

Abstract
Children with an anxious temperament (AT) are at a substantially increased risk to develop anxiety and depression. The young rhesus monkey is ideal for studying the origin of human AT because it shares with humans the genetic, neural, and phenotypic underpinnings of complex social and emotional functioning. Heritability, functional imaging, and gene expression studies of AT in young monkeys revealed that the central nucleus of the amygdala (Ce) is a key environmentally sensitive substrate of this at risk phenotype. Because epigenetic marks (e.g., DNA methylation) can be modulated by environmental stimuli, these data led us to hypothesize a role for DNA methylation in the development of AT. To test this hypothesis, we used reduced representation bisulfite sequencing to examine the cross-sectional genome-wide methylation levels in the Ce of 23 age-matched monkeys (1.3 ± 0.2 years) phenotyped for AT. Because AT reflects a continuous trait-like variable, we used an analytical approach that is consistent with this biology to identify genes in the Ce with methylation patterns that predict AT. Expression data from the Ce of these same monkeys were then used to find differentially methylated candidates linked to altered gene regulation. Two genes particularly relevant to the AT phenotype were BCL11A and JAG1. These transcripts have well-defined roles in neurodevelopmental processes, including neurite arborization and the regulation of neurogenesis. Together, these findings represent a critical step toward understanding the effects of early environment on the neuromolecular mechanisms that underlie the risk to develop anxiety and depressive disorders.
Keywords: anxious temperament, BCL11A, Ce, DNA methylation, JAG1
Introduction
The childhood expression of high levels of dispositional anxiety, also termed anxious temperament (AT), is an important risk factor for the later development of anxiety and depression (Kagan et al., 1988, Degnan et al., 2010). We have developed a nonhuman primate model of childhood AT that allows for the investigation of mechanisms underlying the development of this at risk phenotype (Oler et al., 2010). Rhesus monkeys are ideally suited to study the development of pathological anxiety and other forms of psychopathology because of their similarities to humans in brain structure and function, social organization and the significance of relationships, as well as the critical role of prolonged maternal rearing on normal development (Kaas, 1987, Davidson et al., 2006, Diekhof et al., 2011). Like children, young monkeys with high AT show increased threat-induced behavioral inhibition (freezing), decreased spontaneous vocalizations, and increased levels of the stress hormone cortisol (Kalin and Shelton, 2003). AT in both children and monkeys is identifiable early in life, stable across development, and heritable (Fox et al., 2008, Rogers et al., 2008).
High-resolution (18F-labeled fluoro-deoxyglucose PET) scanning of a large single multigenerational pedigree of young rhesus monkeys indicated that the extended amygdala (central nucleus of the amygdala [Ce]/bed nucleus of the stria terminalis) and anterior hippocampus are more active in monkeys with high AT, suggesting that they are components of the neural circuit underlying AT (Kalin et al., 2004). A causal role for the Ce has been clearly demonstrated such that highly specific neurotoxic Ce lesions decrease primate AT (Kalin et al., 2004). Importantly, AT-related brain metabolism in the Ce and the anterior hippocampus significantly differ in their heritability (anterior hippocampus > Ce), suggesting that, even though these structures are closely linked, and both involved in AT, they are differentially influenced by genetic and environmental factors. Because epigenetics marks (e.g., DNA methylation) can be modulated by environmental factors and can have important functional consequences, these data led us to hypothesize a role for DNA methylation in the development and expression of AT.
To investigate the role of DNA methylation in the development and expression of AT, we performed genome-wide DNA methylation and mRNA expression analyses in Ce tissue collected from young monkeys repeatedly phenotyped for AT and its associated brain metabolism. This unique approach combines the power of functional brain imaging with the potential of methylome and transcriptome analyses to characterize the genes that could underlie the risk for developing anxiety and depression. We hypothesized that high-AT individuals would have differential methylation and/or expression of genes in the Ce that reflect and likely mediate the influences of experience on the persistent expression of high levels of dispositional anxiety. Such alterations are of particular interest because manipulations of these substrates could result in interventions for high-AT children that would facilitate their ability to modify and adaptively regulate their anxiety. These findings are also likely to provide insights into novel treatment targets for individuals that have already developed clinically significant anxiety and depressive disorders.
Materials and Methods
Tissue acquisition and DNA/RNA extraction.
The whole brains from 23 young male monkeys with an average age of 1.3 ± 0.2 years and a broad range of AT levels (−1.48 to 1.43) were sectioned into 4.5 mm slabs, and functionally guided tissue biopsies of the Ce were conducted following animal housing and experimental procedures that are in accordance with institutional guidelines (UW Institutional Animal Care and Use Committee protocol G00181). Ce regions were identified, thawed briefly on wet ice, and placed on an inverted glass Petri dish on top of wet ice. A circular 3 mm punch tool was used to biopsy the region best corresponding to the Ce. Ce regions were identified from the coronal plane as the most dorsal portions of the amygdalar gray matter that were both (1) medial and ventral to the white matter of the anterior commissure and (2) lateral to the medial temporal convexity that houses the amygdalar cortical nuclei and the entorhinal cortex on the surface of the brain. The tissue punches were collected into 1.5 ml Microfuge tubes and placed on dry ice. Once acquired, ∼30 mg of tissue was homogenized with glass beads (Sigma), and DNA and RNA extraction was performed using AllPrep DNA/RNA mini kit (QIAGEN).
DNA methylation profiling.
Genome-wide methylation data were generated for each monkey using an enrichment method: reduced representation bisulfite sequencing (RRBS), which provided the methylation status of ∼1 million CpG dinucleotides in each MspI-digested genome. Briefly, genomic DNA (1 μg) was digested with MspI, and the fragments were end-repaired and ligated to Illumina Truseq adapters (methylated) following the manufacturer's protocol (Illumina). Adapter-ligated DNA fragments, ranging from 200 to 400 bp, were purified by gel electrophoresis (2.5% NuSieve GEG agargose) and then treated with sodium bisulfite (ZymoResearch EZ DNA methylation gold kit), which converts unmethylated cytosines to uracil and leaves methylated cytosines unchanged. Converted libraries of DNA fragments were then amplified using TruSeq adapter primers (Illumina), and amplicons were purified (MiniElute PCR purification kit; QIAGEN 28004) and sequenced on a Next-Generation Sequencer (Illumina HiSeq 2000).
QC and analysis of DNA methylation data.
Sequence reads were mapped to the human genome reference sequence using Bismark (version 0.11.1; NCBI Genome build 37). CpG dinucleotides were annotated to genes within 10 kb and to CpG Islands by downloading the CpG island track from the University of California, Santa Cruz table browser. The data were filtered through two criteria for each CpG location: the sample size should be >16 (i.e., the number of NAs should be <7); and the minimum read depth should be >5 and <1000 (i.e., C + T is >5 and <1000 for each sample). This filter reduced the number of CpGs to 444,312. An additional filter was used to remove CpGs that had minimal variation (SD < 0.001) across the 23 samples, as these are considered uninformative. This final filter reduced our set of CpGs to 433,770. The methylation data were then log transformed and regressed on the age of each monkey as follows:
where i = 1, 2,…, 23 is the index about each monkey, j = 1, 2…, and m is the index about each CpG location. The residuals of the regression were used to age-adjust the methylation data at each CpG.
AT was treated as a continuous variable to identify genes with methylation patterns that are correlated with AT. To achieve this goal, a Pearson's correlation between methylation percentage and AT severity was used to transform the methylation data into z-scores (i.e., Fisher's transform), as follows:
 |
which under the null hypothesis the correlation = 0 and the data are distributed as standard normal.
The z-values were then plugged into a Hidden Markov Model to determine a false discovery rate (FDR), and the model parameters were achieved by expectation maximization, which aims to minimize FDR (i.e., discover the most significant sites possible while keeping false positives low). Here local index of significance (LIS) is, to some extent, an extension of p values, and it is defined (Sun and Cai, 2009) as follows:
Let LIS(1),…, LIS(m) be the ranked LIS values and H(j) the corresponding hypotheses at CpG location j. Then the oracle testing procedure:
 |
controls FDR at α.
For simplicity, we used:
 |
as the FDR control statistic and the “NHMMFDR” package to generate this statistic (Kuan and Chiang, 2012).
Notably, the Hidden Markov Model model only used the absolute value of z-scores. This design allowed the nearby CpGs with z-scores of a different sign to be grouped together so that three contiguous CpGs, whose gap is <500 bp, will have z-scores of the same sign. Notably, the average number of monkeys and minimum read depth for each of the AT-associated CpGs was 20.8 and 76.5, respectively.
Gene expression profiling.
The extracted RNA was used to generate cRNA, which was then labeled and prepped using the 3′ IVT Express Kit (Affymetrix) and then hybridized to Affymetrix GeneChip Rhesus Macaque Genome arrays. Gene expression data were analyzed using R (http://cran.r-project.org) and the bioconductor libraries for microarray analysis (http://www.bioconductor.org; version 2.7) (Gentleman et al., 2004), as described previously (Fox et al., 2012). Briefly, microarray expression values were corrected for background noise using the RMA algorithm, levels of gene expression were normalized across chips with a constant, mismatch probes were ignored, and expression data were summarized across probes using the median-polish technique (Gautier et al., 2004). Gene expression levels were visually inspected using MA plots. Resulting expression estimates for each probe set were filtered using mean expression levels; subthreshold probe sets [<log2(100)] were excluded. Genes were annotated using publicly available annotations verified by BLAST, using the human genome reference sequence constructed by the University of Nebraska NonHuman Primate Genomics Center, which developed the Rhesus Monkey microarray chip in collaboration with Affymetrix (http://www.unmc.edu/rhesusgenechip/).
Functional analysis of DNA methylation data.
Genes on the Affymetrix chip that lack methylation data were removed from this analysis. A fixed-effect regression model was used on the remaining gene expression data (N = 7187 genes corresponding to 256,079 unique CpGs), using age of the monkeys as a covariate, to find CpG methylation levels that were most predictive of gene expression. Similarly, we used a fixed-effect regression model, treating AT as a continuous variable and using age of the monkeys as a covariate, to find the gene expression probes that were correlated with AT. For both of these analyses, we consider a reduced set of CpG dinucleotides (N = 256,079); thus, the methylation data LIS was recalculated on this smaller set of CpGs, which resulted in the inclusion of 70 genes that were not previously identified using the full set of methylation data.
Permutation p value calculations.
Over-representation of AT-significant CpGs that have a mean methylation >0.6: of 5489 AT-significant CpGs, there were 3183 CpGs with a mean methylation >0.6. These 3183 CpGs are the “actual number.” For a permutation, we randomly selected 5489 CpGs (from the full set of 433,770) and counted how many CpGs had a mean methylation value >0.6. We termed this the “permuted number.” We did this 10,000 times. The permutation p value is the number of times the set of “permuted numbers” exceeded the “actual number,” divided by 10,000. Over-representation of AT-methylated and AT-demethylated loci on each chromosome: The proportion of overlap between the CpGs in a chromosome and all AT-methylated (or AT-demythlated) loci (called “actual overlap proportion”) was compared with that obtained if an equal number of loci (i.e., equal in number to the AT-methylated loci) was selected randomly (called “permuted overlap proportion”). To correct for multiple hypotheses (i.e., 24 chromosomes), the actual proportion of each chromosome was compared with the permuted proportions of any chromosome for each permutation. Thus, the permutation p value is the number of times a permuted proportion is greater than the actual proportion, divided by the number of permutations (10,000). Permutations for gene structure and CpG island analysis: The overlap between the AT-methylated (or demethylated) loci and the loci belonging to a particular gene region (e.g., 5′-untranslated region [UTR], body, etc., called the “actual overlap”) was compared with those obtained by randomly selecting an equal number (i.e., equal in number to the AT-methylated loci) of loci and evaluating the overlap (“permuted overlap”). The permutation p value is the number of times the permuted overlap was greater than the actual overlap, divided by the number of permutations (10,000).
Gene ontologies.
All Gene Ontology (GO) analyses were conducted in R using the “GOstats” package. This package calculates the p values for over-representation of a set of genes to specific GO terms. It uses a hypergeometric test to calculate the p values, given a set of genes and a gene universe (i.e., the super set from which the smaller gene set is drawn). The gene universe used for the GO analysis of the AT-associated methylated loci was the 15,465 genes that corresponded to the 433,770 CpGs used in the methylation analysis. The gene universe used for the GO analysis of the functional AT-associated methylated loci was the 7187 genes that passed the QC and were used in the methylation and gene expression analysis.
Results
The methylome of the Ce in young rhesus monkeys
To characterize the methylome of the Ce of young primates and reveal the epigenetic basis of anxious temperament, we extracted genomic DNA from the Ce of 23 rhesus macaques. All 23 monkeys were young males (mean age = 1.3 ± 0.2 years) with a broad range of AT levels (−1.48 to 1.43; see supplementary figure at http://alischlab.psychiatry.wisc.edu). AT is computed as a composite measure among vocalizations, cortisol levels, and time freezing (mean AT score) assessed during the no eye contact condition of the human intruder paradigm (Fox et al., 2012). In this study, AT levels were assessed twice, and the mean score for each monkey was used for analysis. The Ce genome of each monkey was enriched for CpG-rich regions of the genome, treated with sodium bisulfite, and sequenced on a Next-generation Sequencer (see Materials and Methods). This approach generated DNA methylation information at 4,567,765 CpG dinucleotides from the Ce of rhesus macaques.
To investigate comparisons across the 23 individual monkey genomes, the high-quality methylation data were filtered for CpG data that had a read depth of >5 and <1000 occurring in a minimum of 17 monkeys (N = 444,312). These data were further trimmed by removing CpGs that lacked variation in methylation levels across all 23 monkeys (N = 433,770). This final dataset revealed a bimodal distribution of 5-mC in monkey Ce, with the majority (>60%) of CpGs having <10% methylation. We next annotated the methylation data based on its relation to the nearest gene, meaning that the CpG dinucleotides were assigned to the following genomic elements: upstream10,000, TSS1500, TSS200, 5′UTR, first exon, gene body, 3′UTR, downstream10,000, or intergenic regions (Fig. 1a). The distribution of the data indicates that the largest fraction of data is in the gene bodies (Fig. 1b). To characterize the overall monkey Ce methylome, we plotted the mean methylation level of each genomic element. Although genomic regions close to the transcription start site (TSS) averaged low methylation levels (9% for TSS1500, 4% for TSS200, 9% for 5′UTR, and 6% for first exon), regions further away from the TSS averaged much higher methylation levels (36% for gene body, 36% for 3′UTR, and 47% for intergenic regions; Fig. 1c). We also annotated the Ce methylation data with respect to CpG islands, CpG island shores (0–2 kb from the island's edges), CpG island shelves (2–4 kb from the island's edges), and the “open sea” (>4 kb from any island's edges; Fig. 1d,e) and found that CpG islands are hypomethylated (mean methylation ∼10%), shores have moderate methylation (mean methylation ∼30%), and shelves and open sea are hypermethylated (mean methylation >55%; Fig. 1f). These distributions coincide with previous studies that have shown a relative depletion of methylation on CpG islands and a substantial amount of variation in regions flanking the island (Irizarry et al., 2009).
Figure 1.

Methylation differs across the regions of the genome (a–c) and CpG islands (d–f). Standard regions of the genome (a) were used for analyses of density and methylation. Specifically, methylation data distributions were classified depending on how they fell in regard to: the TSS, regions defined as 200 bp upstream of the TSS (TSS200), 1500 bp upstream of the TSS (TSS1500), the 5′ and 3′UTRs, the 10 kb upstream of the gene body (Up10K), and 10 kb downstream of the gene body (Down10K). Distributions of methylation data were binned into these groups based on both density of reads (b) and mean percentage DNA methylation (c), revealing genome region-specific methylation patterns. A similar strategy was used to assess patterns of methylation across CpG islands, which were defined as at least 200 bp stretch of DNA with a C+G content of >50% and an observed CpG/expected CpG in excess of 0.6. CpGs were grouped (d) as those that fell into: CpG island north and south shores (0–2 kb from the island edges), CpG island north and south shelves (2–4 kb from the island edges), and the “open sea” (>4 kb from any island edge). Distributions of methylation data were binned into these groups based on both density of reads (e) and mean percentage DNA methylation (f), revealing CpG-island specific methylation patterns.
AT-associated methylation marks
To examine whether the Ce harbors differential DNA methylation that is related to individual differences in AT, the methylation data were subjected to a fixed-effects linear model and a statistical algorithm that accounts for the underlying dependence between adjacent CpG sites (Materials and Methods) (Zhang et al., 2011). After correcting for multiple comparisons using an FDR analysis, we identified 5489 CpG sites (corresponding to N = 1363 genes; see supplementary figure at http://alischlab.psychiatry.wisc.edu) with AT-associated methylation levels (FDR p value <0.05). Whereas 4766 of the CpG sites (87%) were negatively correlated with the AT phenotype (i.e., methylation levels decreased with increasing AT severity), 723 sites (13%) were positively correlated with AT (Fig. 2a, inset). The majority (>80%) of AT-associated methylation marks had methylation levels >10%. We also found that there was a significant over-representation of AT marks in regions that had >60% methylation (N = 3183; permutation p value <0.0001; Fig. 2a). These data suggest that, in general, hypermethylated CpGs in the Ce are more susceptible to change in relation to AT.
Figure 2.
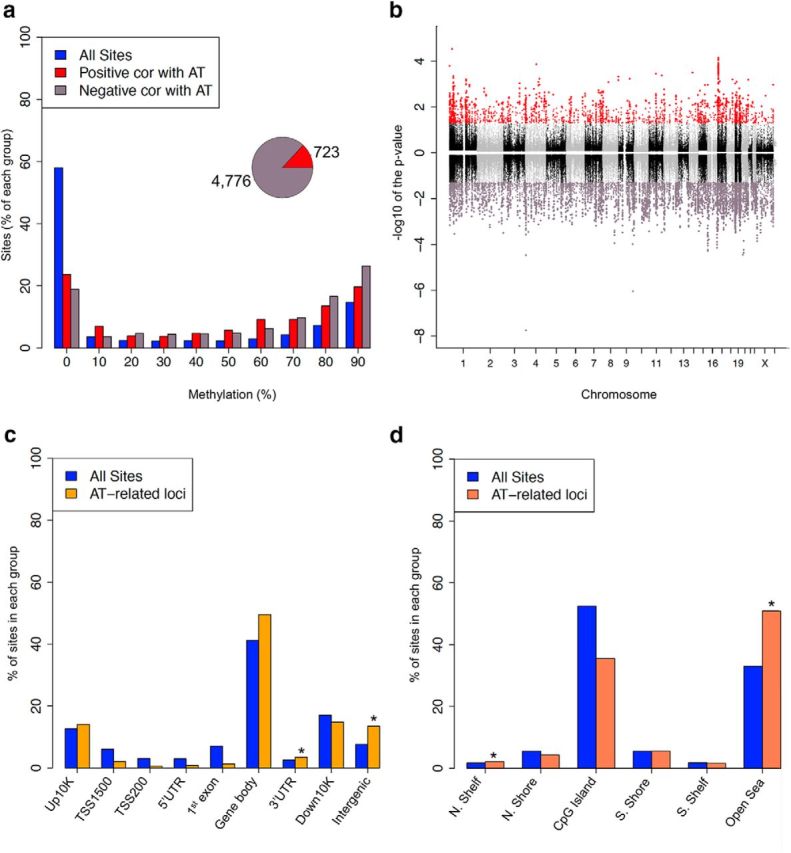
The distribution of AT-associated methylation levels across standard genomic structures. a, The percentage of all CpGs (blue), AT-methylated loci (red), and AT-demethylated loci (purple) for different methylation levels (x-axis) revealed a tendency for highly methylated sites to be more AT-related. Notably, more CpGs were negatively correlated with AT than positively correlated with AT (pie chart inset). b, Manhattan plot of AT-associated loci in the monkey Ce reveals AT-related loci to be distributed across the genome. Positively correlated loci are displayed with a positive log10 (p value), and negatively correlated loci are displayed with a negative log10 (p value). Significant loci are displayed as either AT-methylated (red) or AT-demethylated (purple; FDR < 0.05), whereas loci that are not significant alternate between black and gray to indicate each chromosome. p values shown are all aLIS p values. c, d, The percentage of all CpGs (blue) and AT-related within different genomic regions (c), and CpG islands (d) revealed a tendency for an enrichment in some genomic structures. *Permutation p value <0.05.
It is well known that specific regions of the genome are differentially methylated based on the biological functions of the genes contained within the region, with the most striking example being X inactivation in females. To determine whether AT-associated differentially methylated CpG sites are specific to certain regions of the genome, we displayed their genomic locations using a Manhattan plot (Fig. 2b). This plot indicated that AT-associated loci are located throughout the genome, but a closer examination suggested an excess of AT-associated methylation on some chromosomes. To test the significance of these observations and to determine whether any chromosomes contain a disproportionate number of AT-associated loci, we compared the proportions of AT-associated loci for each chromosome using permutation testing (Materials and Methods). This analysis revealed that chromosomes 1 and 19 have disproportionately more AT-methylated or demethylated loci, respectively, than expected by chance alone (permutation p value = 0.023 and 0.048, respectively; Fig. 2b). These data suggest that chromosomes 1 and 19 carry more AT-related genes that are sensitive to DNA methylation changes than the rest of the genome.
Recent studies suggest that psychiatric-related differential DNA methylation primarily occurs in hypermethylated CpGs that are not associated with CpG islands (Dempster et al., 2011, Klengel et al., 2013). To determine the distribution of AT-associated differential methylation relative to each annotated gene region, we compared, via permutation testing, the proportion of AT-associated CpG loci that reside in or around each gene to the total number of CpG loci interrogated by the RRBS method (see Materials and Methods). This analysis found that the gene body contained the majority of AT-associated loci (55%) and the regions surrounding the gene promoter were under-represented for AT-associated loci (0%–2% of loci). However, the only significant enrichments were located in the 3′UTR (permutation p = 0.0037) and the intergenic regions (permutation p value < 0.001; Fig. 2c). Notably, the 3′UTR and intergenic regions were significantly enriched for both methylated and demethlyated events. Unlike in cancer where differentially methylated loci closely flank CpG islands (i.e., in the shores), <10% of the AT-associated sites are located in the shores and the majority (>50%) are in the open sea where there was a significant over-representation for demethylated loci (permutation p value <0.0001; Fig. 2d). Demethylated loci were also significantly enriched in the upstream CpG island shelf (permutation p = 0.0164). Together, these data indicate that AT-associated changes in DNA methylation primarily reside in nongenic regions of the genome, suggesting preferences in genomic location and that AT-associated changes are governed by nonstochastic mechanisms.
We next examined the gene ontologies (GOs) of the 1363 AT-associated loci and found several biologically relevant ontological terms that were significantly over-represented among the differentially methylated genes in the Ce, including collateral sprouting, neuron generation, axonogenesis, and synaptyic transmission (see Materials and Methods). Many of the AT-associated genes that contributed to the over-representation of these ontological terms have been previously implicated in psychiatric-related disorders, such as GRIN1, GRM5, HTT, ADCYAP1, and SHANK3 (Rodrigues et al., 2002, Krishnan and Nestler, 2008, Labrie et al., 2009, Gauthier et al., 2010, Stevens et al., 2014). Thus, together, these findings suggest that differential methylation levels in the Ce are associated with relevant neurodevelopmental pathways that likely contribute to the AT phenotype.
Functional AT-associated methylation marks
To gain insight into the potential mechanism(s) for AT-associated DNA methylation changes, we used microarrays to profile gene expression in the Ce region of the same monkeys included in the DNA methylation analysis (see Materials and Methods). Gene expression data were filtered such that RNA levels had to pass a stringent quality control (e.g., mean expression levels > log2(100); see Materials and Methods), and that adequate methylation data were available. Thus, the expression levels of 7187 transcripts were used to identify the AT-associated methylation marks that also altered transcription. Notably, only 60% of the methylation data (N = 256,079 CpG dinucleotides) were annotated to the 7187 genes with available expression data. For an appropriate investigation of the functionality of the DNA methylation marks at these genes, we recalculated the AT-associated methylation changes using this reduced set of methylation data and found 1735 CpG loci with methylation levels predictive of individual differences in AT (FDR p value <0.05). These 1735 CpGs corresponded to 269 genes, which included 199 of the 1363 genes that were identified in the full set of methylation data. The mean correlation between DNA methylation and gene expression across all genes was similar to the correlations found in previous studies (r = −0.09) (Zeng et al., 2012). From these data, we identified 22 genes with a significant correlation between AT-associated methylation levels and gene expression (p < 0.05; Table 1), including two glutamate receptors, GRIN1 and GRM5, both of which have reported roles in fear and anxiety-like behaviors (Rodrigues et al., 2002, Labrie et al., 2009). Examination of the GOs of these 22 AT-associated genes found a significant enrichment of neuronal ontological terms in the Ce, including developmental processes, nervous system development, sensory perception, and locomotor behavior (see Materials and Methods). Because we are most interested in genes for which we have evidence that individual differences in gene expression also predict AT, we subjected the expression data of these 22 transcripts to a robust regression analyses correlating AT with mRNA levels and identified two transcripts (BCL11A and JAG1) with expression levels significantly correlated to the AT phenotype (p < 0.05; Fig. 3). These transcripts have well-defined roles in brain plasticity that includes neurite arborization (BCL11A) and the regulation of astrogenesis and neurogenesis (JAG1), which is consistent with a role for BCL11A and JAG1 in the neurodevelopmental processes underlying AT. Moreover, a closer inspection of the AT-predictive methylation marks for each of these genes found that each gene has several adjacent CpGs with methylation levels that are each predictive of individual differences in AT (Fig. 3d,h). These tight clusters of significant differentially methylated loci related to AT further support the strength of this finding. Together, these data indicate that BCL11A and JAG1 are novel targets of the mechanisms mediating how early life experience influences the expression of AT and its underlying neural substrate. Approaches focused on modifying the expression of these neuroplasticity genes could form the basis of novel early life interventions with the potential of preventing the later development of anxiety and depressive disorders.
Table 1.
Genes with functional AT-associated methylationa
| Gene symbol | p* | Chromosome | Gene name |
|---|---|---|---|
| PIK3R1 | 0.00215 | 5 | Phosphoinositide-3-kinase, regulatory subunit 1 (α) |
| KLHDC9 | 0.00232 | 1 | kelch domain containing 9 |
| JAG1 | 0.00312 | 20 | jagged 1 |
| URM1 | 0.00363 | 9 | Ubiquitin-related modifier 1 |
| SHKBP1 | 0.00395 | 19 | SH3KBP1 binding protein 1 |
| ZNF521 | 0.00796 | 18 | Zinc finger protein 521 |
| GRIN1 | 0.00823 | 9 | Glutamate receptor, ionotropic, N-methyl-d-aspartate 1 |
| PIP5K1B | 0.01401 | 9 | Phosphatidylinositol-4-phosphate 5-kinase, Type I, β |
| ERC2 | 0.01435 | 3 | ELKS/RAB6-interacting/CAST family member 2 |
| ABCB1 | 0.01504 | 7 | ATP-binding cassette, subfamily B (MDR/TAP), member 1 |
| SCAMP3 | 0.0166 | 1 | Secretory carrier membrane protein 3 |
| SPTBN4 | 0.02181 | 19 | Spectrin, β, nonerythrocytic 4 |
| GDF11 | 0.0245 | 12 | Growth differentiation factor 11 |
| ALDH7A1 | 0.0325 | 5 | Aldehyde dehydrogenase 7 family, member A1 |
| FGD1 | 0.0329 | X | FYVE, RhoGEF, and PH domain containing 1 |
| GRM5 | 0.03411 | 11 | Glutamate receptor, metabotropic 5 |
| CDH2 | 0.03508 | 18 | Cadherin 2, Type 1, N-cadherin (neuronal) |
| JAG1 | 0.03596 | 20 | jagged 1 |
| YIPF2 | 0.03885 | 19 | Yip1 domain family, member 2 |
| CLK2 | 0.04226 | 1 | CDC-like kinase 2 |
| CECR6 | 0.04271 | 22 | Cat eye syndrome chromosome region, candidate 6 |
| ALDH7A1 | 0.04369 | 5 | Aldehyde dehydrogenase 7 family, member A1 |
| BCL11A | 0.04376 | 2 | B-cell CLL/lymphoma 11A (zinc finger protein) |
| KL | 0.04877 | 13 | klotho |
aThe 22 genes (gene symbol or gene name) with AT-associated methylation marks that are significantly (p value) correlated with gene expression.
*p value based on MExGE correlation.
Figure 3.

The correlations between AT, DNA methylation, and gene expression for BCL11A (a–d) and JAG1 (e–h). For each of these genes, we identified methylation sites where the methylation levels were correlated with individual differences in AT (a, e; p values shown are aLIS p values) and gene expression (b, f). Moreover, expression levels of BCL11A and JAG1 were also correlated with individual differences in AT (c and g, respectively). Importantly, a cluster of CpGs demonstrated correlated methylation differences to AT (d, h; red represents positive; black represents negative) that reached statistical significance (FDR <0.05, dashed line; and FDR <0.01, solid line).
Discussion
Here we report the identification of AT-associated methylation differences in DNA from the Ce of monkeys that were well characterized for their anxious temperament disposition, a key risk factor for developing anxiety and depression. An important strength of this study was the ability to compare methylome and transcriptome data from the Ce of the same monkeys. This comparison revealed genes with functional AT-associated DNA methylation and provides a list of potentially modifiable substrates that ultimately could be targeted to prevent children with high-AT from developing full-blown anxiety and depressive disorders.
This study has several advantages from previous methylation studies of psychiatric-related disorders, including the use of an analytical approach that is consistent with AT reflecting a continuous trait-like variable and that the methylation and expression profiling were examined in a functionally defined brain region within 23 primate brains. With this design, we identified gene expression that is correlated with AT-associated changes in DNA methylation. A common function of many of these genes involves their role in general cell growth processes, including cell maturation (GDF11 and SPTBN4) and cellular developmental processes (GRIN1, BCL11A, JAG1, GDF11, CDH2, SPTBN4, ZNF521, FGD1, GRM5, and PIK3R1). The biological relevance of these genes supports their role in AT. Because neuroplasticity is a fundamental process involved in adapting to a changing environment, altered function of these genes is consistent with our proposed maladaptive neurodevelopmental hypothesis of AT (Fox et al., 2012). Despite the biological relevance of the genes identified in this cross-sectional study, it should be noted that cell populations in the brain are heterogeneous and that the reported methylation in the Ce is likely to reflect contributions from multiple cell types. It will be important to develop monkey brain cell epigenotype-specific markers to determine the impact of cell type on the AT-associated methylation levels identified in this study.
Because in our secondary analyses we restricted the methylation data so that they could be compared with the gene expression data, it is likely that the 22 AT-associated genes are an underestimate of the identifiable functional methylation marks that contribute to the phenotype. Indeed, several genes with AT-associated methylation changes that previously were shown to contribute to psychiatric-related disorders were not included in the functional-gene expression analysis because they either were not on the expression array or did not pass QC. These included HTT, SHANK1, and AVPR1B (Krishnan and Nestler, 2008, Sato et al., 2012, Stoop, 2012). Because functional methylation marks were found in ∼10% of this restricted gene set, we can extrapolate that the full set of methylation data may harbor >100 methylation marks that significantly regulate gene expression. The GO analysis supports this conservative notion, as the full list of genes predictive of AT were enriched in biologically relevant ontological pathways known to be mediated by psychiatric-related genes. It is also noteworthy that we did find several common GO pathways between this study and our previous study of AT-related gene expression (Fox et al., 2012), which included tyrosine kinase, signal transduction, and hormone stimulus pathways. However, we did not find overlap of the methylated genes identified here with those genes identified in that study. Further support that the list of candidate AT-associated genes revealed from this study is conservative is in the use of a restricted methylation detection method (i.e., RRBS). Although this method is more accurate than other affinity-based and whole-genome sequencing approaches, RRBS does not survey all the genes in the genome and does not provide methylation information on CpG-poor regions of the genome (Bock et al., 2010). Thus, future studies that use whole-genome sequencing or array-based methods will provide a more comprehensive survey of the genome, which undoubtedly will find additional methylation levels associated with AT.
BCL11A and JAG1 are of particular interest because their AT-associated methylation patterns predicted gene expression, which in turn predicted individual differences in AT. BCL11A encodes a C2H2-type zinc-finger protein that is an important downstream effector of glutamate receptors in neurite arborization. BCL11A was recently found to contain a frame-shift mutation in a boy with autism (Iossifov et al., 2012) that results in reduced BCL11A expression. Similarly, we found a negative correlation between BCL11A expression and AT severity, which indicates that monkeys with extreme AT had reduced BCL11A expression. Because AT-associated methylation patterns are negatively correlated with BCL11A expression, these data suggest that increased DNA methylation reduces BCL11A expression and contributes to the AT phenotype. JAG1 is a NOTCH receptor and plays a critical role in brain development, adult synaptic plasticity, and memory formation; the loss of JAG1 results in impaired spatial memory (Sargin et al., 2013). We found a negative correlation between JAG1 expression and AT severity, indicating that monkeys with severe AT had less JAG1 expression. Because AT-associated methylation patterns are negatively correlated with JAG1 expression, these data suggest that increased DNA methylation reduces JAG1 expression and contributes to the AT phenotype.
A descriptive analysis of the methylation events found in the Ce revealed that approximately two-thirds of the methylation data were associated with CpG islands; however, there was a surprisingly large fraction of data found in the open sea. In contrast to the CpG loci found in the islands, the CpGs in the open sea were hypermethylated and appear to contribute to the majority of AT-associated changes. Previous studies have reported that psychiatric disorder-related DNA methylation changes are predominantly demethylation events that occur outside of CpG islands in regions of the genome that are hypermethylated (Dempster et al., 2011, Klengel et al., 2013). Although this scenario is consistent with the majority of the AT-associated methylation changes found here, both BCL11A and JAG1 AT-associated changes were found in a CpG Island and the JAG1 AT-associated locus had <10% methylation. Similar characteristics were found for the other 22 AT-candidate genes, as the majority of predictive CpGs were associated with islands and several of them had <10% methylation.
The distribution of DNA methylation in monkey Ce with relation to gene structure was consistent with previous reports that found that methylation levels increase with distance from a gene promoter, indicating that the promoter remains relatively hypomethylated. In regard to the AT phenotype, we found that the 3′UTR and intergenic regions were particularly enriched for changes in this mark. This finding may indicate that AT-associated methylation changes in these regions contribute to direct and/or indirect molecular mechanism(s) that include altering factor binding sites or regulatory sites for noncoding RNA. Noncoding RNAs are highly expressed in the brain and are involved in neurodevelopment and neuroplasticity-related functions; their dysregulation has been described in numerous neurological diseases (Qureshi and Mehler, 2012). Hence, future studies investigating the AT-associated marks that were not correlated to changes in gene expression may reveal that these methylation levels mediate the expression of a noncoding, but functional, RNA found in intergenic regions of the genome.
As we begin to unravel the links between DNA methylation, gene expression, and complex phenotypes, such as psychiatric disorders, studies will need to expand their surveys to the entire genome and to other DNA modifications. A recent report found that an accumulation of 5-hydroxymethylcytosine (5-hmC) promotes rapid behavioral adaptation in the prefrontal cortex (Li et al., 2014), suggesting that it will be informative to investigate the contributions of both 5-mC and 5-hmC in relation to AT. The AT-related functional methylation that we identified in the Ce of monkeys represents the first steps in understanding how epigenetic mechanisms are involved in modifying the expression of genes that function in neurodevelopmental processes underlying the childhood risk to develop anxiety and depressive disorders.
Notes
We have submitted the data generated from the monkey methylation data for this study to the Gene Expression Omnibus, which can be found under the Gene Series GSE58687. Supplemental material for this article is available at http://alischlab.psychiatry.wisc.edu. This material has not been peer reviewed.
Footnotes
This work was supported in part by the University of Wisconsin-Madison Department of Psychiatry (to R.S.A.), the Conti Center, and National Institutes of Health Grants MH081884, MH084051, and MH046729 to N.H.K. We thank Marie Adams and the University of Wisconsin Biotechnology DNA sequencing facility for technical services and expertise.
The authors declare no competing financial interests.
References
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28:1106–1114. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RJ, Fox AS, Kalin NH. Neural bases of emotion regulation in non-human primates and humans. New York: Guildford; 2006. [Google Scholar]
- Degnan KA, Almas AN, Fox NA. Temperament and the environment in the etiology of childhood anxiety. J Child Psychol Psychiatry. 2010;51:497–517. doi: 10.1111/j.1469-7610.2010.02228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, Kalidindi S, Picchioni M, Kravariti E, Toulopoulou T, Murray RM, Mill J. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekhof EK, Geier K, Falkai P, Gruber O. Fear is only as deep as the mind allows: a coordinate-based meta-analysis of neuroimaging studies on the regulation of negative affect. Neuroimage. 2011;58:275–285. doi: 10.1016/j.neuroimage.2011.05.073. [DOI] [PubMed] [Google Scholar]
- Fox AS, Shelton SE, Oakes TR, Davidson RJ, Kalin NH. Trait-like brain activity during adolescence predicts anxious temperament in primates. PLoS One. 2008;3:e2570. doi: 10.1371/journal.pone.0002570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AS, Oler JA, Shelton SE, Nanda SA, Davidson RJ, Roseboom PH, Kalin NH. Central amygdala nucleus (Ce) gene expression linked to increased trait-like Ce metabolism and anxious temperament in young primates. Proc Natl Acad Sci U S A. 2012;109:18108–18113. doi: 10.1073/pnas.1206723109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, Lapointe M, Peng H, Côté M, Noreau A, Hamdan FF, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Joober R, Fathalli F, Mouaffak F, Haghighi AP, Neri C, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas JH. The organization of neocortex in mammals: implications for theories of brain function. Annu Rev Psychol. 1987;38:129–151. doi: 10.1146/annurev.ps.38.020187.001021. [DOI] [PubMed] [Google Scholar]
- Kagan J, Reznick JS, Snidman N. Biological bases of childhood shyness. Science. 1988;240:167–171. doi: 10.1126/science.3353713. [DOI] [PubMed] [Google Scholar]
- Kalin NH, Shelton SE. Nonhuman primate models to study anxiety, emotion regulation, and psychopathology. Ann N Y Acad Sci. 2003;1008:189–200. doi: 10.1196/annals.1301.021. [DOI] [PubMed] [Google Scholar]
- Kalin NH, Shelton SE, Davidson RJ. The role of the central nucleus of the amygdala in mediating fear and anxiety in the primate. J Neurosci. 2004;24:5506–5515. doi: 10.1523/JNEUROSCI.0292-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, Nemeroff CB, Holsboer F, Heim CM, Ressler KJ, Rein T, Binder EB. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan PF, Chiang DY. Integrating prior knowledge in multiple testing under dependence with applications to detecting differential DNA methylation. Biometrics. 2012;68:774–783. doi: 10.1111/j.1541-0420.2011.01730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie V, Clapcote SJ, Roder JC. Mutant mice with reduced NMDA-NR1 glycine affinity or lack of d-amino acid oxidase function exhibit altered anxiety-like behaviors. Pharmacol Biochem Behav. 2009;91:610–620. doi: 10.1016/j.pbb.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D'Alessio A, Zhang Y, Bredy TW. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A. 2014;111:7120–7125. doi: 10.1073/pnas.1318906111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler JA, Fox AS, Shelton SE, Rogers J, Dyer TD, Davidson RJ, Shelledy W, Oakes TR, Blangero J, Kalin NH. Amygdalar and hippocampal substrates of anxious temperament differ in their heritability. Nature. 2010;466:864–868. doi: 10.1038/nature09282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012;13:528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci. 2002;22:5219–5229. doi: 10.1523/JNEUROSCI.22-12-05219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Shelton SE, Shelledy W, Garcia R, Kalin NH. Genetic influences on behavioral inhibition and anxiety in juvenile rhesus macaques. Genes Brain Behav. 2008;7:463–469. doi: 10.1111/j.1601-183X.2007.00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargin D, Botly LC, Higgs G, Marsolais A, Frankland PW, Egan SE, Josselyn SA. Disrupting Jagged1-Notch signaling impairs spatial memory formation in adult mice. Neurobiol Learn Mem. 2013;103:39–49. doi: 10.1016/j.nlm.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, O'Connor I, Russell C, Drmic IE, Hamdan FF, Michaud JL, Endris V, Roeth R, Delorme R, Huguet G, Leboyer M, Rastam M, Gillberg C, Lathrop M, Stavropoulos DJ, et al. SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet. 2012;90:879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JS, Almli LM, Fani N, Gutman DA, Bradley B, Norrholm SD, Reiser E, Ely TD, Dhanani R, Glover EM, Jovanovic T, Ressler KJ. PACAP receptor gene polymorphism impacts fear responses in the amygdala and hippocampus. Proc Natl Acad Sci U S A. 2014;111:3158–3163. doi: 10.1073/pnas.1318954111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop R. Neuromodulation by oxytocin and vasopressin. Neuron. 2012;76:142–159. doi: 10.1016/j.neuron.2012.09.025. [DOI] [PubMed] [Google Scholar]
- Sun W, Cai T. Large-scale multiple testing under dependence. J R Stat Soc B. 2009;71:393–424. [Google Scholar]
- Zeng J, Konopka G, Hunt BG, Preuss TM, Geschwind D, Yi SV. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet. 2012;91:455–465. doi: 10.1016/j.ajhg.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Fan J, Yu T. Multiple testing via FDR for large scale imaging data. Ann Stat. 2011;39:613–642. doi: 10.1214/10-AOS848. [DOI] [PMC free article] [PubMed] [Google Scholar]