

Abstract
The common neurodegenerative syndromes exhibit age-related incidence, and many Mendelian neurodegenerative diseases exhibit age-related penetrance. Mutations slowing aging retard age related pathologies. To assess whether delayed aging retards the effects of a mutant allele causing a Huntington's disease (HD)-like syndrome, we generated compound mutant mice, placing a dominant HD knock-in polyglutamine allele onto the slow-aging Snell dwarf genotype. The Snell genotype did not affect mutant huntingtin protein expression. Bigenic and control mice were evaluated prospectively from 10 to 100 weeks of age. Adult HD knock-in allele mice lost weight progressively with weight loss blunted significantly in male bigenic HD knock-in/Snell dwarf mice. Impaired balance beam performance developed significantly more slowly in bigenic HD knock-in/Snell dwarf mice. Striatal dopamine receptor expression was diminished significantly and similarly in all HD-like mice, regardless of the Snell genotype. Striatal neuronal intranuclear inclusion burden was similar between HD knock-in mice with and without the Snell genotype, whereas nigral neuropil aggregates were diminished in bigenic HD knock-in/Snell dwarf mice. Compared with control mice, Snell dwarf mice exhibited differences in regional benzodiazepine and cannabinoid receptor binding site expression. These results indicate that delaying aging delayed behavioral decline with little effect on the development of striatal pathology in this model of HD but may have altered synaptic pathology. These results indicate that mutations prolonging lifespan in mice delay onset of significant phenotypic features of this model and also demonstrate dissociation between striatal pathology and a commonly used behavioral measure of disease burden in HD models.
Keywords: aging, neurodegeneration, polyglutamine, striatum
Introduction
Aging increases incidence of organ system pathologies, including the great majority of human neurodegenerative disorders. Sporadic Alzheimer disease (AD), Parkinson disease, frontotemporal dementias, and motor neuron disease are restricted largely to late adult life, with incidence and prevalence rising markedly with age. Aging increases vulnerability to the mutant alleles associated with Mendelian forms of these disorders, as penetrance is age-related. This is true for several other neurodegenerations, such as the polyglutamine (polyQ) diseases. The basis for the effect of age on neurodegenerative disease incidence-penetrance is unknown. The strong relationship between aging and manifestation of sporadic and Mendelian neurodegenerative diseases raises the possibility that modulating aging may be a pathway to ameliorate the underlying pathogenesis of neurodegenerative disorders (Taylor and Dillin, 2011; Fontana et al., 2014; Herskovits and Guarente, 2014).
Some single-gene mutations slow aging in invertebrates and rodents. Several of these mutations implicate pathways involving insulin or insulin-like growth factors (IGF) signaling (IIS) in modulation of aging (Brown-Borg et al., 1996; Flurkey et al., 2001; Kenyon, 2011; Bartke, 2012). The role of IIS signaling in modulating aging is well established in Caenorhabditis elegans with considerable data indicating that this pathway modulates aging in mammals. Snell and Ames dwarf mice, for example, exhibit deficient growth hormone (GH) and IGF-1 production, live ∼40% longer than controls, and show retardation of aging effects in many organs (Brown-Borg et al., 1996; Flurkey et al., 2001). Prior work in C. elegans models of polyQ disease and AD demonstrated that retarding aging by manipulation of IIS ameliorated pathologic effects of polyQ and AD transgenes, including reduced protein aggregate burden (Morley et al., 2002; Hsu et al., 2003; Cohen et al., 2006). Efforts to reproduce these experiments with murine genetic models of human neurodegenerative diseases report similar positive results, but these experiments generally used murine lines whose delayed aging has been difficult to reproduce (Holzenberger et al., 2003; Taguchi et al., 2007; Selman et al., 2008; Cohen et al., 2009; Freude et al., 2009; Killick et al., 2009; Bokov et al., 2011; Sadagurski et al., 2011). One of these experiments used an aggressive transgenic fragment model of the polyQ disorder Huntington disease (HD; Sadagurski et al., 2011). These lines exhibit early mortality with death by 15 weeks of age and may not be relevant to neurodegenerative diseases that develop in middle age or later.
More recently developed murine HD knock-in models exhibit excellent construct and face validity with considerably later onset of phenotypic features, generally in the second year of life (Heng et al., 2008, 2010; Pouladi et al., 2013). We took advantage of this feature to generate mice combining two mutations: one causing a HD-like phenotype (HdhQ200, dominant knock-in allele) and one (Pit1dw/dw; recessive Snell dwarf mutation; Flurkey et al., 2001) slowing aging. Prospective evaluation of these bigenic mice and littermate controls allowed us to assess whether manipulating a pathway that modulates age-related pathologies reduces susceptibility to the pathogenic effects of mutant alleles causing HD-like age-associated neuronal dysfunction and pathology.
Materials and Methods
Animals.
All experiments were performed on F2 progeny generated with a two-tiered breeding scheme, beginning with crossing HdhQ200 mice on a C57BL/6J background with Pit1dw/dw mice on a mixed (C3H/HeJ × DW/J) background. An initial cross of homozygous Pit1dw/dw males with heterozygous HdhQ200 females was used to generate a F1 generation of four genotypes segregating at both the Pit1 and the HdhQ200 loci. F1 female progeny heterozygous for both the Pit1dw and HD alleles were then crossed to homozygous Pit1dw/dw males to generate the four genotypes used in this study: homozygous dwarf (Pit1dw/dw) mice with (dwHD; N = 36) and without (dw; N = 37) a HdhQ200 allele, and normal size control mice heterozygous for the dwarf mutation (Pit1dw/+) with (HD; N = 38) and without (WT; N = 44) an HdhQ200 allele (Table 1). Mice were genotyped using PCR as described previously (Heng et al., 2010). HdhQ200 alleles were sized at Laragen). The median repeat numbers for HD-like mice were 187 (range,167–219; Table 1). Groups were balanced by gender and genotype. Animals were housed in a specific pathogen free room at 23°C under a 12 h light/dark cycle. They were given ad libitum access to food and water. All procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animals as adopted by the NIH and approved by the Veterinary Medical Unit at the Veterans Affairs Ann Arbor Health System. This colony is tested quarterly for virological evidence of infection, and all such tests were negative during the experimental interval. To confirm the effects of the Snell mutation, liver IGF-1 expression was quantified in all genotypes with quantitative real-time PCR. IGF-1 transcripts were undetectable in dwarf mice, without or without the HD-like mutation.
Table 1.
Genotypes of prospectively evaluated mice
| Genotype | Nomenclature | Size | Aging effect | Median repeats | Repeat range |
|---|---|---|---|---|---|
| Hdh200-Pit1dw/+ | HD | Normal | Normal aging | 187 | 167–219 |
| Pit1dw/+ | WT | Normal | Normal aging | NA | NA |
| HdhQ200-Pit1dw/dw | dwHD | Dwarf | Delayed aging | 187 | 167–219 |
| Pit1dw/dw | dw | Dwarf | Delayed aging | NA | NA |
NA, Not applicable.
Behavioral testing.
Body weights were measured every 10 weeks, starting at age 10 weeks. Motor function was evaluated every 10 weeks, from 10 to 100 weeks of age, using a balance beam test with 5 and 11 mm beams as described previously (Heng et al., 2010). Balance beam testing was videotaped and scored off-line by viewers blind to genotype, though complete blinding is impossible due to size differences in mice. Seconds to cross the beam and number of hind-limb slips while traversing the beam were recorded. Any mouse unable to cross the beam in 25 s was recorded as a 25 s crossing time and removed from the apparatus.
Tissue harvesting.
Animals were killed by decapitation at 100 weeks of age. Brains were dissected and divided in the sagittal plane. One hemisphere was coated with M-1 embedding matrix (Thermo Fisher Scientific), frozen quickly in dry ice, and stored at −70°C until processed for receptor binding or biochemical studies. The other hemisphere was immersion fixed in 4% paraformaldehyde (72 h at 4°C), cryoprotected in 20% sucrose (48 h at 4°C), frozen in chilled isopentane, and stored at −70°C until processed for immunohistochemistry. Quadriceps muscle was collected, coated in M-1 embedding matrix, frozen in dry ice, and stored at −70°C.
Western blot analysis.
Semiquantitative analysis of normal (htt) and expanded huntingtin (mhtt) protein was performed as described previously (Zeng et al., 2013). Flash-frozen mouse brains (3 animals per genotype) were homogenized in RIPA buffer with protease inhibitors. Homogenates were subjected to centrifuge at 13,000 × g for 20 min at 4° to separate supernatants (RIPA soluble) and pellets. Supernatants proteins were separated by SDS-PAGE. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes, which were stained using the following primary antibodies: anti-polyQ 1C2 (1:10,000, Millipore), anti-htt 2166 (1:500, Millipore), anti-tubulin-α (1:10,000, Cell Signaling Technology). Pellets were resuspended in 1% SDS in PBS (RIPA insoluble fractions). Proteins from RIPA insoluble fractions were separated by 3% SDS-PAGE, transferred to PVDF membranes, and stained with anti-polyQ 1C2. Peroxidase-conjugated secondary antibodies were used at 1:10,000 (Jackson Immunoresearch) and blots developed using blue basic autorad films (GeneMate). ImageJ software was used for semiquantification of band intensities.
Immunohistochemistry.
Free-floating 40-μm-thick sagittal sections were processed for immunohistochemistry using the ImmPRESS polymer system with DAB immPACT substrate (Vector Laboratories). Primary antibodies used were generated against NeuN, a pan-neuronal marker (1:1000; no. MAB377, Millipore), N-terminal huntingtin (1:200; no. sc8767, Santa Cruz Biotechnology), the microglial marker Iba1 (1:500; no. 019-19741, Wako Chemicals), the astrocyte intermediate filament protein glial fibrillary acidic protein (GFAP; 1:2000; no. AB5804, Millipore), and peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α; 1:600; no. AB3242, Lot no. 239229, Millipore).
Quantitative receptor autoradiography.
Fresh frozen hemispheres were cut on a cryostat in parasagittal plane into 14 μm serial sections, thaw mounted on Superfrost slides, and stored until use at −70°C (N = 5–7 animals per genotype). Autoradiographic studies on D1 and D2 dopamine receptors and GABA-A/benzodiazepine receptors were performed as described previously using radioligands purchased from PerkinElmer (Heng et al., 2010). Briefly, D1 receptors were labeled with 0.55 nm [3H]SCH23390, and nonspecfic binding determined in the presence of 1 μm cis-flupenthixol. Total binding to D2 receptors was established by incubating sections with 0.75 nm [3H]Spiperone, and nonspecific binding was assessed by blocking with 50 μm dopamine. GABA-A/benzodiazepine receptors were identified with 5 nm [3H]flunitrazepam and nonspecific binding determined in the presence of 2 μm clonazepam. 10 nm [3H]CP55,940 (PerkinElmer) was used to label cannabinoid receptors with 10 μm WIN55,212-2 (Sigma-Aldrich) as the blocking agent as described previously (Richfield and Herkenham, 1994).
Autoradiograms were generated by apposing the [3H]-labeled sections and [14C] standards (ARC) to a tritium sensitive plate (BAS-TR2025; GE Healthcare). A digital image of the plate was made with a BAS-5000 Imaging Plate Scanner (GE Healthcare). Autoradiograms were analyzed by quantitative densitometry using an MCID image analysis system (Interfocus). Optical density measurements were converted to tissue radioactivity with a calibration curve generated from measurements of coexpressed standards of known radioactivity. Specific binding for all ligands was calculated by subtracting nonspecific from total binding.
BDNF and PPARGC1A gene expression.
Brain derived neurotrophic factor (BDNF) is produced by corticostriate neurons, is important for trophic support of striatal neurons, and is deficient in HD (Zuccato and Cattaneo, 2014). We assessed forebrain BDNF gene expression as an index of corticostriate neuron dysfunction. Peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) is an important regulator of mitochondrial and metabolic function, abnormal PGC-1α function has been implicated in HD and other polyglutamine diseases (Tsunemi et al., 2012), and PGC-1α is suggested to regulate aging. Total RNA was extracted from forebrains (5 animals/genotype) was analyzed using quantitative real-time PCR. RNA was isolated using the QIAGEN RNeasy Kit (Qiagen), which was combined with the RNase-Free DNase Set (Qiagen). RNA was reversely transcribed with High Capacity cDNA RT Kit and amplified using TaqMan Universal PCR-Master Mix, NO AmpErase UNG with TaqMan Assay-on-demand kits (Applied Biosystems). Relative expression of target mRNAs was adjusted for total RNA content by β-actin RNA quantitative PCR. Quantitative PCR was performed on an ABI-PRISM 7900 HT Sequence Detection system (Applied Biosystems). Each reaction was performed in triplicates as previously described (Sadagurski et al., 2012). BDNF second exon, forward: TGGCCCTGCGGAGGCTAAGT, BDNF, reverse: AGGGTGCTTCCGAGCCTTCCT and PGC-1α gene (PPARGC1A, forward: ATGACCCTCCTCACACCAAACCCACAG, PPARGC1A, reverse: CTTGAGCATGTTGCGACTGCGGTTGTG) expression. β-actin, forward: GACATGGAGAAGATCTGGCA, β-actin, reverse: GGTCTCAAACATGATCTGGGT. BDNF and PPARGC1A expression were normalized to β-actin expression for statistical analysis.
Stereology.
Striatal cell counts and striatal volume were obtained in 100-week-old animals (4–6 animals/sex/genotype) using StereoInvestigator software (Micro-Brightfield). Striatal counts were taken unilaterally in four 40 μm sections from each brain corresponding approximately to levels 3.44, 2.64, 1.8, and 0.96 mm from the midline (Paxinos and Franklin, 2001). The optical fractionator method was used to generate an estimate of total striatal neuron number with positively stained NeuN-immunoreactive cells. Volume was reconstructed by the StereoInvestigator software. The number of GFAP- and Iba1-reactive cells and htt-immunoreactive neuronal intranuclear inclusions (NIIs) in striatum was estimated according to the optical fractionator method combined with the dissector principle with unbiased counting rules. To assess cytoplasmic htt-immunoreactive aggregates, we measured neuropil aggregate burden in the substantia nigra (both pars reticulata and pars compacta). In aged animals of this knock-in line, neuropil aggregates are particularly abundant in striatal efferent projection targets, the substantia nigra and globus pallidus, where they probably reside in striatal neuron terminals (Tallaksen-Greene et al., 2005). The same sections used for quantifying striatal NIIs were counterstained with methyl green to stain nuclei and the number of htt-immunoreactive nigral neuropil aggregates and NIIs was estimated using the optical fractionator method combined with the dissector principle with unbiased counting rules.
Muscle histology.
Cross sections (10 μm; cryocut 1800, Leica) were cut from fresh frozen quadriceps (−80°C, n = 16). For hematoxylin and eosin (H&E) staining, sections were rehydrated in a graded ethanol series. Sections were stained with hematoxylin (Shandon Gill 3 hematoxylin, 6765010, Fisher Thermo Scientific) and counter stained with eosin (01602, Surgipath). After dehydration and coverslipping (DPX mountant, 06522, Sigma-Aldrich), slides were examined in bright field (Zeiss Axio Imager Z.1). For NADH staining, sections were incubated at 37°C for 30 min in NADH (1.6 mg/ml, N8129, Sigma-Aldrich) and nitroblue tertrazolium (NBT; 2 mg/ml, N6876, Sigma-Aldrich), dissolved in Tris-HCl (0.05 m, pH 7.6, T3253, Sigma-Aldrich). Unbound NBT was removed with washing in ascending and descending concentrations of acetone and coverslips affixed (DPX mountant, 06522, Sigma-Aldrich). Sections were examined in bright-field (Zeiss Axio Imager Z.1).
Statistics.
Intergroup comparisons of balance beam data, striatal dopamine receptor data, GABA-A/benzodiazepine receptor data, and BDNF gene expression data were analyzed using two-way ANOVA (STATA), in which the dependent variable was the behavioral outcome or pathologic outcome measure and the independent variables were genotype at Pit1, genotype at HdhQ200, and their interaction. A significant interaction term (p < 0.05) was taken as evidence that the effect of HdhQ200 differed between dwarf and control mice. Effects on body weight were quantified as the change in weight after 40 weeks, and expressed as a percentage of initial weight, to compensate for the effects of different body size between dwarf and full size mice. Genders were evaluated separately. Weight percentage changes were evaluated with two-way ANOVA to assess effects of age and genotype. Cannabinoid receptor, stereology, and Western blot studies of mhtt expression were analyzed with one-way ANOVAs and post hoc Tukey's tests. Survival was assessed with the log-rank test.
Results
Huntingtin expression
Mutant huntingtin (mhtt) expression did not differ between HD (normal size; no retarded aging phenotype) and dwHD (dwarf; retarded aging phenotype) mice. Figure 1 shows results of Western blots with 1C2 antibody against expanded polyQ domains. Similar results were obtained with the N-terminal antibody against htt (results not shown). We also quantified the insoluble fraction of mhtt and found no difference between HD and dwHD mice (data not shown).
Figure 1.

Results of Western blots for mhtt expression. Mice were evaluated at age 100 weeks; N = 3 for each group. Immunoblots with 1C2 mAb for expanded polyQ domains shown. No significant difference in mhtt expression between HD and dwHD mice. Error bars are SEM.
Survival analysis
Survival was assessed with the log-rank test. There were no differences in survival among any of the genotypes (data not shown), although the termination of the experiment at 100 weeks of age, needed to obtain tissue for biochemistry and histopathology, prevents any inference about possible differences in survival at older ages.
Weight
Data were analyzed as percentage of weight loss after 40 weeks of age. Both HD and dwHD mice exhibited age related decline in weight compared with littermate controls (Fig. 2). This was true for mice of both genders. For females, t = −3.66, p = 0.0009. For males, t = −3.18, p = 0.003. The HdhQ200 allele leads to weight loss, from 40 weeks onward, in both genders, and the Snell dwarf genotype blunts this decline, expressed as a percentage of initial weight, in both males (p < 0.003) and females (p < 0.0009) for the contrast between HD and dwHD mice with unpaired t tests (Fig. 2). A formal test of the interaction between the HD and dw genotypes with two-way ANOVAs, however, showed a significant blunting of the HD effect only in the male mice (p < 0.0006) after adjustment for the independent effects of HdhQ200 and Snell dwarf genotypes.
Figure 2.

Prospectively obtained weight results for all genotypes. Data presented as percentage change in weight after 40 weeks. Female and male data displayed separately. In both females and males, the presence of an HD-like allele was associated with age-related weight loss. **Indicates significant difference relative degree of weight loss between dw and dwHD mice with unpaired t tests (see Results). Age-related weight loss was apparently ameliorated by the Snell dwarf genotype significantly in both genders but analysis with two-way ANOVA revealed a significant interaction term only for male mice (see Results). Error bars are SEM.
Motor behavior
WT and dwarf mice without an HD-like allele (dw) exhibited a modest and nonsignificant age-related increase in time to cross the 5 mm balance beam and associated paw slips (Fig. 3). HD (nondwarf; no retarded aging phenotype) mice exhibited a progressive increase in time to cross the 5 mm balance beam and increasing number of paw slips after 50 weeks of age. This progressive decline in balance beam performance is similar to that documented previously with HdhQ200 heterozygous mice on a different genetic background (Heng et al., 2010). In contrast, dwHD mice exhibited less age-related decline in 5 mm balance beam performance than HD mice. With two-way ANOVA, there was a significant interaction term between the effects of the HD allele and the dwarf allele (p < 0.05), both for time to cross the beam and for the number of hindlimb slips during beam passage, at each age from 80 to 100 weeks, indicating that in dwHD mice, decline in balance beam performance was slowed compared with HD mice. For slips, for example, at 80 weeks of age: F = 10.7, p = 0.014; at 90 weeks of age: F = 10.2, p = 0.002; at 100 weeks of age, F = 11.8, p = 0.0008. For the time measurement, at 80 weeks of age: F = 19.6, p < 0.001; at 90 weeks of age: F = 9.34, p = 0.003; at 100 weeks of age: F = 12.91, p < 0.001. Similar results were obtained with the 11 mm balance beam (data not shown).
Figure 3.
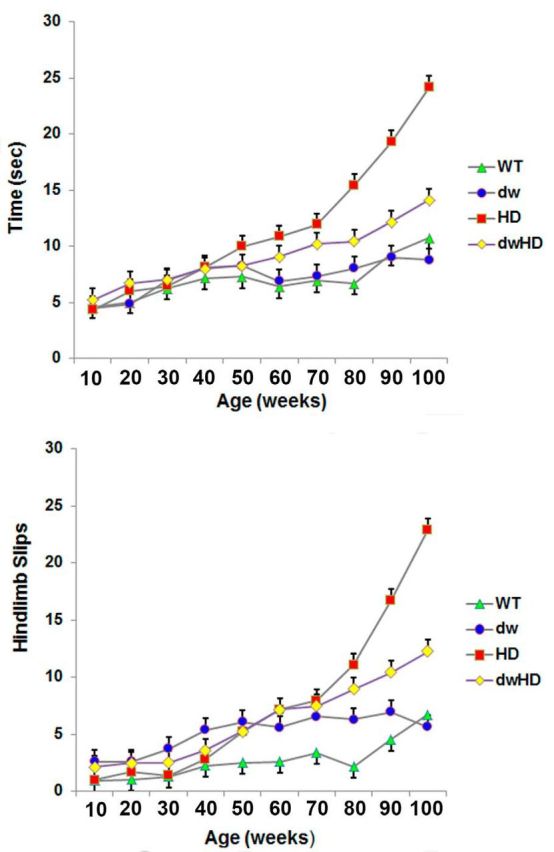
Balance beam performance on the 5 mm beam. Animals evaluated longitudinally. N values described in text. WT = Pitdw/−; dw = Pitdw/dw; HD = HdhQ200—Pitdw/−; dwHD = HdhQ200—Pitdw/dw. A, Time to cross beam. B, Number of hindlimb slips when crossing beam. Control (WT and dw) mice exhibit insignificant decline in balance beam performance over time. HD mice exhibit marked decline in performance in the second year of life. dwHD mice also exhibit significant decline in balance beam performance in the second year of life but of lesser magnitude and delayed in comparison with HD mice. Error bars are SEM.
Striatal pathology
Both dwHD and HD mice exhibited marked, ∼50%, reductions of striatal dopamine D1 and D2 receptors (Table 2; Fig. 4). These results are essentially identical with prior results in HdhQ200 heterozygous mice (Heng et al., 2010). Two-way ANOVA did not reveal a significant interaction term, confirming that the magnitude of the D1 and D2 receptor reductions was similar in dwHD and HD mice. To broadly survey extrastriatal regions, we performed GABAA/benzodiazepine receptor autoradiography (Fig. 5). As in a recent study, we found an effect of the HdhQ200 allele on GABAA/benzodiazepine receptor binding site expression with higher levels in mice carrying the HdhQ200 allele (Zeng et al., 2013; Table 2). There was also an effect of the dw genotype; dw mice expressing increased GABAA/benzodiazepine receptor binding sites in several brain regions regardless of the HdhQ200 genotype (Table 2). Two-way ANOVA did not reveal a significant interaction between HD and dw genotypes. NIIs were abundant within striatal neurons of all mice with HdhQ200 alleles and were absent in mice lacking the HdhQ200 allele. There was no difference in striatal NII burden between dwHD and HD mice (Fig. 6). Total striatal neurons were quantified with NeuN staining. As expected from prior work in HdhQ200 heterozygous mice, there was no loss of striatal neurons in mice with HdhQ200 alleles (data not shown; Heng et al., 2010). We found no differences in striatal GFAP-immunoreactive glia and Iba1-immunoreactive microglia between dwHD and HD mice (data not shown).
Table 2.
Results of receptor autoradiography
| Binding site/ligand | Region |
Genotype |
HD effect, p | dw effect, p | |||
|---|---|---|---|---|---|---|---|
| WT | dw | HD | dwHD | ||||
| D1 receptor | Dorsal striatum | 195 ± 9 (6) | 183 ± 1 (6) | 74 ± 9 (6) | 79 ± 1 (7) | 0.0001 ↓ | |
| [3H]-SCH23390 | Ventral striatum | 187 ± 2 (6) | 158 ± 2 (6) | 76 ± 9 (6) | 82 ± 1 (7) | 0.0001 ↓ | |
| Substantia nigra | 96 ± 8 (6) | 97 ± 1 (6) | 46 ± 6 (6) | 72 ± 1 (7) | 0.001 ↓ | ||
| Frontoparietal cortex | 9 ± 1 (6) | 12 ± 1 (6) | 9 ± 0 (6) | 16 ± 1 (7) | 0.0001 ↑ | ||
| D2 receptor | Dorsal striatum | 78 ± 1 (6) | 57 ± 2 (6) | 48 ± 1 (6) | 34 ± 5 (7) | 0.005 ↓ | 0.053 ↓ |
| [3H]-Spiperone | Ventral striatum | 104 ± 7 (6) | 100 ± 7 (6) | 71 ± 9 (6) | 60 ± 6 (7) | 0.0001 ↓ | |
| Frontoparietal cortex | 6 ± 2 (6) | 13 ± 3 (6) | 11 ± 3 (6) | 12 ± 3 (7) | |||
| Cannabinoid receptor | Whole-brain | 15 ± 1 (6) | 13 ± 1 (5) | 15 ± 2 (6) | 14 ± 2 (5) | ||
| [3H]-CP55,940 | Substantia nigra | 167 ± 4 (6) | 30 ± 3 (5) | 60 ± 1 (6) | 24 ± 4 (5) | 0.05 ↓ | 0.02 ↓ |
| Cerebellar cortex | 38 ± 3 (6) | 47 ± 4 (5) | 41 ± 4 (6) | 40 ± 3 (5) | |||
| GABAA receptor | Whole-brain | 103 ± 5 (6) | 110 ± 7 (6) | 118 ± 4 (6) | 141 ± 5 (6) | 0.0003 ↑ | 0.009 ↑ |
| [3H]-Flunitrazepam | Dorsal striatum | 66 ± 6 (6) | 80 ± 8 (6) | 82 ± 4 (6) | 110 ± 6 (6) | 0.002 ↑ | 0.003 ↑ |
| Ventral striatum | 87 ± 9 (6) | 113 ± 1 (6) | 117 ± 3 (6) | 157 ± 1 (6) | 0.0004 ↑ | 0.001 ↑ | |
| Substantia nigra | 199 ± 7 (6) | 226 ± 1 (5) | 231 ± 4 (6) | 282 ± 1 (5) | 0.001 ↑ | 0.003 ↑ | |
| Frontoparietal cortex | 168 ± 1 (6) | 192 ± 2 (6) | 202 ± 1 (6) | 244 ± 6 (6) | 0.003 ↑ | 0.02 ↑ | |
| Hippocampus | 187 ± 6 (6) | 191 ± 2 (6) | 199 ± 8 (6) | 237 ± 6 (6) | 0.03 ↑ | 0.0007 ↑ | |
| Cerebellum | 56 ± 4 (6) | 44 ± 6 (6) | 82 ± 4 (6) | 80 ± 5 (6) | 0.0001 ↑ | ||
Values listed are specific binding (nCi/g) ± SEM (n).
Figure 4.

Results of striatal dopamine D1 and D2 receptor autoradiography. WT = Pitdw/+; dw = Pitdw/dw; HD = HdhQ200-Pitdw/−; dwHD = HdhQ200-Pitdw/dw. A, Striatal D1 receptor binding. B, Striatal D2 receptor binding. Striatal D1 and D2 receptor binding markedly reduced in all HD-like mice regardless of Snell dwarf phenotype. *Indicates effect of HD genotype. **Indicates effect of dw genotype (Table 2). For N values, see Table 2. Error bars are SEM.
Figure 5.

Results of GABA-A/Benzodiazepine and nigral cannabinoid receptor autoradiography. WT = Pitdw/; dw = Pitdw/dw; HD = HdhQ200-Pitdw/−; dwHD = HdhQ200-Pitdw/dw. A, GABA-A/Benzodiazepine receptor binding modestly increased in several regions of HD and dwarf mice (see Results; Table 2). B, Nigral cannabinoid receptor binding is markedly reduced in HD mice and in dwarf mice of both (dw and dwHD) genotypes. *Indicates effect of HD genotype. **Indicates effect of dw genotype (Table 2). For N values, see Table 2. Error bars are SEM.
Figure 6.

Striatal NII burden. WT = Pitdw/−; dw = Pitdw/dw; HD = HdhQ200-Pitdw/−; dwHD = HdhQ200-Pitdw/dw. A, Representative photomicrographs of NII expression in all four genotypes. NIIs are abundant in mice with an HD-like allele and absent in control mice. Scale bar, 25 μm. B, Histographs of NII abundance as determined with stereology. No difference between HD and dwHD mice. *Indicates different from wt and dw mice. Error bars are SEM.
Nigral pathology
Stereologic quantification of nigral neuropil aggregates revealed a significant reduction in neuropil aggregate burden in dwHD compared with HD mice (p < 0.0001, F = 143.99 for overall ANOVA; p < 0.01 for dwHD versus HD [Tukey's HSD]; Fig. 7) without any difference in nigral NIIs (Fig. 7). Nigral dopamine D1 receptors, which are largely expressed on striatal efferent terminals, were reduced equally in both dwHD and HD mice (Table 2; Fig. 4). We also assessed cannabinoid receptor binding within the substantia nigra (Table 2; Fig. 5). These binding sites are associated with striatonigral efferent terminals and can be used as a measure of striatal neuron dysfunction. As expected, nigral cannabinoid receptor binding was quite high in Pit1dw/− (WT) mice and markedly reduced in substantia nigra of HD mice (Fig. 5). Surprisingly, cannabinoid receptor binding was reduced in all dwarf mice (dw and dwHD), preventing its use as an indicator of whether the Pit1dw/dw genotype alters the effects of HdhQ200 alleles on striatal neuron terminal integrity (Fig. 5).
Figure 7.

Nigral cytoplasmic inclusion and NII burden. WT = Pitdw/−; dw = Pitdw/dw; HD = HdhQ200-Pitdw/−; dwHD = HdhQ200-Pitdw/dw. A, Representative photomicrographs of cytoplasmic inclusion and NII burden in HD and dwHD mice. Nigral NII burden is similar in HD and dwHD mice but cytoplasmic inclusion burden is reduced in dwHD mice. B, Histographs of NII and cytoplasmic inclusion burden. *Indicates different from wt and dw. **Indicates dwHD different from HD. Scale bar, 25 μm. Error bars are SEM.
Muscle pathology
Neither H&E stained sections nor NADH histochemistry revealed any evidence of muscle pathology (Fig. 8). Snell dwarf mice had smaller fibers than normal size mice but there was no apparent effect of the HD-like genotype.
Figure 8.

NADH histochemistry of quadriceps muscle. Muscle fibers are smaller in dwarf mice, with or without HD-like alleles. No pathologic changes were detected in any mice. Scale bar, 200 μm.
BDNF and PPARGC1A transcript expression
Forebrain BDNF transcript expression was reduced in mice carrying HD-like alleles (HD and dwHD) without differences between dwarf and nondwarf mice (F = 8.77, p = 0.006 for the HD effect in two-way ANOVA; F = 0.2, p = 0.66 for the interaction term; Fig. 9). PPARGC1A forebrain transcript expression was reduced in dwarf mice, regardless of the presence of an HD-like allele (data not shown). Immunohistochemistry did not reveal any clear changes in PGC-1α distribution or expression among all genotypes examined (data not shown).
Figure 9.

Forebrain BDNF gene expression. Two-way ANOVA indicates an effect of HD-like alleles reducing BDNF gene expression but no significant effect of the Snell dwarf genotype and no interaction between HD-like alleles and Snell dwarf genotype. Bar indicates significant difference (t = −2.53, p = 0.02) in contrast of WT and HD mice. Error bars are SEM.
Discussion
To determine whether the Snell dwarf genotype, which delays many aspects of aging and extends lifespan, would ameliorate the features of a genetically faithful model of HD, we constructed double-mutants by placing the dominant HdhQ200 allele onto the Snell dwarf (Pit1dw/dw) genotype. Motor deficits, as measured by a balance beam task, developed more slowly in double mutant animals than in HdhQ200 control littermates. Male double-mutant mice exhibited proportionately less weight loss than HdhQ200 control littermates and there was a trend toward a similar effect in female double mutant mice. We found a decrease in nigral neuropil aggregates in double mutant mice, suggesting diminished striatal terminal pathology. In contrast, the Snell dwarf genotype had no effect on the development of striatal pathology as measured by striatal neuron dopamine receptor expression and NII accumulation. Nigral D1 receptors, expressed primarily on striatal terminals, were equivalently reduced in both HD and dwHD mice, indicating that the Snell genotype had no effect on this marker of striatal terminal integrity. Similarly, forebrain BDNF mRNA, produced by corticostriate neurons and reduced in HD and other HD murine models, was reduced in mice with HdhQ200 alleles regardless of the presence of the Snell genotype. The motor, weight, and neuropil inclusion data suggest amelioration of the HD-like features of this model, but there was no corresponding effect on most markers of neuronal dysfunction.
Our results extend prior efforts assessing the effects of delayed aging on mouse models of HD and AD (Cohen et al., 2009; Freude et al., 2009; Killick et al., 2009; Sadagurski et al., 2011). The prior experiment with HD-like mice used R6/2 mhtt fragment transgenic mice, which exhibit essentially complete mortality by age 15 weeks (Stack et al., 2005). The early onset and aggressive pathology of R6/2 mice may reflect effects of expanded polyQ fragments at least partly during periods of active brain development and maturation, diminishing the relevance of this model for typical midlife to late-life onset of HD where neurodegeneration develops in a mature brain. The delayed aging models used in these experiments exhibit problematic features. Cohen et al. (2009) used heterozygous IGF-1 receptor knock-out mice. This stock was reported originally to exhibit 33% extension of lifespan in females and 16% extension of lifespan in males (Holzenberger et al., 2003). The original report was notable for relatively short lifespan of control animals and a recent replication study revealed only modest (4%) lifespan extension in females and no effects in males (Bokov et al., 2011). Similarly, some prior experiments with HD-like and AD-like mice used Insulin Receptor Substrate 2 (IRS2) knock-out mice to delay aging (Freude et al., 2009; Killick et al., 2009; Sadagurski et al., 2011). There is evidence that delayed aging of these mice relative to controls is demonstrable only when animals are fed a high fat diet, though this is controversial (Taguchi et al., 2007; Selman et al., 2008, 2011).
Our results differ somewhat from results of prior experiments in C. elegans models of polyQ disease and AD (Morley et al., 2002; Hsu et al., 2003; Cohen et al., 2006). Delaying aging with mutants that alter IIS in C. elegans significantly prolongs survival, improves motor function, and reduces inclusion burden in polyQ transgenic worms, results our murine experiments replicate only partly. We found an effect of delayed aging on motor function but no effect on NII accumulation. Beyond the obvious differences between mammals and nematodes, there are other differences in these experiments. The C. elegans models use polyQ fragment transgenes expressed in body wall muscle, not actual htt-like genes expressed in neurons. Altering IIS in C. elegans profoundly alters aging and reduces protein aggregation accompanying aging, but results in nematode models may not predict results in analogous mammalian experiments (David et al., 2010). Altering IIS is not, of course, the only way to manipulate aging and it is possible that other modifications influencing the rate of aging may have more beneficial effects in polyQ disease models. Caloric restriction, for example, is reported to have beneficial effects in a C. elegans polyQ disease model and the R6/2 model of HD (Duan et al., 2003). It is also possible that reducing IGF-1 production may retard aging and slow neurodegenerative processes generally but have countervailing negative effects in the specific context of HD or other polyQ disorders. Humbert et al. (2002) found that IGF-1 activation of Akt reduced neuronal death and NII formation in an in vitro model of HD. Systemic IGF-1 administration is reported to improve hyperglycemia, weight loss, and limb clasping abnormalities in the R6/2 model of HD. Intranasal IGF-1 administration is reported to improve biochemical and some motor features in the YAC128 transgenic model of HD (Duarte et al., 2011; Lopes et al., 2014). Both muscle-specific overexpression and systemic administration of IGF-1 in murine models of another polyQ disease, spinobulbar muscular atrophy (Kennedy syndrome) reduced disease effects (Palazzolo et al., 2009; Rinaldi et al., 2012). Excitotoxic neuronal injury is a likely important mechanism of neuronal death in HD, and IGF-1 administration was reported to reduce quinolinate-induced neurodegeneration (Escartin et al., 2004). On the other hand, IGF-1 administration potentiated neuronal death in another toxin model of HD, using the mitochondrial inhibitor and indirect excitotoxin 3-nitropropionic acid (Escartin et al., 2007). There is little clinical data on the relationship between IGF-1 and features of polyQ diseases, but Saleh et al. (2010) reported a positive correlation between circulating IGF-1 levels and rates of cognitive decline in HD.
We found that the progressive decline of HdhQ200 heterozygous mice in balance beam performance was ameliorated by the Snell dwarf genotype. Given the lack of effect of the Snell dwarf genotype on our readouts of striatal pathology; our results suggest that balance beam performance in these mice is not closely linked to the measures of striatal pathophysiology we evaluated. It is possible that amelioration of the decline in balance beam performance reflects a beneficial effect of the Snell dwarf genotype in one or more extrastriatal brain regions. These results may be analogous to prior results reported by Jafar-Nejad et al. (2012) in a model of the polyQ disease spinocerebellar ataxia type 1 (SCA1). SCA1 knock-in mice haploinsufficient for the 14-3-3ε protein exhibited reduced cerebellar pathology and improved motor performance, but there were no changes in trajectories of weight loss, respiratory abnormalities, or premature mortality. 14-3-3ε haploinsufficiency reduced Ataxin-1 (ATXN1) protein levels and recruitment of expanded repeat ATXN1 into toxic protein complexes in the cerebellum but not the brainstem. The pathogenic effects of expanded polyQ proteins differ regionally, and these regionally different effects may be modulated differentially by delayed aging.
It is possible as well that the Snell dwarf genotype had a beneficial effect on some peripheral organ function. We assessed muscle pathology without finding evidence of muscle dysfunction in any of the evaluated genotypes.
The Snell dwarf genotype did affect GABA-A/benzodiazepine receptor expression in some brain regions, and we found reduced nigral cannabinoid receptor binding sites in Snell dwarf mice lacking the HdhQ200 allele. Little is known about the effects of Pit1-deficiency on brain function, though age-related changes in some cognitive tests are delayed in the closely related Ames dwarf (Prop1df/df) mouse stock (Sun et al., 2005). Prior limited studies of Snell dwarfs suggested modest abnormalities of cortical neuron morphology and delayed myelination (Noguchi et al., 1983, 1985), though our studies indicate normal myelination in mature Snell dwarf mice (data not shown). Prior neurochemical studies suggest developmental abnormalities in cholinergic and monoaminergic neurotransmission in Snell dwarf mice (Fuhrmann et al., 1985; Kempf et al., 1985). Further work will be required to understand the effects of this mutation on brain structure and function.
The dissociation between the effects of the Snell dwarf genotype on balance beam performance and weight loss, and indices of striatal neuron dysfunction-pathology are a warning that results of some commonly used behavioral tests should be interpreted cautiously. An ideal readout would predict accurately the results of human clinical intervention trials. The paucity of completed clinical trials precludes systematic comparison of mouse model studies with human intervention studies. Considerable further preclinical and clinical work is needed to establish the predictive validity of murine genetic HD models and to determine the best ways to evaluate various functional tests of the underlying neuropathology.
In summary, we found good evidence of an effect of the Snell dwarf genotype on some phenotypic features but little evidence that delaying aging reduced striatal neuron dysfunction-pathology in this model of HD with strong face and construct validity. These results undermine the notion that this form of delaying aging by reducing IIS will ameliorate neurodegeneration, at least for HD. It is possible that this intervention had some beneficial effects on neuronal dysfunction in extrastriatal brain regions. Our results also suggest caution in interpreting a commonly used behavioral outcome measure in HD-like mice and underscore the need for more work to validate these models.
Footnotes
This work was supported by RO1 NS 038712 (H.L.P.), RO1 AG034228 (H.L.P.), P30-AG024824 (R.A.M.), R01-AG019899 (R.A.M.), P30-AG013283 (R.A.M.), R21 NS NS059537 (R.L.A.), and a VA Merit Review Grant (R.L.A.). We thank Sabrina Van Roekel and Amanda Keedle for husbandry assistance, and the anonymous reviewers for helpful criticisms.
The authors declare no competing financial interests.
References
- Bartke A. Healthy aging: is smaller better? A mini-review. Gerontology. 2012;58:337–343. doi: 10.1159/000335166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokov AF, Garg N, Ikeno Y, Thakur S, Musi N, DeFronzo RA, Zhang N, Erickson RC, Gelfond J, Hubbard GB, Adamo ML, Richardson A. Does reduced IGF-1R signaling in Igf1r+/- mice alter aging? PLoS One. 2011;6:e26891. doi: 10.1371/journal.pone.0026891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte AI, Petit GH, Ranganathan S, Li JY, Oliveira CR, Brundin P, Björkqvist M, Rego AC. IGF-1 protects against diabetic features in an in vivo model of Huntington's disease. Exp Neurol. 2011;231:314–319. doi: 10.1016/j.expneurol.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Escartin C, Boyer F, Bemelmans AP, Hantraye P, Brouillet E. Insulin growth factor-1 protects against excitotoxicity in the rat striatum. Neuroreport. 2004;15:2251–2254. doi: 10.1097/00001756-200410050-00022. [DOI] [PubMed] [Google Scholar]
- Escartin C, Boyer F, Bemelmans AP, Hantraye P, Brouillet E. IGF-1 exacerbates the neurotoxicity of the mitochondrial inhibitor 3NP in rats. Neurosci Lett. 2007;425:167–172. doi: 10.1016/j.neulet.2007.08.031. [DOI] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Kennedy BK, Longo VD, Seals D, Melov S. Medical research: treat aging. Nature. 2014;511:405–407. doi: 10.1038/511405a. [DOI] [PubMed] [Google Scholar]
- Freude S, Hettich MM, Schumann C, Stöhr O, Koch L, Köhler C, Udelhoven M, Leeser U, Müller M, Kubota N, Kadowaki T, Krone W, Schröder H, Brüning JC, Schubert M. Neuronal IGF-1 resistance reduces Aβ accumulation and protects against premature death in a model of Alzheimer's disease. FASEB J. 2009;23:3315–3324. doi: 10.1096/fj.09-132043. [DOI] [PubMed] [Google Scholar]
- Fuhrmann G, Durkin T, Thiriet G, Kempf E, Ebel A. Cholinergic neurotransmission in the central nervous system of the Snell dwarf mouse. J Neurosci Res. 1985;13:417–430. doi: 10.1002/jnr.490130308. [DOI] [PubMed] [Google Scholar]
- Heng MY, Detloff PJ, Albin RL. Rodent genetic models of Huntington disease. Neurobiol Dis. 2008;32:1–9. doi: 10.1016/j.nbd.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Heng MY, Duong DK, Albin RL, Tallaksen-Greene SJ, Hunter JM, Lesort MJ, Osmand A, Paulson HL, Detloff PJ. Early autophagic response in a novel knock-in model of Huntington disease. Hum Mol Genet. 2010;19:3702–3720. doi: 10.1093/hmg/ddq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits AZ, Guarente L. SIRT1 in neurodevelopment and brain senescence. Neuron. 2014;81:471–483. doi: 10.1016/j.neuron.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntington phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/S1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad P, Ward CS, Richman R, Orr HT, Zoghbi HY. Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci U S A. 2011;108:2142–2147. doi: 10.1073/pnas.1018748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf E, Fuhrmann G, Thiriet G, Ebel A. Dopamine and serotonin metabolism in striatum and in the septohippocampal pathway of the Snell dwarf mouse. Neurochem Res. 1985;10:969–981. doi: 10.1007/BF00964633. [DOI] [PubMed] [Google Scholar]
- Kenyon C. The first long-lived mutants: discovery of the insulin/IGF-1 pathway for aging. Philos Trans R Soc Lond B Biol Sci. 2011;366:9–16. doi: 10.1098/rstb.2010.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killick R, Scales G, Leroy K, Causevic M, Hooper C, Irvine EE, Choudhury AI, Drinkwater L, Kerr F, Al-Qassab H, Stephenson J, Yilmaz Z, Giese KP, Brion JP, Withers DJ, Lovestone S. Deletion of Irs2 reduces amyloid deposition and rescues behavioral deficits in APP transgenic mice. Biochem Biophys Res Comm. 2009;386:257–262. doi: 10.1016/j.bbrc.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes C, Ribeiro M, Duarte AI, Humbert S, Saudou F, Pereira de Almeida L, Hayden M, Rego AC. IGF-1 intranasal administration rescues Huntington's disease phenotypes in YAC128 mice. Mol Neurobiol. 2014;49:1126–1142. doi: 10.1007/s12035-013-8585-5. [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi T, Sekiguchi M, Sugisaki T, Tsukada Y, Shimai K. Faulty development of cortical neurons in the Snell dwarf cerebrum. Brain Res. 1983;312:125–138. doi: 10.1016/0165-3806(83)90127-x. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Sekiguchi M, Sugisaki T, Tsukada Y, Shimai K. Cerebral myelinogenesis in the Snell dwarf mouse: stimulatory effects of GH and T4 restricted to the first 20 days of postnatal life. Neurochem Res. 1985;10:767–778. doi: 10.1007/BF00964534. [DOI] [PubMed] [Google Scholar]
- Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci. 2013;14:708–721. doi: 10.1038/nrn3570. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Herkenham M. Selective vulnerability in Huntington's disease: preferential loss of cannabinoid receptors in lateral globus pallidus. Ann Neurol. 1994;36:577–584. doi: 10.1002/ana.410360406. [DOI] [PubMed] [Google Scholar]
- Rinaldi C, Bott LC, Chen KL, Harmison GG, Katsuno M, Sobue G, Pennuto M, Fischbeck KH. Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol Med. 2012;18:1261–1268. doi: 10.2119/molmed.2012.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Cheng Z, Rozzo A, Palazzolo I, Kelley GR, Dong X, Krainc D, White MF. IRS2 increases mitochondrial dysfunction and oxidative stress in a mouse model of Huntington disease. J Clin Invest. 2011;121:4070–4081. doi: 10.1172/JCI46305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Leshan RL, Patterson C, Rozzo A, Kuznetsova A, Skorupski J, Jones JC, Depinho RA, Myers MG, Jr, White MF. IRS2 signaling in LepR-b neurons suppresses FoxO1 to control energy balance independently of leptin action. Cell Metab. 2012;15:703–712. doi: 10.1016/j.cmet.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh N, Moutereau S, Azulay JP, Verny C, Simonin C, Tranchant C, El Hawajri N, Bachoud-Lévi AC, Maison P Huntington French Speaking Group. High insulinlike growth factor I is associated with cognitive decline in Huntington disease. Neurology. 2010;75:57–63. doi: 10.1212/WNL.0b013e3181e62076. [DOI] [PubMed] [Google Scholar]
- Selman C, Lingard S, Gems D, Partridge L, Withers DJ. Comment on “Brain IRS2 signaling coordinates life span and nutrient homeostasis”. Science. 2008;320:1012. doi: 10.1126/science.1152366. [DOI] [PubMed] [Google Scholar]
- Selman C, Partiridge L, Withers DJ. Replication of extended phenotype lifespan in mice with deletion of insulin receptor substrate 1. PLoS One. 2011;6:e16144. doi: 10.1371/journal.pone.0016144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Sun LY, Al-Regaiey K, Masternak MM, Wang J, Bartke A. Local expression of GH and IGF-1 in the hippocampus of GH-deficient long-lived mice. Neurobiol Aging. 2005;26:929–937. doi: 10.1016/j.neurobiolaging.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Tallaksen-Greene SJ, Crouse AB, Hunter JM, Detloff PJ, Albin RL. Neuronal intranuclear inclusions and neuropil aggregates in HdhCAG(150) knockin mice. Neuroscience. 2005;131:843–852. doi: 10.1016/j.neuroscience.2004.10.037. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3:a004440. doi: 10.1101/cshperspect.a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Science Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Tallaksen-Greene SJ, Wang B, Albin RL, Paulson HL. The de-ubiquitinating enzyme ataxin-3 does not modulate disease progression in a knock-in mouse model of Huntington disease. J Huntingtons Dis. 2013;2:201–215. doi: 10.3233/JHD-130058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Huntington's disease. Handb Exp Pharmacol. 2014;220:357–409. doi: 10.1007/978-3-642-45106-5_14. [DOI] [PubMed] [Google Scholar]