recently approved cephalosporin. (Figure 4D). Ceftaroline is the active form of the prodrug ceftaroline fosamil. It is resistant to the S. aureus β-lactamase activity. However, ceftaroline binds to S. aureus PBP1, PBP2, PBP3, and PBP2a (IC50 <1 μg/mL in most cases) avidly, with PBP4 showing lower affinity (37, 38). It has favorable low MICs (0.5–1 μg/mL) against MRSA strains (37, 39, 40). It has been approved by the FDA for use in the treatment of community-acquired pneumonia and acute bacterial skin and skin structure infections. Current treatment is an intravenous dosage of 600 mg every 12 h for 5–14 days depending on the infection. A second new cephalosporin, ceftobiprole (Figure 4D), seems to have comparable properties. It has activity against MRSA and vancomycin-resistant strains of MRSA. It has been approved for use in Europe against hospital- and community-acquired pneumonia, but has not been approved by the FDA for use in the United States. It has MIC values in the range of 0.5 – 4 μg/mL against MRSA (41–43). As with ceftaroline, it is a poor substrate for the PC1 β-lactamase (41), but it is a good inhibitor of all PBPs of S. aureus, with IC50 values of <1 μg/mL (42).
PBP2a-ceftaroline complex reveals allosteric regulation of resistance
The active site of PBP2a is impervious to inhibition by β-lactam antibiotics because it exists in a closed conformation, with the active-site serine deep in a tight groove (32). However, it would have to open up to accommodate the two peptidoglycan strands in order to carry out the transpeptidation reaction in cell-wall synthesis. This process requires an active site with a volume of more than 1000 Å3 (44, 45), much larger than that needed for interaction with an antibiotic. There is precedence that proteins experience conformational change by allosteric modulation, occasionally with the two binding sites separated by a large distance (46–48). In fact, when the PBP2a structure was analyzed with the CASTp server (49)—which identifies surface-accessible and interior pockets that could be associated with substrate binding—two major grooves were identified: one at the active site, where binding of antibiotic or peptidoglycan is known to take place, and another within the non-penicillin-binding domain. This study was prescient. As it turns out, both sites are involved in catalysis (33), as explained below.
We first reported the possibility of an allosteric site in PBP2a in 2005 after observing an increase in the acylation rate of PBP2a in the presence of a synthetic peptidoglycan fragments (50). These results indicated that the active site becomes more available to the antibiotic, and in principle, the process could also enable catalysis, when the task is performed by the allosteric trigger. We proposed the existence of a second peptidoglycan-binding site outside of the active site. Recently, we reported a crystal structure of PBP2a in complex with the synthetic peptidoglycan fragment (PDB 3ZG5) and two structures in complex with ceftaroline (PDB 3ZFZ and 3ZG0) (Figure 4B) (33). The synthetic peptidoglycan fragment was non-covalently bound in the non-penicillin-binding domain. In the complexes with ceftaroline, as expected, we observed that ceftaroline not only acylates the active-site serine, but a second ceftaroline molecule was also found non-covalently bound in the non-penicillin-binding domain, between the N-terminal extension and Lobe 2 (33). This supports the Tipper-Strominger hypothesis (14), that ceftaroline is recognized not only at the active site, but also at a second site as a result of the similarity of its core structure to the peptidoglycan peptide stem. The β-lactam backbone of this ceftaroline molecule interacts with Y105 and Y297, and salt bridge interactions unique to the PBP2a-ceftaroline complex are formed around this second binding site. The function of this domain was unknown prior to this study. We now know that binding at this domain can trigger a conformational change that activates the protein for the transpeptidase activity, indicative of allostery. For the remainder of this review, this domain will be referred to as the allosteric domain (Figure 4).
The allosteric domain is found 60 Å distant from the active site. In the X-ray structure of PBP2a with ceftaroline bound at both the allosteric and active sites, a large conformational change was seen through the protein when compared to the structure of the apo enzyme (PDB 1VQQ) (33). A smaller conformational change occurred when the peptidoglycan fragment was bound at the allosteric site, but the active site remained closed. Ceftaroline binding at the allosteric site translated to a set of conformational changes that resulted in a twist of the β3 strand near the active site, enabling its acylation with the antibiotic.. This results in an active-site volume increase from approximately 500 Å3 to 1300 Å3, allowing not only acylation by the antibiotic, but also accommodation of the peptidoglycan strands for the transpeptidation event.
This allosteric triggering propagates to the active site through a network of salt bridges, akin to dominoes falling from one site to the other (a span of 60 Å) (33). A series of single, double, and triple mutations of the residues implicated in this salt-bridge network was produced and the effect on acylation was tested. Only four of the twelve mutants had no impact on acylation of PBP2a by ceftaroline. Three mutants with substitutions in the allosteric site (K188A, K188A-K219A, and K219A-D367A) show diminished acylation ability (3–4-fold decrease in k2/Ks (33)). Two mutants (K387A-D635A and D343A-E389A-D635A) were unable to be acylated by nitrocefin. The final three mutants showed a modest decrease in acylation by ceftaroline. As we reported previously, the presence of a synthetic peptidoglycan fragment increases the acylation efficiency of PBP2a (50). When we investigated the ability of this compound to facilitate acylation in the mutants, K287A-D635A was still unable to be acylated and the acylation of D343A-E389A-D635A was diminished compared to wild-type PBP2a. These results indicate that mutations in the salt-bridge network closer to the active site have a larger effect on the signal transmission. It would appear that there are several routes for the propagation of the conformational changes from the allosteric site to the active site through the different lobes of the allosteric domain, with residues near the active site being critical as the bottleneck.
While ceftaroline and a few other β-lactam antibiotics, including ceftobiprole and L-695,256 (51) have good activity against MRSA, some resistance has emerged. Clinical isolates showing decreased susceptibility to ceftaroline, with MIC values of 4 μg/mL, have led to the identification of mutations in PBP2a (52). These include two mutations in the allosteric site, N146K and E150K, and a third mutation outside the allosteric site, H351N. Likewise, strains with mutations around the allosteric site have shown decreased susceptibility to ceftobiprole and L-695,256. These mutations include E150K, E237K, and E239K (53, 54). While it has been suggested that these mutations may disrupt the interaction of PBP2a with other proteins (53), we propose that these mutations either affect binding at the allosteric site or the signal propagation that leads to the opening of the active site.
Comparison of PBP2a in complex with ceftaroline and ceftobiprole
A crystal structure of PBP2a acylated by ceftobiprole was recently published (34) in which structural changes in the β3 strand and the α2 helix, at the N-terminus of which one finds the active-site serine, were similar to those observed in the PBP2a-ceftaroline complex (33). One notable difference between the acyl-enzyme structures with ceftaroline and ceftobiprole is the absence of the E602-R612 salt bridge that is made as a result of interaction with the R1 group of ceftaroline seen in the PBP2a-ceftaroline complex. In addition, the R2 group of ceftobiprole is stacked between Y446 and M641 in the PBP2a-ceftobiprole structure (34), while upon acylation by ceftaroline, the two residues interact to close the active site (33) (Figure 4A–C). Also absent from the ceftobiprole acyl-enzyme structure is the salt-bridge interaction between K387 and D635 due to a different conformation of D635. This residue appears to play an important role in the conformational change seen in the ceftaroline acyl-enzyme structure, as mutants with an alanine substitution at this position K387A-D635A and D343A-E389A-D635A completely abolished acylation activity (33).
New treatments against MRSA
Despite success in discoveries of new β-lactam antibiotics for treatment of infections by MRSA, resistance is already beginning to emerge. The inexorable emergence of resistance to any antibiotic will always be with us, hence the need for new antibiotics. Recent examples include non-β-lactam antibiotics that target PBP2a and other PBPs (55), new drug scaffolds (56), new antibiotic targets such as the isoprenoid biosynthesis (57), and combination therapy, in which synthetic compounds of various types synergize with conventional antibiotics to fight resistance (58–61). The identification of the allosteric site provides an activation mechanism for PBP2a to facilitate the transpeptidase activity. The allosteric site is an unexploited drug target, as its function is critical to catalysis in cell-wall biosynthesis.
References
- 1.Jevons MP. Celbenin resistant staphylococci. BMJ. 1961;1:124–125. [Google Scholar]
- 2.Walsh C. Deconstructing vancomycin. Science. 1999;284:442–443. doi: 10.1126/science.284.5413.442. [DOI] [PubMed] [Google Scholar]
- 3.French GL. Bactericidal agents in the treatment of MRSA infections--the potential role of daptomycin. J. Antimicrob. Chemother. 2006;58:1107–1117. doi: 10.1093/jac/dkl393. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez PO, Lema S, Tyring SK, Mendoza N. Ceftaroline in complicated skin and skin-structure infections. Infect. Drug Resist. 2012;5:23–35. doi: 10.2147/IDR.S17432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilcox MH. Update on linezolid: The first oxazolidinone antibiotic. Expert Opin. Pharmacother. 2005;6:2315–2326. doi: 10.1517/14656566.6.13.2315. [DOI] [PubMed] [Google Scholar]
- 6.Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, et al. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997;40:135–136. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- 7.Appelbaum PC. The emergence of vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2006;12(Suppl 1):16–23. doi: 10.1111/j.1469-0691.2006.01344.x. [DOI] [PubMed] [Google Scholar]
- 8.Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 2003;348:1342–1347. doi: 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- 9.Stryjewski ME, Corey GR. Methicillin-resistant Staphylococcus aureus: An evolving pathogen. Clin. Infect. Dis. 2014;58:S10–S19. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- 10.Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, et al. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4404–4409. doi: 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugg TD, Walsh CT. Intracellular steps of bacterial cell wall peptidoglycan biosynthesis: Enzymology, antibiotics, and antibiotic resistance. Nat. Prod. Rep. 1992;9:199–215. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- 12.Scheffers DJ, Pinho MG. Bacterial cell wall synthesis: New insights from localization studies. Microbiol. Mol. Biol. Rev. 2005;69:585–607. doi: 10.1128/MMBR.69.4.585-607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 14.Tipper DJ, Strominger JL. Mechanism of action of penicillins: A proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. U.S.A. 1965;54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox G, Wright GD. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013;303:287–292. doi: 10.1016/j.ijmm.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Hawkey PM. The origins and molecular basis of antibiotic resistance. BMJ. 1998;317:657–660. doi: 10.1136/bmj.317.7159.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher JF, Mobashery S. 8.13 - Enzymology of bacterial resistance. In: Liu HW, Mander L, editors. Comprehensive Natural Products II. Oxford: Elsevier; 2010. pp. 443–487. [Google Scholar]
- 18.Lowy FD. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Invest. 2003;111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blazquez B, Llarrull LI, Luque-Ortega JR, Alfonso C, Boggess B, et al. Regulation of the expression of the beta-lactam antibiotic-resistance determinants in methicillin-resistant Staphylococcus aureus (MRSA) Biochemistry. 2014;53:1548–1550. doi: 10.1021/bi500074w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha J, Vakulenko SB, Mobashery S. Characterization of the beta-lactam antibiotic sensor domain of the MecR1 signal sensor/transducer protein from methicillin-resistant Staphylococcus aureus. Biochemistry. 2007;46:7822–7831. doi: 10.1021/bi7005459. [DOI] [PubMed] [Google Scholar]
- 21.McKinney TK, Sharma VK, Craig WA, Archer GL. Transcription of the gene mediating methicillin resistance in Staphylococcus aureus (mecA) is corepressed but not coinduced by cognate mecA and beta-lactamase regulators. J. Bacteriol. 2001;183:6862–6868. doi: 10.1128/JB.183.23.6862-6868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosato AE, Kreiswirth BN, Craig WA, Eisner W, Climo MW, et al. mecA-blaZ corepressors in clinical Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 2003;47:1460–1463. doi: 10.1128/AAC.47.4.1460-1463.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryffel C, Kayser FH, Berger-Bachi B. Correlation between regulation of mecA transcription and expression of methicillin resistance in staphylococci. Antimicrob. Agents Chemother. 1992;36:25–31. doi: 10.1128/aac.36.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger-Bachi B, Barberis-Maino L, Strassle A, Kayser FH. femA a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: Molecular cloning and characterization. Mol. Gen. Genet. 1989;219:263–269. doi: 10.1007/BF00261186. [DOI] [PubMed] [Google Scholar]
- 25.de Lencastre H, Tomasz A. Reassessment of the number of auxiliary genes essential for expression of high-level methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1994;38:2590–2598. doi: 10.1128/aac.38.11.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuda C, Suvorov M, Shi Q, Hesek D, Lee M, et al. Shared functional attributes between the mecA gene product of Staphylococcus sciuri and penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. Biochemistry. 2007;46:8050–8057. doi: 10.1021/bi7004587. [DOI] [PubMed] [Google Scholar]
- 27.Tsubakishita S, Kuwahara-Arai K, Sasaki T, Hiramatsu K. Origin and molecular evolution of the determinant of methicillin resistance in staphylococci. Antimicrob. Agents Chemother. 2010;54:4352–4359. doi: 10.1128/AAC.00356-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu SW, de Lencastre H, Tomasz A. Expression of high-level methicillin resistance in Staphylococcus aureus from the Staphylococcus sciuri mecA homologue: Role of mutation(s) in the genetic background and in the coding region of mecA. Microbial. Drug Resist. 2005;11:215–224. doi: 10.1089/mdr.2005.11.215. [DOI] [PubMed] [Google Scholar]
- 29.Finan JE, Rosato AE, Dickinson TM, Ko D, Archer GL. Conversion of oxacillin-resistant staphylococci from heterotypic to homotypic resistance expression. Antimicrob. Agents Chemother. 2002;46:24–30. doi: 10.1128/AAC.46.1.24-30.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niemeyer DM, Pucci MJ, Thanassi JA, Sharma VK, Archer GL. Role of mecA transcriptional regulation in the phenotypic expression of methicillin resistance in Staphylococcus aureus. J. Bacteriol. 1996;178:5464–5471. doi: 10.1128/jb.178.18.5464-5471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuda C, Suvorov M, Vakulenko SB, Mobashery S. The basis for resistance to beta-lactam antibiotics by penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. J. Biol. Chem. 2004;279:40802–40806. doi: 10.1074/jbc.M403589200. [DOI] [PubMed] [Google Scholar]
- 32.Lim D, Strynadka NC. Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002;9:870–876. doi: 10.1038/nsb858. [DOI] [PubMed] [Google Scholar]
- 33.Otero LH, Rojas-Altuve A, Llarrull LI, Carrasco-Lopez C, Kumarasiri M, et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. U.S.A. 2013;110:16808–16813. doi: 10.1073/pnas.1300118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovering AL, Gretes MC, Safadi SS, Danel F, de Castro L, et al. Structural insights into the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of ceftobiprole. J. Biol. Chem. 2012;287:32096–32102. doi: 10.1074/jbc.M112.355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu WP, Sun Y, Bauer MD, Paule S, Koenigs PM, et al. Penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus: Kinetic characterization of its interactions with beta-lactams using electrospray mass spectrometry. Biochemistry. 1999;38:6537–6546. doi: 10.1021/bi990025e. [DOI] [PubMed] [Google Scholar]
- 36.Wu CY, Hoskins J, Blaszczak LC, Preston DA, Skatrud PL. Construction of a water-soluble form of penicillin-binding protein 2a from a methicillin-resistant Staphylococcus aure us isolate. Antimicrob. Agents Chemother. 1992;36:533–539. doi: 10.1128/aac.36.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosowska-Shick K, McGhee PL, Appelbaum PC. Affinity of ceftaroline and other β-lactams for penicillin-binding proteins from Staphylococcus aureus and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2010;54:1670–1677. doi: 10.1128/AAC.00019-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, Mobashery S. Co-opting the cell wall in fighting methicillin-resistant Staphylococcus aureus : Potent inhibition of PBP 2a by two anti-MRSA beta-lactam antibiotics. J. Am. Chem. Soc. 2008;130:9212–9213. doi: 10.1021/ja8029448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saravolatz LD, Stein GE, Johnson LB. Ceftaroline: A novel cephalosporin with activity against methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2011;52:1156–1163. doi: 10.1093/cid/cir147. [DOI] [PubMed] [Google Scholar]
- 40.Jones RN, Mendes RE, Sader HS. Ceftaroline activity against pathogens associated with complicated skin and skin structure infections: Results from an international surveillance study. J. Antimicrob. Chemother. 2010;65(Suppl 4):iv17–iv31. doi: 10.1093/jac/dkq252. [DOI] [PubMed] [Google Scholar]
- 41.Hebeisen P, Heinze-Krauss I, Angehrn P, Hohl P, Page MG, et al. In vitro and in vivo properties of Ro 63-9141, a novel broad-spectrum cephalosporin with activity against methicillin-resistant staphylococci. Antimicrob. Agents Chemother. 2001;45:825–836. doi: 10.1128/AAC.45.3.825-836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davies TA, Page MG, Shang W, Andrew T, Kania M, et al. Binding of ceftobiprole and comparators to the penicillin-binding proteins of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2007;51:2621–2624. doi: 10.1128/AAC.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farrell DJ, Flamm RK, Sader HS, Jones RN. Activity of ceftobiprole against methicillin-resistant Staphylococcus aureus strains with reduced susceptibility to daptomycin, linezolid or vancomycin, and strains with defined SCCmec types. Int. J. Antimicrob. Agents. 2014;43:323–327. doi: 10.1016/j.ijantimicag.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 44.Lee W, McDonough MA, Kotra L, Li ZH, Silvaggi NR, et al. A 1.2-Å snapshot of the final step of bacterial cell wall biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2001;98:1427–1431. doi: 10.1073/pnas.98.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Q, Meroueh SO, Fisher JF, Mobashery S. A computational evaluation of the mechanism of penicillin-binding protein-catalyzed cross-linking of the bacterial cell wall. J. Am. Chem. Soc. 2011;133:5274–5283. doi: 10.1021/ja1074739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ansari A, Berendzen J, Bowne SF, Frauenfelder H, Iben IE, et al. Protein states and proteinquakes. Proc. Natl. Acad. Sci. U.S.A. 1985;82:5000–5004. doi: 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 48.Changeux J-P. Allostery and the Monod-Wyman-Changeux model after 50 years. Annu. Rev. Biophys. 2012;41:103–133. doi: 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]
- 49.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, et al. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl. Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuda C, Hesek D, Lee M, Morio K-i, Nowak T, et al. Activation for catalysis of penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus by bacterial cell wall. J. Am. Chem. Soc. 2005;127:2056–2057. doi: 10.1021/ja0434376. [DOI] [PubMed] [Google Scholar]
- 51.Chambers HF. In vitro and in vivo antistaphylococcal activities of L-695,256, a carbapenem with high affinity for the penicillin-binding protein PBP 2a. Antimicrob. Agents Chemother. 1995;39:462–466. doi: 10.1128/aac.39.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendes RE, Tsakris A, Sader HS, Jones RN, Biek D, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J. Antimicrob. Chemother. 2012;67:1321–1324. doi: 10.1093/jac/dks069. [DOI] [PubMed] [Google Scholar]
- 53.Banerjee R, Gretes M, Basuino L, Strynadka N, Chambers HF. In vitro selection and characterization of ceftobiprole-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2008;52:2089–2096. doi: 10.1128/AAC.01403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katayama Y, Zhang H-Z, Chambers HF. PBP 2a mutations producing very-high-level resistance to beta-lactams. Antimicrob. AgentsChemother. 2004;48:453–459. doi: 10.1128/AAC.48.2.453-459.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Daniel PI, Peng Z, Pi H, Testero SA, Ding D, et al. Discovery of a new class of non-beta-lactam inhibitors of penicillin-binding proteins with Gram-positive antibacterial activity. J. Am. Chem. Soc. 2014;136:3664–3672. doi: 10.1021/ja500053x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh CT, Wencewicz TA. Prospects for new antibiotics: A molecule-centered perspective. J. Antibiot. 2014;67:7–22. doi: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 57.Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, et al. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2013;110:123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan CM, Therien AG, Lu J, Lee SH, Caron A, et al. Restoring methicillin-resistant Staphylococcus aureus susceptibility to beta-lactam antibiotics. Sci. Transl. Med. 2012;4:126ra135. doi: 10.1126/scitranslmed.3003592. [DOI] [PubMed] [Google Scholar]
- 59.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, et al. Discovery of wall teichoic acid inhibitors as potential anti-MRSA beta-lactam combination agents. Chem. Biol. 2013;20:272–284. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SH, Jarantow LW, Wang H, Sillaots S, Cheng H, et al. Antagonism of chemical genetic interaction networks resensitize MRSA to beta-lactam antibiotics. Chem. Biol. 2011;18:1379–1389. doi: 10.1016/j.chembiol.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 61.Guignard B, Vouillamoz J, Giddey M, Moreillon P. A positive interaction between inhibitors of protein synthesis and cefepime in the fight against methicillin-resistant Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2013;32:899–907. doi: 10.1007/s10096-013-1824-x. [DOI] [PubMed] [Google Scholar]