

Supplemental Digital Content is available in the text.
Keywords: apoptosis, chemokine CX3CL1, monocytes, oxidative stress
Abstract
Objective—
The CX3C chemokine fractalkine (CX3CL1) has a critical role in the development of atherogenesis because apolipoprotein-E–deficient mice lacking CX3CL1 or its receptor CX3CR1 develop smaller plaques and polymorphisms in CX3CR1 are associated with altered risk of cardiovascular disease. CX3CR1 is found on numerous cell types involved in atherogenesis but seems to have a key role in monocyte function. We aimed to elucidate the role of CX3CL1 in human monocyte survival and determine the mechanism by which CX3CL1 spares monocytes from apoptosis.
Approach and Results—
Primary human monocytes were prepared from healthy donors and subjected to serum-starvation to induce spontaneous apoptosis. The addition of CX3CL1, but not other chemokines tested, promoted monocyte survival in a dose-dependent manner with full-length CX3CL1 (including the mucin stalk) having a more potent antiapoptotic effect than chemokine-domain CX3CL1. The prosurvival effect of CX3CL1 was evident in both monocyte subsets although nonclassical monocytes were more prone to spontaneous apoptosis. In addition, we found that the effect of CX3CL1 was independent of CX3CR1 genotype. Serum-starvation increased the level of intracellular reactive oxygen species, and this was reduced by the addition of CX3CL1. Inhibition of oxidative stress with an antioxidant prevented monocyte apoptosis, indicating that this is the dominant mechanism of cell death targeted by CX3CL1.
Conclusions—
CX3CL1 has a substantial and highly reproducible antiapoptotic effect on human monocytes, via a mechanism involving a reduction in oxidative stress. This suggests that CX3CL1 is likely to play a key role in human atherogenesis and may provide a novel therapeutic target in cardiovascular disease.
Chemokines are a family of low molecular weight soluble proteins, which orchestrate the migration of cells expressing their cognate G-protein–coupled receptors.1 Divided into 4 subfamilies on the basis of structure, chemokines regulate cell trafficking under normal conditions and in response to inflammatory or injurious agents. Dysregulated or excessive chemokine expression is a hallmark of multiple chronic inflammatory diseases, including atherosclerosis.2 Fractalkine (CX3CL1) is the only member of the CX3C family of chemokines and is expressed as a membrane-bound molecule with the chemokine domain (CKD) attached to the cell surface via a mucin-like stalk.3 Cleaved from the cell surface under homeostatic and inflammatory conditions by the metalloproteases ADAM10 and ADAM17, respectively, CX3CL1 expression has been described on neurons, epithelial cells, endothelial, and smooth muscle cells.4–9 CX3CL1 is the unique ligand for the 7 transmembrane G-protein–coupled receptor CX3CR1 expressed on monocytes, natural killer cells, T cells, and smooth muscle cells.10–12 CX3CR1 has 2 nonsynonymous single-nucleotide polymorphisms, V249I and T280M, which are associated with an altered risk of cardiovascular disease.13,14 The CX3CL1-CX3CR1 axis has been implicated in multiple pathologies, including atherosclerosis,15 multiple sclerosis,16 rheumatic diseases,17 and neuropathic pain.18,19
In monocytes, the level of CX3CR1 expression helps to define the 2 major subsets in both humans and mice with classical monocytes (CD14++ CD16- in humans, LyChi in mice), expressing relatively lower levels of CX3CR1 than nonclassical monocytes (CD14+ CD16++ in humans, Ly6Clo in mice).20 Classical monocytes (previously referred to as inflammatory monocytes) are known to be short-lived, respond rapidly to infection or inflammation whereupon they migrate to tissue and differentiate into inflammatory macrophages or dendritic cells capable of producing high levels of effector cytokines.21,22 Nonclassical (known formerly as resident) monocytes have been shown to crawl along the luminal side of endothelial cells and display a patrolling behavior, which allows rapid entry into sites of infection.23 Recent evidence has demonstrated that in mice, nonclassical monocytes in the blood are derived from circulating classical monocytes, with their abundance directly regulated by that of their precursor.24 The 2 subsets also differ in their size (nonclassical monocytes being smaller), their abundance (nonclassical monocytes comprise ≈10% of circulating monocytes in humans), and their expression of the chemokine receptor CCR2: classical monocytes express CCR2; nonclassical monocytes do not.20
Deletion of CX3CL1 and its receptor in murine models of atherosclerosis has shown a nonredundant role for this ligand–receptor pair in atherogenesis. Cx3cr1−/− Apoe−/− mice develop smaller lesions with fewer macrophages when fed either a normal chow or a high-fat diet.25,26 Cx3cl1−/− Apoe−/− or Ldlr−/− mice also develop smaller lesions than single knockout animals.27 In murine models of atherosclerosis, hypercholesterolaemia induces monocytosis, which is dominated by the classical subset and these cells give rise to macrophages within atherosclerotic plaques.28,29 The reversal of hypercholesterolaemia via statin treatment abolishes monocytosis in these animals.28 Interestingly, deficiency of CX3CR1 in Apoe−/− mice has no effect on classical monocyte numbers in the blood but prevents the increase in circulating nonclassical monocytes.30 Deletion of both CCL2 and CX3CR1 abrogates monocytosis of both subsets. These findings were extended by a study which showed that in Apoe−/− mice fed a high-fat diet, transfer of Cx3cr1gfp/gfp bone marrow (which lacks functional CX3CR1) led to reduced monocyte numbers in blood when compared with mice receiving wild-type bone marrow.31 These mice also developed smaller plaques that contained higher numbers of apoptotic cells, suggesting a role for CX3CR1 in promoting intraplaque macrophage survival. These authors also demonstrated that CX3CL1 could prevent apoptosis of human monocytes in vitro although no mechanism for this activity was proposed.
Previous work from our laboratory has demonstrated that CX3CL1 promotes the survival and proliferation of primary human vascular smooth muscle cells via an epidermal growth factor receptor–dependent pathway.32 CX3CL1 has also been implicated in the survival of other cells, including microglia, in a Fas-induced apoptosis model, T cells in a murine asthma model, and monocytes in a murine liver fibrosis model.33–35 Despite the number of published studies implicating CX3CL1 in cell survival, a clear mechanism for this function is lacking.
In this study, we initially aimed to confirm the effect of CX3CL1 on primary human monocyte survival then to extend these findings by establishing the role of the stalk region of CX3CL1 and determining whether the 2 common single-nucleotide polymorphisms in CX3CR1 influence the response to CX3CL1. We sought to elucidate the mechanism further by which CX3CL1 regulates human monocyte survival.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
In the absence of growth factors, monocytes undergo spontaneous apoptosis within hours.36 Using serum deprivation as a reproducible method to induce apoptosis, we first sought to establish whether CX3CL1 was able to prevent this phenomenon. Monocytes from anonymous human donors were isolated using CD14 labeling and positive selection (to generate cells predominantly from the classical monocyte subset unless otherwise specified) and then incubated for 4 hours in serum-free medium in the presence of 10% fetal calf serum (positive control) or increasing doses of CX3CL1 CKD or full-length (FL) CX3CL1, including the mucin stalk (FL). Apoptosis was then quantified by staining with annexin V and 7-aminoactinomycin D. Approximately 30% of monocytes died in this timeframe, and this was significantly reduced by treatment with fetal calf serum (P<0.001), 100 nmol/L CKD CX3CL1 (P<0.001), and 10 to 100 nmol/L FL CX3CL1 (P<0.01–0.001), implying that both forms of CX3CL1 are capable of suppressing monocyte apoptosis but that FL CX3CL1 can do so at lower doses (Figure 1A and 1B). To confirm that there was no difference in quality between the 2 forms of recombinant CX3CL1, we used a second assay—receptor internalization. Monocytes were incubated with an equivalent dose (50 nmol/L) of CKD or FL CX3CL1 then stained for CX3CR1 surface expression. Both forms of CX3CL1 induced an equivalent internalization of receptor when compared with untreated cells (Figure I in the online-only Data Supplement). To assess whether monocytes were capable of producing any endogenous CX3CL1 in response to serum-starvation, we quantified soluble CX3CL1 in cell-free supernatants generated from monocytes starved for 0 to 4 hours in a sandwich ELISA. No CX3CL1 was detectable at any time point (data not shown).
Figure 1.
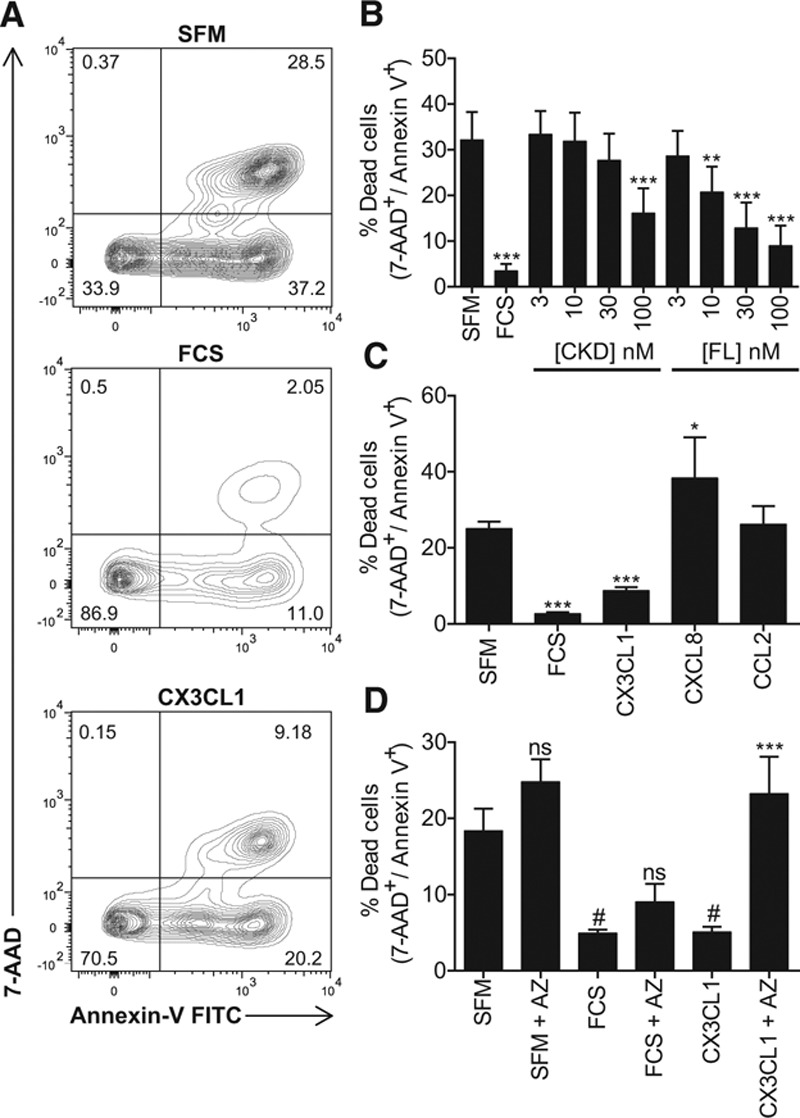
CX3C chemokine fractalkine (CX3CL1) prevents monocyte apoptosis in response to serum-starvation. A, Primary human monocytes were incubated for 4 hours in serum-free medium (SFM; left), in SFM+10% fetal calf serum (FCS; middle), or SFM+100 nmol/L CX3CL1 then stained with annexin V fluorescein isothiocyanate (FITC) and 7-aminoactinomycin D (AAD), and analyzed by flow cytometry. Dead cells were defined as those in the upper right quadrant. B, Monocytes were treated as in A with the indicated doses of chemokine-domain (CKD) CX3CL1 or full-length (FL) CX3CL1, including the mucin stalk. C, Monocytes were treated as in A with the indicated chemokines at a dose of 100 nmol/L. D, Monocytes were pretreated ± AZ12201182 (AZ) and then treated as in A. Data shown as mean+SEM of n=6 donors (B), 5 to 59 donors (C), and 5 donors (D). Data were analyzed with 1-way ANOVA and Dunnett post hoc test (B and C), *P<0.05, **P<0.01, and ***P<0.001 relative to SFM or Sidak’s multiple comparison test (D), #P<0.05 relative to SFM, ns=not significant, ***P<0.001 relative to agonist without AZ.
Because monocytes are reported to express a variety of chemokine receptors implicated in atherosclerosis, we also tested interleukin-8 (CXCL8) and monocyte chemoattractant protein-1 (CCL2) for their ability to promote monocyte survival. At a dose of 100 nmol/L, neither chemokine could rescue monocytes from apoptosis, in fact CXCL8 significantly increased cell death compared with control (P<0.05; Figure 1C). To confirm that the CCL2 used in these experiments was active, we tested it in a real-time monocyte chemotaxis assay where it induced a robust migratory response (Figure II in the online-only Data Supplement). Interestingly, CX3CL1 had no chemotactic effect on these cells, despite robust receptor expression and in contrast to previous reports37 (Figure II in the online-only Data Supplement and data not shown).
To address whether the effect of CX3CL1 on monocyte apoptosis is specifically mediated via CX3CR1, we used a receptor antagonist (AZ12201182 compound 18a, referred to here as AZ), which inhibits CX3CL1 responses in multiple assays.32,38 Pretreatment with AZ blocked the antiapoptotic activity of CX3CL1 but did not increase apoptosis in cells treated with serum-free medium or fetal calf serum (P<0.001; Figure 1D).
We next assessed whether CX3CL1 was able to promote survival of the 3 known human monocyte subsets.39 Human monocytes are divided into 3 subsets based on their expression of CD14 and CD16: classical monocytes (CD14++ CD16−), intermediate monocytes (CD14++ CD16+), and nonclassical monocytes (CD14+ CD16++).20 Monocytes isolated by CD14 selection included the classical and intermediate subsets, whereas those isolated using a CD16 monocyte selection kit included the intermediate and nonclassical subsets of cells (Figure 2A). Confirming a previous report, the extent of apoptosis differed between the 2 subsets (CD16 cells showed increased cell death compared with CD14 cells; P<0.05),40 but CX3CL1 had an equally potent antiapoptotic effect in both populations (P<0.01; Figure 2B).
Figure 2.
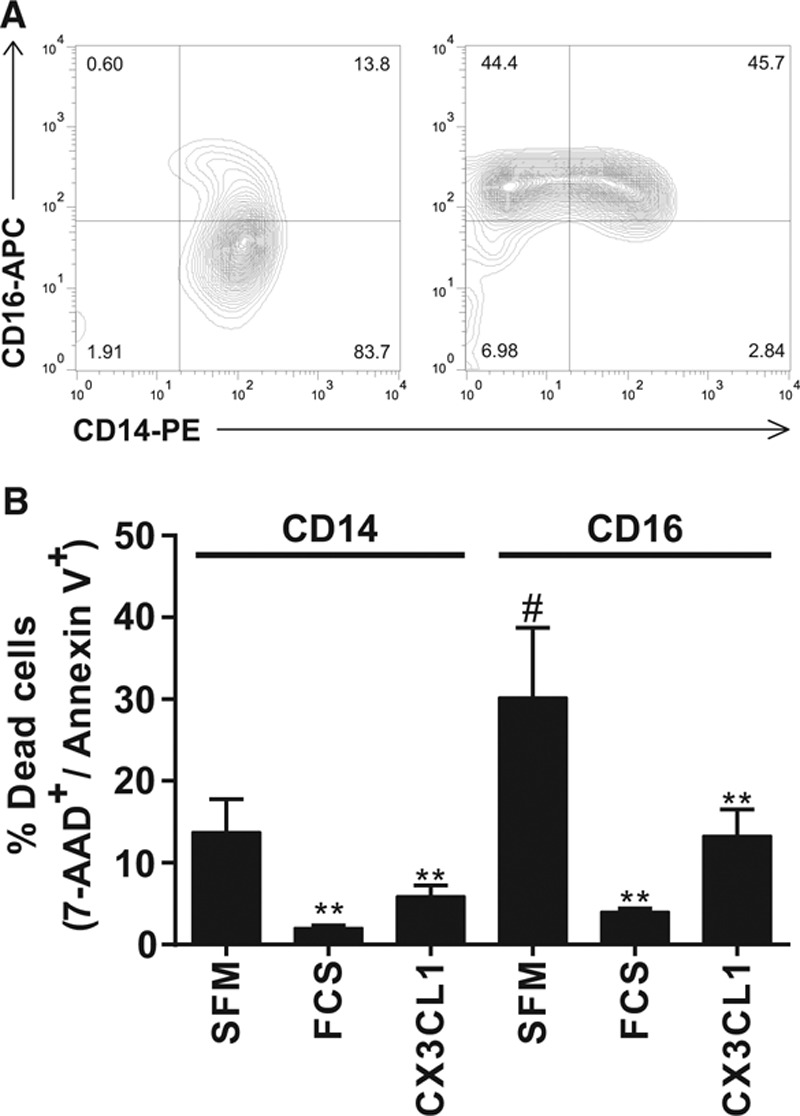
CX3C chemokine fractalkine (CX3CL1) prevents apoptosis of both monocyte subsets. A, Monocytes were isolated with either CD14 labeling and magnetic selection to isolate monocytes of the classical and intermediate subsets (left) or with a CD16 monocyte isolation kit to isolate cells of the nonclassical and intermediate subsets (right). Cells were stained with antibodies against CD14 and CD16 and analyzed by flow cytometry. B, Monocytes of each subset were incubated as in Figure 1 with serum-free medium (SFM), fetal calf serum (FCS), or CX3CL1 and apoptosis quantified by annexin V/7-aminoactinomycin D (AAD), staining. Data shown as mean+SEM of 3 donors from 3 experiments. Data were analyzed by 1-way ANOVA and Tukey multiple comparison test, **P<0.01 relative to SFM of each subset, #P<0.05 relative to SFM of CD14 subset.
Because CX3CL1 seems to have such a potent effect on monocyte survival, we investigated whether the 2 known polymorphisms of CX3CR1 had any effect on the antiapoptotic effect of CX3CL1. DNA from all donors was stored at the time of assay, and we were therefore able to genotype all donors subsequently for both polymorphisms (V249I and T280M) to establish whether CX3CR1 genotype affected response to CX3CL1. Using an allelic discrimination polymerase chain reaction–based assay, we found that 47% of donors possessed the VV/TT genotype, 15% VI/TT, and 38% were heterozygote at both loci VI/TM. We did not identify any donors with the rarer homozygote haplotypes, as would be expected in this modest (n=34) sample size. These frequencies are in line with other published studies although we did see an over-representation of the VI/TM genotype in our sample.41 To minimize variation between experiments, the response to serum-starvation was normalized to 100% and the response to CX3CL1 expressed as a fraction of this. We found that the antiapoptotic effect of CX3CL1 was independent of CX3CR1 genotype in donors with the most common genotypes (Figure 3).
Figure 3.
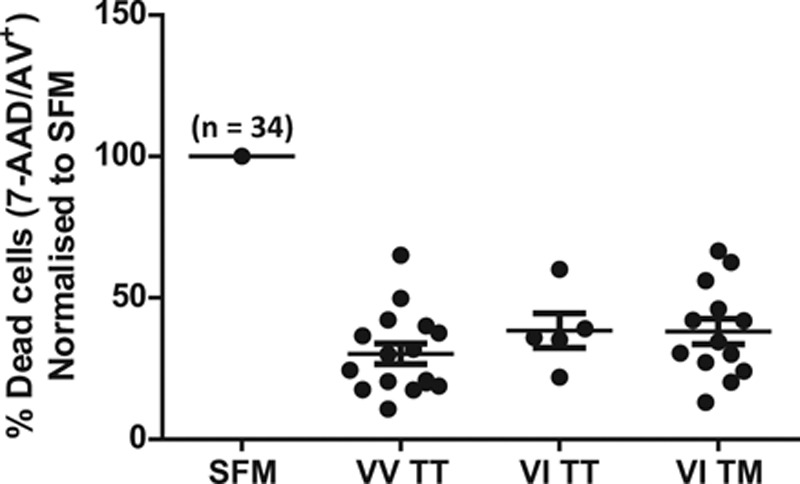
The antiapoptotic effect of CX3C chemokine fractalkine (CX3CL1) is independent of CX3CR1 genotype. Genomic DNA was isolated from a subgroup of donors used in Figure 1C and genotyped for the 2 nonsynonymous single-nucleotide polymorphisms (SNPs) V249I and T280M in CX3CR1 using a polymerase chain reaction–based allelic discrimination assay. Only the 3 most common haplotypes were identified, and the response to CX3CL1 was analyzed by genotype. For each donor in each experiment, the response to serum-starvation was normalized to 100% and the effect of CX3CL1 expressed as a fraction of this to minimize technical variation between experiments. Data shown as mean±SEM, n=34 donors. No significant difference between genotypes was detected using 1-way ANOVA. AAD indicates 7-aminoactinomycin D.
We next sought to address the mechanism by which CX3CL1 mediates monocyte survival. Previous studies have demonstrated that spontaneous apoptosis of monocytes and neutrophils involves an increase in intracellular reactive oxygen species (ROS).40,42 Oxidative stress is known to drive opening of the mitochondrial permeability transition pore, leading to induction of apoptosis via the intrinsic pathway.43 To establish whether growth factor withdrawal induced monocyte oxidative stress in our hands, we used the fluorogenic CellROX green probe, which exhibits bright green fluorescence once oxidized and bound to DNA. Compared with cells incubated with fetal calf serum, monocytes serum-starved for 30 minutes demonstrated an increase in ROS as determined by flow cytometry (Figure 4A; Figure III in the online-only Data Supplement). CX3CL1 was able to block the increase in ROS induced by serum-starvation (P<0.01; Figure 4A), whereas CCL2 used at an equivalent dose had no effect (Figure 4A). We next assessed whether FL CX3CL1 could reduce levels of intracellular ROS. As shown in Figure 4B, FL CX3CL1 also significantly reduced the level of oxidative stress induced by serum-starvation (P<0.05). We used a second assay to measure intracellular superoxide specifically, namely incubation of cells with the superoxide indicator dihydroethidium and quantification of 2-hydroxyethidium (measuring superoxide) and ethidium (measuring other ROS) by high performance liquid chromatography. Serum-starvation increased the generation of 2-hydroxyethidium, and this was blocked by the addition of CX3CL1 (P<0.01; Figure 4C). Levels of ethidium were unaffected by serum-starvation or the addition of CX3CL1 (data not shown).
Figure 4.
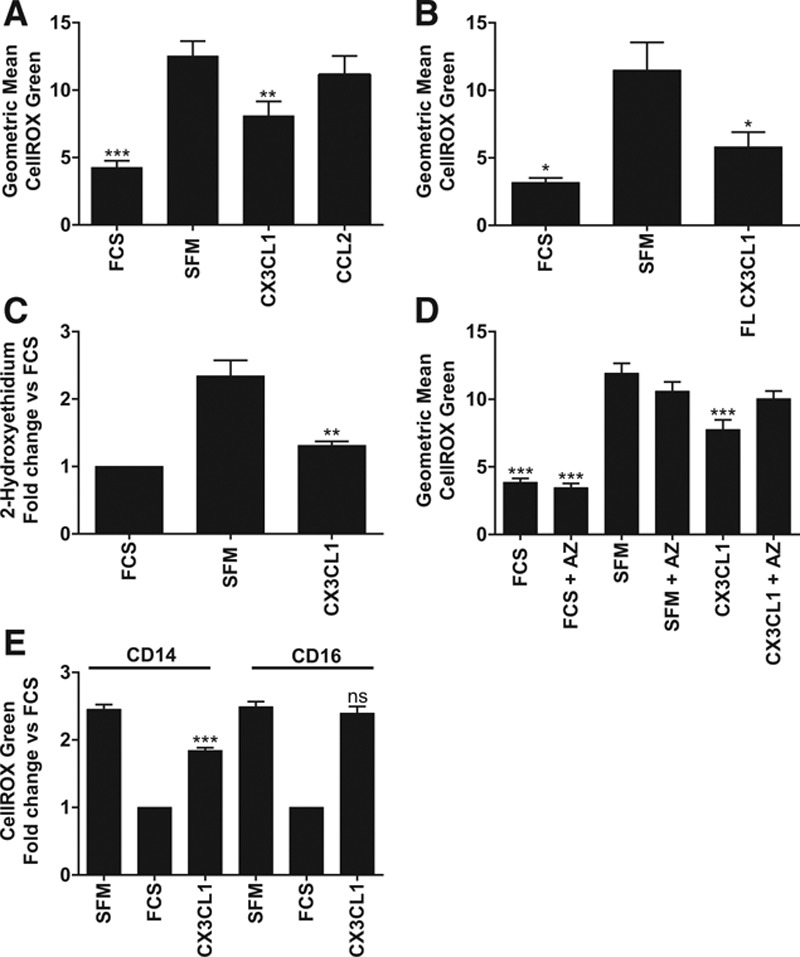
Serum-starvation induces production of reactive oxygen species, which is blocked by CX3C chemokine fractalkine (CX3CL1) in classical monocytes. A, Monocytes were loaded with CellROX green reagent, and then incubated for 30 minutes with serum-free medium (SFM), fetal calf serum (FCS), CX3CL1, or CCL2 then analyzed by flow cytometry. Data are shown as geometric mean of 3 to 12 donors from 2 to 6 experiments. B, Cells were treated as in A with SFM, FCS, or full-length (FL) CX3CL1. n=4 donors from 2 experiments. C, Monocytes were loaded with dihydroethidium then treated with agonists as in A. Levels of 2-hydroxyethidium were analyzed by high performance liquid chromatography (HPLC), data shown as fold change relative to FCS sample for each donor, n=4 donors from 2 experiments. D, Cells were loaded with CellROX green±2 μmol/L AZ12201182 (AZ) then incubated with SFM, FCS, or CX3CL1. n=8 donors from 4 experiments. E, Classical and nonclassical monocytes were isolated from the same donor as described in Figure 2 then treated as in A. Data shown as fold change relative to FCS sample for each donor. n=5 donors from 2 experiments. Data shown as mean+SEM. Data were analyzed by 1-way ANOVA and Dunnett (A–D) or Sidak (E) post hoc test, *P<0.05, **P<0.01, ***P<0.001, ns=not significant relative to SFM.
To ensure that the effect of CX3CL1 on intracellular ROS levels was mediated via CX3CR1, we pretreated cells for 30 minutes with the AZ compound before serum-starvation or CX3CL1 treatment. Addition of AZ blocked the ability of CX3CL1 to reduce ROS levels but did not affect ROS levels in cells that were not treated with CX3CL1 (Figure 4D).
As mentioned previously, the nonclassical monocyte subset has been reported to undergo a greater degree of spontaneous apoptosis and have a higher basal level of intracellular ROS when compared with classical monocytes.40 To confirm this report and address whether CX3CL1 was capable of reducing ROS in both subsets, we isolated the 2 predominant subsets as described above from the same donor and used them in the CellROX assay. In contrast to Zhao et al,40 we found no significant difference in the basal level of ROS between the 2 subsets (geometric mean for classical monocytes was 12.7±5 versus 11.2±2 for nonclassical monocytes; data not shown). Interestingly, despite its ability to prevent apoptosis in both subsets, CX3CL1 was unable to reduce intracellular ROS levels in nonclassical monocytes (Figure 4E).
To confirm that the CellROX green assay specifically measured ROS production induced by serum-starvation, we used the antioxidant enzyme pegylated-superoxide dismutase (PEG-SOD), which is cell permeant and catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide. Pretreatment of cells with PEG-SOD reduced intracellular ROS production induced by serum-starvation (Figure 5A). To assess whether increased oxidative stress is absolutely required for monocyte apoptosis, we pretreated cells with increasing doses of PEG-SOD and then measured apoptosis induced by serum-starvation. PEG-SOD induced a dose-dependent reduction in monocyte apoptosis, with a dose of 400 U/mL completely ablating cell death induced by serum withdrawal (P<0.05–0.001; Figure 5B). To prove that CX3CL1 acts via an antioxidant mechanism to reduce monocyte apoptosis, we pretreated cells with a submaximal dose of PEG-SOD (100 U/mL), a submaximal dose of CX3CL1 (50 nmol/L), or the 2 combined. As shown in Figure 5C, neither PEG-SOD alone nor CX3CL1 could significantly block apoptosis at these submaximal doses, but the combination of both had an additive and significant antiapoptotic effect (P<0.05). This suggests that CX3CL1 and PEG-SOD are likely to act via the same mechanism because they show an additive rather than synergistic or opposite effect.
Figure 5.
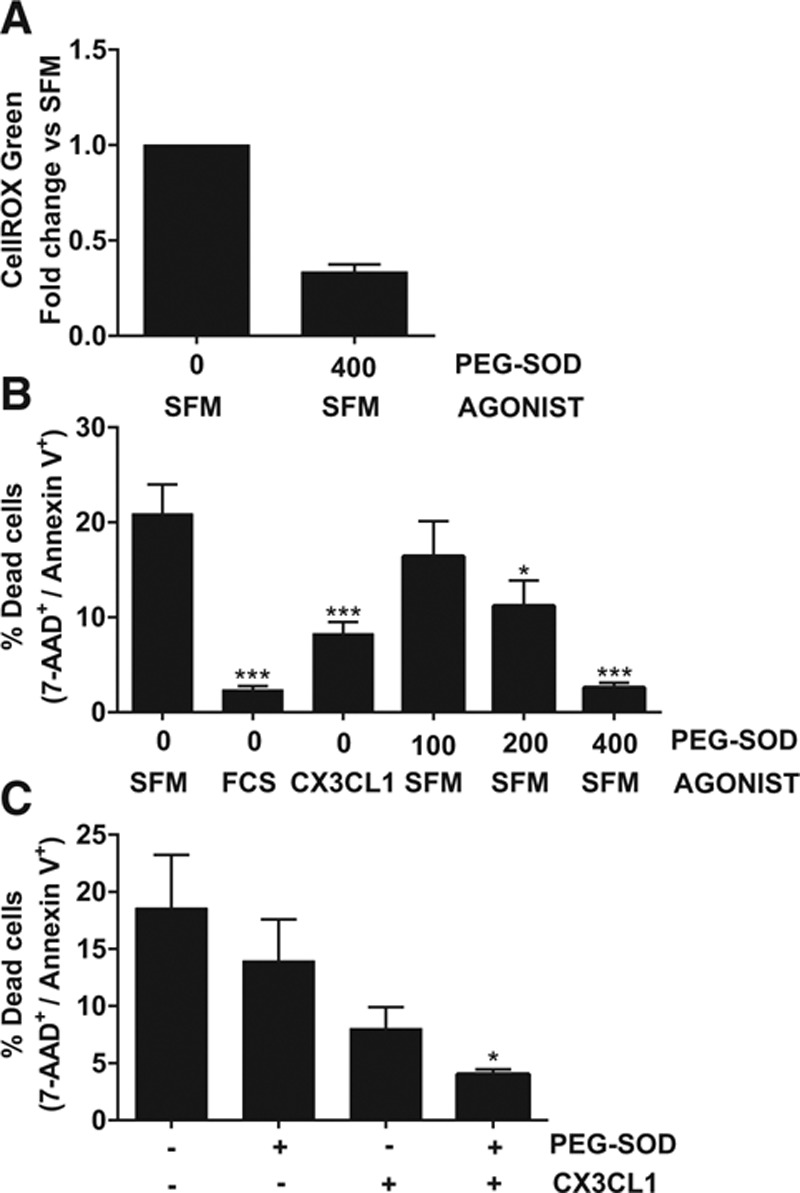
Inhibition of superoxide prevents monocyte apoptosis. A, Cells were preincubated with the indicated dose of pegylated-superoxide dismutase (PEG-SOD U/mL) at the same time as CellROX green loading, then serum-starved for 30 minutes and analyzed by flow cytometry. Data shown as fold change in CellROX green geometric mean relative to serum-free medium (SFM) for each donor, n=4 donors from 2 experiments. B, Cells were pretreated for 30 minutes±the indicated dose of PEG-SOD (U/mL) then treated with the indicated agonist or serum-starved for 4 hours and apoptosis quantified as in figure 1. n=6 donors from 3 experiments. C, Cells were pretreated for 30 minutes±a submaximal dose of PEG-SOD (100 U/mL) then treated±a submaximal dose of CX3CL1 (50 nmol/L). n=4 donors from 2 experiments. Data shown as mean+SEM. Data were analyzed by 1-way ANOVA and Dunnett post hoc test, *P<0.05, ***P<0.001 relative to SFM. AAD indicates 7-aminoactinomycin D.
Discussion
In this study, we have demonstrated that CX3CL1 has a substantial and highly reproducible prosurvival function in primary human monocytes. This effect was apparent in both monocyte subsets and was independent of CX3CR1 genotype. Growth factor withdrawal was found to increase intracellular ROS, and this effect was absolutely required for apoptosis induction. In classical monocytes, CX3CL1 was found to reduce ROS in 2 independent assays, suggesting that the reduction in oxidative stress is the predominant mechanism by which CX3CL1 promotes monocyte survival. Interestingly, although CX3CL1 can block apoptosis of both predominant monocyte subsets, it did not affect ROS levels in nonclassical monocytes, suggesting a differential mechanism operates in the 2 subsets. To our knowledge, this is the first report of a chemokine reducing intracellular ROS levels to increase cell survival.
Our data show that FL CX3CL1 has a more potent effect on monocyte survival than chemokine-domain alone. Although the stalk has no known independent signaling function, we could speculate that the presence of the stalk causes the CKD to adopt a different structure that could stabilize a receptor conformation, which promotes more potent antiapoptotic signaling. Indeed the only other chemokine known to be expressed as a membrane-bound molecule, CXCL16,44 has been shown to have differential effects depending on whether the stalk is present. Petit et al45 demonstrated that a single amino acid substitution in the CXCL16 receptor CXCR6 (E274Q) abrogated binding of soluble (CKD) CXCL16 but had no effect on adhesion mediated by FL membrane-bound CXCL16. Thus, the stalk may induce stabilization of an alternative receptor conformation, which is unaffected by the point mutation.The effect of CX3CL1 on monocyte apoptosis in our study was found to be independent of CX3CR1 genotype in donors with the most common genotypes. The most recent meta-analysis of the 2 common CX3CR1 polymorphisms has shown that there is a demonstrable link between CX3CR1 genotype and susceptibility to coronary artery disease, with the M280 allele associated with reduced risk.41 The mechanism for altered disease susceptibility remains unclear because the M280 allele has been associated with both increased and decreased adhesion to CX3CL146, 47. Radioligand binding assays in transfected cells demonstrated that all variants of the receptor have similar affinity for CX3CL148. These mutations are predicted to fall in transmembrane domains 6 and 7 of CX3CR1; it remains to be seen exactly how the polymorphisms in CX3CR1 might affect receptor activation and cell function.The 2 predominant human monocyte subsets (classical and nonclassical) both express CX3CR1 on their surface (>10-fold above isotype control levels), but the nonclassical subset have higher levels: ≈2-fold more in our experiments (data not shown); other authors have shown 5-fold higher levels in nonclassical versus classical.39 Despite this, both subsets were found to respond equally to CX3CL1 in apoptosis assays although the nonclassical subset was found to be more sensitive to serum-starvation, as previously reported.40 These same authors found that nonclassical monocytes produce higher levels of ROS than classical monocytes in response to growth factor withdrawal and that blocking oxidative stress leads to a substantial reduction of apoptosis in nonclassical monocytes but a relatively minor effect in the classical subset. In contrast, we found that the induction of oxidative stress is the dominant mechanism of cell death in the classical subset (which predominate in our experiments) because blocking ROS via the addition of PEG-SOD ablated monocyte apoptosis. Furthermore, we observed no difference in the basal level of ROS in the 2 subsets. This may reflect donor variability or differences in the way ROS was measured between our study and that by Zhao et al.40 Interestingly, although CX3CL1 could block apoptosis of both subsets, a reduction in intracellular ROS levels was only seen in the classical subset. Because we have clearly demonstrated that this reduction in ROS is required for classical monocyte survival, a second mechanism must exist in nonclassical monocytes to mediate the antiapoptotic effects of CX3CL1.
A recent study in mice showed that an antagonist at CX3CR1 (F1) blocked survival of bone marrow–derived monocytes of both the 7/4high (equivalent to classical) and the 7/4low (equivalent to nonclassical) subsets in vitro.15 This suggests that CX3CR1 has some constitutive prosurvival signaling activity because there is no exogenous source of CX3CL1 in these experiments. The mechanism for this remains unclear. In vivo, antagonism of CX3CR1 only affected the classical subset, in contrast to a previously published study where only the nonclassical subset was affected.31 Thus, there is a discrepancy between in vivo and in vitro studies and between 2 in vivo studies both in Apoe−/− mice. Possible differences include the timepoint of analysis, the length of time on a high-fat diet, the way the subsets were identified by flow cytometry, and the method of CX3CL1 inhibition—an acute antagonism of CX3CR1 versus genetic ablation.
The effects of CX3CL1 on intracellular ROS levels in our studies were replicated in 2 independent assays: CellROX green staining that measures multiple species and dihydroethidium loading that specifically measures superoxide when 2-hydroxyethidium production is quantified by high performance liquid chromatography. Because CX3CL1 had a similar effect in both assays and PEG-SOD dramatically reduced CellROX green fluorescence, we hypothesize that the predominant ROS affected by CX3CL1 is superoxide. Sources of superoxide in monocytes include the mitochondrial respiratory chain and several isoforms of NADPH oxidase.49 Given that submaximal doses of CX3CL1 and PEG-SOD were shown to have an additive effect (rather than synergistic or opposite), it seems likely that they act via a common mechanism, namely a reduction in intracellular superoxide levels.
Our results demonstrating a reduction in ROS induced by CX3CL1 are in direct contrast with those in a recently published study showing that CX3CL1 can induce ROS.19 Using a model of vincristine-induced pain, Old et al19 demonstrated that vincristine increases adhesion molecule expression leading to infiltration of CX3CR1+ monocytes into the sciatic nerve These cells then differentiate into macrophages and are activated by CX3CL1 produced by local endothelial cells leading to an increase in ROS. This leads to the activation of the TRP1A channel in sensory nerves, generating pain. We could speculate on possible reasons for the apparently contradictory finding of ROS production in response to CX3CL1 compared with the decrease shown in our study. These factors could include the fact that these cells have been recruited to a site of injury, which may select a subpopulation of monocytes that respond in a different way to CX3CL1. Because we have shown only an effect of CX3CL1 on ROS production by classical monocytes, it may be that the cells recruited are nonclassical monocytes. Alternatively, the fact that these cells have matured to macrophages may have altered the signaling pathways induced by CX3CL1 leading to ROS production rather than inhibition. In addition, the in vitro experiments performed by Old et al19 analyzing the effect of CX3CL1 on ROS used Biogel-elicited peritoneal macrophages. Extensive experience in our laboratory has shown that these are not a pure population and may include contaminating cells (eg, neutrophils), which may respond differently to CX3CL1. Finally, we cannot ignore the fact that these experiments were performed in mice and that there are already known signaling differences that exist between human and murine CX3CR1.48 The mechanism by which CX3CL1 is able to reduce intracellular ROS levels remains to be determined. The rapidity of the effect (within 30 minutes) rules out de novo transcription of an antioxidant factor, which suggests that it must rely on changes in abundance, post-translational modification, or localization of existing proteins. It has been known for 20 years that the expression of the antiapoptotic protein Bcl-2 can decrease the net concentration of intracellular ROS and thus prevent cell death.50,51 Multiple mechanisms have been postulated for this effect, which seems to be indirect.52. Furthermore, Bcl-2 is transcriptionally regulated but can also be post-translationally modified by phosphorylation at multiple sites,53 which can alter Bcl-2 activity or drive proteasome-mediated degradation.54 However, we have ruled out any effect of CX3CL1 on the total level of antiapoptotic Bcl-2 family members, including Bcl-2, Bcl-xL, and Mcl-1, or on levels of phospho-Bcl-2 in data not presented here. A previous study has shown some effect of CX3CL1 on the Bcl-2 family in microglia although this was over a much longer timecourse than that used in our experiments, which is likely to suggest a different mechanism.33 A recent publication demonstrates that mechanisms that regulate ROS continue to be uncovered, which may in the future allow us to delineate the pathways further that mediate CX3CL1’s effects on ROS.55
Despite its potent antiapoptotic effects on human monocytes, CX3CL1 did not induce monocyte chemotaxis in our studies. This has been a consistent finding in our laboratory using multiple batches of recombinant protein (both CKD and FL) and with both primary monocytes and the monocytic cell line THP-1. In fact, the inability of CX3CL1 to induce chemotaxis has been reported previously.56 Instead CX3CL1 was found to function predominantly as an adhesion molecule. Because in vitro chemotaxis assays using monocytes rely on both migration through the pore and then adhesion to the underside of the membrane, we could speculate that surfaces that support greater monocyte adhesion may show an apparent chemotactic effect of CX3CL1. Another possible reason is differences in cell preparation although primary monocytes generated in our laboratory show robust migration to CCL2. We used the same dose of CX3CL1 (10 nmol/L), which has been shown to promote transendothelial migration of monocytes,12 and a wider dose range did not reveal any chemotaxis (data not shown). Thus, it is hard to reconcile these data with published studies showing a chemotactic effect of CX3CL1.12,37
The use of serum-starvation as a method to induce monocyte apoptosis in vitro is convenient and provides consistent results. It seems likely that the necrotic core of advanced plaques may represent an environment, which is both nutrient poor and hypoxic because of inadequate vascularization, making our model a relevant approach to study the effect of CX3CL1. Numerous pathways have been suggested to promote macrophage apoptosis within plaques although a dominant mechanism seems to be the induction of endoplasmic reticulum stress.57,58 Once the level of macrophage apoptosis exceeds the available mechanisms for clearance of dying cells (a process known as efferocytosis), plaque progression is inevitable because apoptotic macrophages undergo secondary necrosis, a highly inflammatory process that promotes further monocyte recruitment, activation, and cell death.59 It will be interesting to see whether CX3CL1 can prevent macrophage apoptosis induced by endoplasmic reticulum stress, which can be recapitulated in vitro with free cholesterol loading.57
In patients with cardiovascular disease, peripheral leukocyte count is a predictor of mortality60,61 and correlates with the degree of plaque regression in response to statin treatment.62 Thus, agents that promote monocyte/macrophage survival either in the periphery or within atherosclerotic plaques are likely to have a critical role in atherogenesis. Our demonstration that CX3CL1 has a robust antiapoptotic effect on human monocytes suggests a direct means by which it could contribute to atherosclerosis. Furthermore, we provide here a mechanism by which CX3CL1 exerts its effects in monocytes. Treatments inhibiting CX3CL1/CX3CR1 may, therefore, be a novel avenue for therapeutic intervention in cardiovascular disease.
Acknowledgments
We thank Edward A. Fisher for experimental advice. We also thank Howell J. Williams and Suzanne Harrogate for assistance with apoptosis assays.
Sources of Funding
The work in the Greaves and Channon laboratories is funded by the British Heart Foundation, Programme Grant Number RG/10/15/28578. G.E. White is funded by British Heart Foundation project grant number PG/10/60/28496. E. McNeill and K.M. Channon receive core funding from Wellcome Trust Core Award grant number 090532/Z/09/Z.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- CKD
- chemokine domain
- FL
- full-length
- PEG-SOD
- pegylated superoxide dismutase
- ROS
- reactive oxygen species
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.114.304717/-/DC1.
Significance
Monocytes are central to the process of atherogenesis and give rise to macrophage-derived foam cells, which are present from the earliest stages of atherosclerotic plaque development. Factors that drive monocyte recruitment or promote monocyte survival are thus likely to have a critical role in vascular disease. Further understanding of these mechanisms may facilitate future development of therapies targeting atherosclerosis. In this article, we show that a member of the chemokine family, known as fractalkine (CX3CL1), is able to promote the survival of primary human monocytes. Different forms of CX3CL1 have altered antiapoptotic activity, and the function of CX3CL1 is independent of fractalkine receptor genotype. We show that CX3CL1 mediates its effects by opposing the increase in oxidative stress, which would otherwise lead to apoptosis. CX3CL1 seems to be unique among chemokines for its ability to promote monocyte survival and may, therefore, represent a novel therapeutic target.
References
- 1.Bachelerie F, Ben-Baruch A, Burkhardt AM, et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. 2014;66:1–79. doi: 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zernecke A, Weber C. Chemokines in atherosclerosis: proceedings resumed. Arterioscler Thromb Vasc Biol. 2014;34:742–750. doi: 10.1161/ATVBAHA.113.301655. [DOI] [PubMed] [Google Scholar]
- 3.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 4.Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J Biol Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 5.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 6.Lucas AD, Chadwick N, Warren BF, Jewell DP, Gordon S, Powrie F, Greaves DR. The transmembrane form of the CX3CL1 chemokine fractalkine is expressed predominantly by epithelial cells in vivo. Am J Pathol. 2001;158:855–866. doi: 10.1016/S0002-9440(10)64034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwaeble WJ, Stover CM, Schall TJ, Dairaghi DJ, Trinder PK, Linington C, Iglesias A, Schubart A, Lynch NJ, Weihe E, Schäfer MK. Neuronal expression of fractalkine in the presence and absence of inflammation. FEBS Lett. 1998;439:203–207. doi: 10.1016/s0014-5793(98)01384-2. [DOI] [PubMed] [Google Scholar]
- 8.Ludwig A, Berkhout T, Moores K, Groot P, Chapman G. Fractalkine is expressed by smooth muscle cells in response to IFN-gamma and TNF-alpha and is modulated by metalloproteinase activity. J Immunol. 2002;168:604–612. doi: 10.4049/jimmunol.168.2.604. [DOI] [PubMed] [Google Scholar]
- 9.Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, Ludwig A, Lira SA, Jung S. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood. 2011;118:e156–e167. doi: 10.1182/blood-2011-04-348946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucas AD, Bursill C, Guzik TJ, Sadowski J, Channon KM, Greaves DR. Smooth muscle cells in human atherosclerotic plaques express the fractalkine receptor CX3CR1 and undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1). Circulation. 2003;108:2498–2504. doi: 10.1161/01.CIR.0000097119.57756.EF. [DOI] [PubMed] [Google Scholar]
- 11.Combadiere C, Salzwedel K, Smith ED, Tiffany HL, Berger EA, Murphy PM. Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J Biol Chem. 1998;273:23799–23804. doi: 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- 12.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 13.Moatti D, Faure S, Fumeron F, Amara Mel-W, Seknadji P, McDermott DH, Debré P, Aumont MC, Murphy PM, de Prost D, Combadière C. Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood. 2001;97:1925–1928. doi: 10.1182/blood.v97.7.1925. [DOI] [PubMed] [Google Scholar]
- 14.Lavergne E, Labreuche J, Daoudi M, Debré P, Cambien F, Deterre P, Amarenco P, Combadière C GENIC Investigators. Adverse associations between CX3CR1 polymorphisms and risk of cardiovascular or cerebrovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:847–853. doi: 10.1161/01.ATV.0000157150.23641.36. [DOI] [PubMed] [Google Scholar]
- 15.Poupel L, Boissonnas A, Hermand P, Dorgham K, Guyon E, Auvynet C, Charles FS, Lesnik P, Deterre P, Combadiere C. Pharmacological inhibition of the chemokine receptor, CX3CR1, reduces atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2013;33:2297–2305. doi: 10.1161/ATVBAHA.112.300930. [DOI] [PubMed] [Google Scholar]
- 16.Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- 17.Jones BA, Koch AE, Ahmed S. Pathological Role of Fractalkine/CX3CL1 in Rheumatic Diseases: A unique chemokine with multiple functions. Frontiers in Immunology. 2012;2 doi: 10.3389/fimmu.2011.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D’Acquisto F, Malcangio M. Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem. 2010;114:1143–1157. doi: 10.1111/j.1471-4159.2010.06837.x. [DOI] [PubMed] [Google Scholar]
- 19.Old EA, Nadkarni S, Grist J, Gentry C, Bevan S, Kim KW, Mogg AJ, Perretti M, Malcangio M. Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. J Clin Invest. 2014;124:2023–2036. doi: 10.1172/JCI71389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–e80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 21.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 22.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 24.Yona S, Kim K-W, Wolf Y, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Combadière C, Potteaux S, Gao JL, Esposito B, Casanova S, Lee EJ, Debré P, Tedgui A, Murphy PM, Mallat Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–1016. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 26.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–340. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teupser D, Pavlides S, Tan M, Gutierrez-Ramos JC, Kolbeck R, Breslow JL. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci U S A. 2004;101:17795–17800. doi: 10.1073/pnas.0408096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 31.Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113:963–972. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 32.White GE, Tan TC, John AE, Whatling C, McPheat WL, Greaves DR. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc Res. 2010;85:825–835. doi: 10.1093/cvr/cvp341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J Immunol. 2000;165:397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- 34.Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, Trautwein C, Tacke F. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–1782. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 35.Mionnet C, Buatois V, Kanda A, Milcent V, Fleury S, Lair D, Langelot M, Lacoeuille Y, Hessel E, Coffman R, Magnan A, Dombrowicz D, Glaichenhaus N, Julia V. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat Med. 2010;16:1305–1312. doi: 10.1038/nm.2253. [DOI] [PubMed] [Google Scholar]
- 36.Mangan DF, Wahl SM. Differential regulation of human monocyte programmed cell death (apoptosis) by chemotactic factors and pro-inflammatory cytokines. J Immunol. 1991;147:3408–3412. [PubMed] [Google Scholar]
- 37.Chapman GA, Moores KE, Gohil J, Berkhout TA, Patel L, Green P, Macphee CH, Stewart BR. The role of fractalkine in the recruitment of monocytes to the endothelium. Eur J Pharmacol. 2000;392:189–195. doi: 10.1016/s0014-2999(00)00117-5. [DOI] [PubMed] [Google Scholar]
- 38.Karlström S, Nordvall G, Sohn D, et al. Substituted 7-amino-5-thio-thiazolo[4,5-d]pyrimidines as potent and selective antagonists of the fractalkine receptor (CX3CR1). J Med Chem. 2013;56:3177–3190. doi: 10.1021/jm3012273. [DOI] [PubMed] [Google Scholar]
- 39.Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, Kourilsky P, Wong SC. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118:e16–e31. doi: 10.1182/blood-2010-12-326355. [DOI] [PubMed] [Google Scholar]
- 40.Zhao C, Tan YC, Wong WC, Sem X, Zhang H, Han H, Ong SM, Wong KL, Yeap WH, Sze SK, Kourilsky P, Wong SC. The CD14(+/low)CD16(+) monocyte subset is more susceptible to spontaneous and oxidant-induced apoptosis than the CD14(+)CD16(-) subset. Cell Death Dis. 2010;1:e95. doi: 10.1038/cddis.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Apostolakis S, Amanatidou V, Papadakis EG, Spandidos DA. Genetic diversity of CX3CR1 gene and coronary artery disease: new insights through a meta-analysis. Atherosclerosis. 2009;207:8–15. doi: 10.1016/j.atherosclerosis.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 42.Um HD, Orenstein JM, Wahl SM. Fas mediates apoptosis in human monocytes by a reactive oxygen intermediate dependent pathway. J Immunol. 1996;156:3469–3477. [PubMed] [Google Scholar]
- 43.Madesh M, Hajnóczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. The Journal of Cell Biology. 2001;155:1003–1016. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol. 2000;1:298–304. doi: 10.1038/79738. [DOI] [PubMed] [Google Scholar]
- 45.Petit SJ, Chayen NE, Pease JE. Site-directed mutagenesis of the chemokine receptor CXCR6 suggests a novel paradigm for interactions with the ligand CXCL16. Eur J Immunol. 2008;38:2337–2350. doi: 10.1002/eji.200838269. [DOI] [PubMed] [Google Scholar]
- 46.Daoudi M, Lavergne E, Garin A, Tarantino N, Debré P, Pincet F, Combadière C, Deterre P. Enhanced adhesive capacities of the naturally occurring Ile249-Met280 variant of the chemokine receptor CX3CR1. J Biol Chem. 2004;279:19649–19657. doi: 10.1074/jbc.M313457200. [DOI] [PubMed] [Google Scholar]
- 47.McDermott DH, Fong AM, Yang Q, Sechler JM, Cupples LA, Merrell MN, Wilson PW, D’Agostino RB, O’Donnell CJ, Patel DD, Murphy PM. Chemokine receptor mutant CX3CR1-M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest. 2003;111:1241–1250. doi: 10.1172/JCI16790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis CN, Harrison JK. Proline 326 in the C terminus of murine CX3CR1 prevents G-protein and phosphatidylinositol 3-kinase-dependent stimulation of Akt and extracellular signal-regulated kinase in Chinese hamster ovary cells. J Pharmacol Exp Ther. 2006;316:356–363. doi: 10.1124/jpet.105.093039. [DOI] [PubMed] [Google Scholar]
- 49.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 51.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 52.Susnow N, Zeng L, Margineantu D, Hockenbery DM. Bcl-2 family proteins as regulators of oxidative stress. Semin Cancer Biol. 2009;19:42–49. doi: 10.1016/j.semcancer.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E, Martinou JC, Arkinstall S. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–25242. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- 54.Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmeler S. Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol Cell Biol. 2000;20:1886–1896. doi: 10.1128/mcb.20.5.1886-1896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noubade R, Wong K, Ota N, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. 2014;509:235–239. doi: 10.1038/nature13152. [DOI] [PubMed] [Google Scholar]
- 56.Umehara H, Goda S, Imai T, Nagano Y, Minami Y, Tanaka Y, Okazaki T, Bloom ET, Domae N. Fractalkine, a CX3C-chemokine, functions predominantly as an adhesion molecule in monocytic cell line THP-1. Immunol Cell Biol. 2001;79:298–302. doi: 10.1046/j.1440-1711.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- 57.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 58.Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792–2797. doi: 10.1161/ATVBAHA.111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50:S382–S387. doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmerini T, Mehran R, Dangas G, et al. Impact of leukocyte count on mortality and bleeding in patients with myocardial infarction undergoing primary percutaneous coronary interventions: analysis from the Harmonizing Outcome with Revascularization and Stent in Acute Myocardial Infarction trial. Circulation. 2011;123:2829–37. doi: 10.1161/CIRCULATIONAHA.110.985564. 7 p following. [DOI] [PubMed] [Google Scholar]
- 61.Barron HV, Cannon CP, Murphy SA, Braunwald E, Gibson CM. Association between white blood cell count, epicardial blood flow, myocardial perfusion, and clinical outcomes in the setting of acute myocardial infarction: a thrombolysis in myocardial infarction 10 substudy. Circulation. 2000;102:2329–2334. doi: 10.1161/01.cir.102.19.2329. [DOI] [PubMed] [Google Scholar]
- 62.Tani S, Nagao K, Anazawa T, Kawamata H, Furuya S, Takahashi H, Iida K, Matsumoto M, Washio T, Kumabe N, Hirayama A. Association of leukocyte subtype counts with coronary atherosclerotic regression following pravastatin treatment. Am J Cardiol. 2009;104:464–469. doi: 10.1016/j.amjcard.2009.04.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.