

Abstract
Activity-dependent shifts in ionic concentrations and water that accompany neuronal and glial activity can generate osmotic forces with biological consequences for brain physiology. Active regulation of osmotic gradients and cellular volume requires volume-sensitive ion channels. In the vertebrate retina, critical support to volume regulation is provided by Müller astroglia, but the identity of their osmosensor is unknown. Here, we identify TRPV4 channels as transducers of mouse Müller cell volume increases into physiological responses. Hypotonic stimuli induced sustained [Ca2+]i elevations that were inhibited by TRPV4 antagonists and absent in TRPV4−/− Müller cells. Glial TRPV4 signals were phospholipase A2- and cytochrome P450-dependent, characterized by slow-onset and Ca2+ waves, and, in excess, were sufficient to induce reactive gliosis. In contrast, neurons responded to TRPV4 agonists and swelling with fast, inactivating Ca2+ signals that were independent of phospholipase A2. Our results support a model whereby swelling and proinflammatory signals associated with arachidonic acid metabolites differentially gate TRPV4 in retinal neurons and glia, with potentially significant consequences for normal and pathological retinal function.
Keywords: retina, TRP channels, osmoregulation, Müller glia, ganglion cell
Introduction
The ability to sense increases in cell volume represents a primal sensory modality used by cells and organisms as they detect and adapt to activity-dependent changes in their physical environment. Within the CNS, swelling induces compensatory relocation of ions/water and long-term changes in enzyme activation/gene expression in neurons and glia but can also result in excitotoxicity, cytotoxic/vasogenic edema, and irreversible loss of neural function (Hoffmann et al., 2009; Pasantes-Morales and Cruz-Rangel, 2010). Astroglial swelling results in increased intracellular calcium concentration [Ca2+]i, phospholipase A2 (PLA2) activation, release of arachidonic acid (all-cis-5,8,11,14-eicosatetraenoic acid; AA), and production of oxygen-derived free radicals (Staub et al., 1994; Hoffmann et al., 2009; Thrane et al., 2011); however, the detailed molecular mechanisms that transduce cellular swelling within the brain into the physiological response remain to be elucidated.
Müller cells mediate bidirectional ion and water transport between retinal neurons and vascular endothelial cells in part through strategically placed ion and water channels. Under pathological conditions, aquaporin 4 (AQP4)-mediated water fluxes drive glial swelling and AA/eicosanoid release and may contribute to excitotoxic edema and ischemic damage (Da and Verkman, 2004; Verkman et al., 2008; Reichenbach and Bringmann, 2010). Here, we provide evidence that the transient receptor potential vanilloid type 4 (TRPV4) channel mediates Müller cell osmosensing, define its link to PLA2 and its downstream metabolite 5′6′-epoxyeicosatrienoic acid (5,6-EET), and formulate a mechanistic framework for astroglial volume regulation.
Gain/loss of TRPV4 function has been related to deficient osmoregulation, force transduction, and numerous neurological and musculoskeletal phenotypes (Liedtke and Friedman, 2003; Tian et al., 2009; Loukin et al., 2010; Nilius and Voets, 2013). Nonetheless, the gating mechanism of this ubiquitous channel (Strotmann et al., 2000; Nilius et al., 2004; Kunert-Keil et al., 2006; Ryskamp et al., 2011) remains unclear. The canonical view, that TRPV4 activation requires the involvement of PLA2 and cytochrome P450 (CYP450) (Watanabe et al., 2003a; Nilius et al., 2004; Vriens et al., 2004), has been challenged by studies in heterologously transfected yeast, oocytes, endothelial cells, and sensory neurons (Loukin et al., 2009, 2010; Matthews et al., 2010; Lechner et al., 2011). Here we show that activation of astroglial, but not neuronal, TRPV4 channels requires AA, its epoxidation enzyme CYP450, and the downstream metabolite product 5,6-EET. TRPV4-dependent Ca2+ influx enables further swelling in a hypoosmotic gradient and is sufficient to trigger glial reactivity. This work thus expands the known relationships among glial swelling, mechanical stress, proinflammatory signaling, and edema (Pannicke et al., 2006; Reichenbach and Bringmann, 2010; Pinar-Sueiro et al., 2011; Križaj et al., 2014) into a cellular and molecular framework that centers upon the osmosensor, TRPV4.
Materials and Methods
Animals.
All experiments adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committees at the University of Utah and the University of Nebraska Medical Center. Mouse strains C57BL/6J, B6.Cg-Tg(Thy1-CFP)23Jrs/J, and pan-null TRPV4−/− 12 (Liedtke and Friedman et al., 2003) were maintained in a 12 h light/dark cycle with free access to food and water. Data from male and female mice were pooled. No sex differences were noted.
Acutely dissociated retina preparation.
Mice were killed, eye removed, and retinas isolated in cold Leibovitz 15 (L15) medium (Invitrogen) containing 11 mg/ml L15 powder, 20 mm d-glucose, 10 mm Na-HEPES, 2 mm Na-pyruvate, 0.3 mm Na-ascorbate, and 1 mm glutathione. To digest the extracellular matrix, retinas were incubated in L15 containing papain (7 U/ml; Worthington) for 1 h at room temperature. Retinas were rinsed, placed on ice, and cut into 500 μm pieces. One or two of these pieces were triturated and plated on concanavalin A (1 mg/ml)-coated coverslips. As appropriate, dissociated cells were loaded with fura-2 or fura-5F AM (5–10 μm; Invitrogen) for 30–40 min and washed for 10–20 min. Under our experimental conditions, most plated cells maintained homeostasis for several hours at 25°C without substantial shifts in baseline [Ca2+]i or the amplitude of [Ca2+]i responses to agonists or depolarization (Szikra et al., 2009; Ryskamp et al., 2011; Molnar et al., 2012). Müller glia were identified by their distinctive morphology (Dyer and Cepko, 2000; Gaiano et al., 2000) and retinal ganglion cells (RGCs) were identified as in Ryskamp et al. (2011) (e.g., by Thy1:CFP expression). In a subset of experiments, cellular identity was additionally confirmed by postimaging immunocytochemistry (mouse anti-glutamine synthetase [GS]; 1:1000; BD Biosciences) for Müller glia and rabbit anti-III β-tubulin (TuJ1; 1:1000; Sigma) for RGCs.
Retinal slice preparation.
Eye cups were loaded for 70 min with Oregon Green BAPTA-1 AM (Invitrogen; 200 μm in L15 with 7 mg/ml pluronic acid) to load the dye in Müller glia. The isolated retina was flat-mounted (RGC side down) on filter paper using vacuum suction. Retinas were sliced at 200 μm and rotated 90 degrees before mounting by lines of vacuum grease on a BD Cell-Tak-treated coverslip (BD Biosciences).
Superfusion of retinal tissue and cell swelling assays.
During imaging experiments, Ringer's solution was perfused at a rate of 1–2.5 ml/min. It contained in mm the following: 133 NaCl, 2.5 KCl, 1.5 NaH2PO4, 1.5 MgCl2 (6H2O), 2 CaCl2, 10 glucose, 10 HEPES hemisodium salt, 1 pyruvic acid, 1 lactic acid, 0.5 l-glutamine, 0.5 glutathione, and 0.3 Na-ascorbate. pH was set at 7.4 and osmolarity at 300 mOsm. Extracellular osmolarity was set by addition or removal of mannitol, a procedure that minimally disrupts ionic strength of the extracellular solution but also reduces the secondary effects of regulatory volume decrease (RVD) in Müller glia (Fernández et al., 2013). Following hypotonic stimulation (HTS), the x-y cross-sectional area of calcein-loaded cells was determined offline using NIS-Elements AR 3.2 (Ryskamp et al., 2011). To examine the geometric properties of swelling more closely, dissociated retinal cells were labeled with 100 μm sulforhodamine 101. Cell volume is proportional to √ area3 when swelling occurs uniformly in all directions, which we confirmed with confocal z-stacks over time (data not shown). Confocal imaging with a Zeiss LSM 510 was used to acquire z-stacks before and during swelling. 3D volume was analyzed in ImageJ.
Optical imaging.
Ratiometric Ca2+ fluorescence imaging was performed on an inverted Nikon Ti or an upright Nikon E600 FN microscope using 20× (0.75 NA oil), 40× (1.3 NA oil and 0.8 NA water), and 60× (1.0 NA water) objectives. Excitation was provided by the Lambda DG-4 illumination system (Sutter Instruments). Images were captured with a 14-bit CoolSNAP HQ2 camera and processed using NIS-Elements AR 3.2 and Excel. [Ca2+]i was measured as described previously (Ryskamp et al., 2011; Molnar et al., 2012), accounting for the Ca2+ dissociation constant of fura-2 and fura-5F at room temperature. We followed the conventional Tsien protocol to determine the apparent free [Ca2+]i using the equation [Ca2+]i = Kd × β × ((R − Rmin)/(Rmax − R)), where R is the ratio of emission intensity at 510 nm evoked by 340 nm excitation versus emission intensity at 510 nm evoked by 380 nm excitation; Rmin is the ratio at zero free Ca2+; Rmax is the ratio at saturating Ca2+; and β is F380max/F380min; the dissociation constant (Kd) of fura-2 for Ca2+ at room temperature was taken to be 224 nm (Grynkiewicz et al., 1985), whereas the Kd for fura-5F was taken to be 2.29-fold higher (e.g., Invitrogen Handbook). After permeabilization with 10 μm ionomycin, Rmin was established in 0 Ca2+/0.8 mm EGTA saline and Rmax after exposure to ionomycin in 10 mm Ca2+ saline. All calibrated values for Müller glia were obtained from experiments using the lower-affinity indicator fura-5F; nonetheless, calcium signals ∼>2 μm enter the nonlinear range and must be viewed as approximating the actual concentrations. If exposure to calibrating solutions induced dye leakage or cells died during the calibration procedure, ΔR/R (peak ratio − baseline/baseline) was used. Glutamate (100 μm) was perfused to check for neuronal health and responsiveness. Results represent averages of RGC or Müller cell responses from a minimum of three animals.
Electrophysiology.
Retina pieces were incubated in Hibernate A medium with papain (30 U/ml) plus cysteine (0.2 mg/ml) for 25–30 min at room temperature. Tissue was washed in ice-cold, Hibernate A supplemented with 1% BSA and DNase (1 mg/ml) followed by two additional washes in ice-cold Hibernate A alone. Retinal cells were dissociated and plated as before. After 10–15 min of plating, cells were superfused with oxygenated Ames' medium. Upon obtaining a whole-cell recording, the superfusate was switched to Ames' medium containing 5 mm CsCl and 10 mm TEA (minimizes inward and outward K+ currents).
Recording electrodes were pulled using a PP-830 vertical puller (Narishige) from borosilicate glass pipettes (1.2 mm O.D., 0.9 mm I.D.) to obtain tips that were 2 μm in diameter and with resistance values between 8 and 10 mΩ. Müller cells were voltage-clamped at −70 mV using a Multiclamp 700A patch-clamp amplifier. Voltage steps and ramps were applied and membrane currents acquired using pClamp 9.2 software with a Digidata 1322 interface. The pipette solution contained the following (in mm): 125 Cs gluconate, 10 TEACl, 10 HEPES, 3 EGTA, 1 ATP, 0.5 GTP, 3 MgCl2, 1 CaCl2, pH 7.2. Simultaneous ratiometric intracellular Ca2+ measurements were made as before with 0.2 mm fura-2 pentapotassium salt in the pipette.
Immunofluorescence.
Eyes were removed after death, punctured with a needle at the ora serrata, and placed in 4% PFA in 1× PBS for 10 min. In PBS, the anterior eye was cut away and the posterior eye was fixed for another 50 min. After 3 × 10 min washes with PBS, eyecups were soaked in 15% sucrose for 45 min at room temperature and then 30% sucrose overnight at 4°C. Cryoprotected eyecups were embedded in OCT (Ted Pella), frozen at −80°C, sliced at 16 μm with a cryostat, and mounted onto Superfrost Plus slides. Slides were warmed at 40°C for 10 min and circled with a PAP pen. After washing with PBS, tissue was blocked for 30 min (10 ml PBS, 30 μl of Triton X-100, 100 mg BSA, 200 μl 5% w/v Na azide solution). Primary antibodies in PBS were incubated with the tissue overnight at 4°C. Retinas were rinsed with PBS, and secondary antibodies in PBS were applied at 1:1000 for 1 h at room temperature. After rinsing, labeled slices were protected with Fluoromount-G (Southern Biotechnology), coverslipped, and imaged. Dissociated cells were fixed and immunostained as above. The following primary antibodies were used in this study: anti-TRPV4 (Lifespan Biosciences), 1:100–1:1000; anti-GS (BD Biosciences), 1:1000–1:2000; anti-GFAP (Dako), 1:500; and anti-AGB (Signature Immunologics), 1:100. The secondary antibodies were goat anti-mouse or goat anti-rabbit IgG (H+L) conjugated to fluorophores (Alexa-488 and Alexa-594; Invitrogen). Negative controls without a primary antibody showed no staining. For the AGB loading experiment, retinas were incubated in 5 mm AGB (agmatine) for 10 min at 37°C, fixed, and cryoprotected. Immunofluorescence and differential interference contrast images were acquired on a confocal microscope (Zeiss LSM 510 or Olympus FX1000) using 488 nm Ar (10%) and 543 nm He/Ne (100%) lines for fluorophore excitation, suitable filters for emission detection, and 40×/1.2 NA oil objectives.
Reagents.
Salts and reagents were purchased from Sigma, except where noted otherwise. AA and its metabolites were from Cayman Pharmaceuticals. Given the instability of AA and 5,6-EET due to auto-hydrolyzation, the compounds were aliquoted, gassed with liquid nitrogen, and stored at −80°C until use.
Statistics.
GraphPad Prism 6.0 was used to analyze statistics. Data are mean ± SEM. Unless specified, an unpaired t test was used to compare two means and a one-way or two-way ANOVA along with the Holm-Šídák test was used to compare three or more means.
Results
TRPV4 is functional in RGCs and Müller glia
To map the pattern of functional TRPV4 expression in the retina, we incubated intact mouse retinas with AGB+, which permeates most nonselective cation channels (Marc, 1999). Control light-adapted retinas probed with an anti-AGB antibody displayed little endogenous signal (Fig. 1A). AGB incubation (10 min at 37°C) revealed cation accumulation in RGCs and photoreceptors (Fig. 1B,E). Stimulation with the selective TRPV4 agonist GSK1016790A (hereafter GSK101; 100 nm) increased AGB immunoreactivity (ir) in large somata within the ganglion cell layer (GCL) (Fig. 1C, arrowheads) and in radial processes of Müller glia (Fig. 1C,F) marked by GS staining. Consistent with preferential Müller glia and RGC cation loading, GSK101 increased AGB-ir by 14.2 ± 5.3-fold between the outer edge of the GCL and the outer limiting membrane together with a 4.1 ± 1.3-fold increase in the GCL (p < 0.05 for both; Fig. 1G,H). Demonstrating agonist specificity, AGB-ir was blocked by coincubation with the selective TRPV4 antagonist HC-067047 (hereafter HC-06; 1 μm; Fig. 1D,G–H). Thus, RGCs and Müller glia represent predominant transducers of TRPV4 signals in the mouse retina.
Figure 1.

TRPV4 mediates cation influx in Müller glia and RGCs. A–D, Vertical cryosections of mouse retinas immunolabeled for Müller glia marker GS (red) and AGB (green). Freshly isolated retinas were incubated for 10 min at 37°C with the indicated conditions. A, Negative control (L15 media alone; n = 2). B, Retinas incubated with AGB (N = 3). Basal cation (AGB+) influx takes place in RGCs and photoreceptors. C, GSK101 (100 nm) induces cation influx in RGCs (arrowheads) and a subset of Müller glia (n = 3). D, Agonist-induced cation entry is suppressed by HC-06 (1 μm; n = 3). E, F, Additional examples of cation influx in the absence (E) and presence (F) of GSK101. G, H, AGB influx between the outer edge of the GCL and the outer limiting membrane (OLM) (G) or within the GCL (H) was quantified by measuring the mean value (optical density) of AGB-ir. Ii–Iiii, Vertical cryosection of mouse retina immunolabeled for the Müller cell marker GS (red) and TRPV4 (green) (Ii). TRPV4 is preferentially localized to the inner limiting membrane region that contains the processes of protoplasmic astrocytes, Müller cell endfeet (arrows), and RGCs (blue asterisk). Müller cell somata (white asterisk) lack TRPV4 in intact retinas. An example distal stalk/distal end is indicated with a bracket. Iii, Iiii, Close-up of Ii (dashed rectangle) showing TRPV4 (Iii) and the merge (Iiii). J, TRPV4-ir is present throughout acutely dissociated Müller cells and a presumed RGC soma. Proximal stalk indicated by arrowhead. K, In a human retina section, TRPV4 similarly localizes to endfeet and proximal stalks of Müller glia as well as RGC somata and axon bundles (#). Scale bars, 10 μm. I, Inner; O, outer; S, segments; N, nuclear; P, plexiform; L, layer; GC, ganglion cell. *p < 0.05.
A TRPV4 antibody prominently labeled RGCs and Müller cell processes (GS-ir) (Fig. 1I). The specificity of the staining was confirmed by omitting the primary antibody and in sections from TRPV4−/− retinas (Ryskamp et al., 2011). TRPV4-ir was strongest in the perivascular endfoot region (Fig. 1I), suggesting a role for the channel in negotiating ion/water fluxes between the inner retina and blood vessels. Localization to glial endfeet was similarly observed in the human retina (Fig. 1K).
TRPV4 agonists induce sustained elevations in Müller cell [Ca2+]i
We next investigated the properties of TRPV4 signals in Müller glia. Acutely dissociated mouse cells had resting levels of 225 ± 31 nm, similar to previously recorded [Ca2+]MC levels in salamander Müller cells (Keirstead and Miller, 1995). GSK101 elicited robust, reversible, and dose-dependent increases in intracellular Ca2+ ([Ca2+]MC; Fig. 2A–C) with an EC50 of ∼16 nm (Fig. 2D). Hence, 25 nm GSK101 elevated [Ca2+]MC to 2547 ± 269 nm (p < 0.0001; Tukey test; Fig. 2B). In our hands, the amplitudes of TRPV4-mediated [Ca2+]MC signals markedly exceeded calcium signals evoked by other known [Ca2+]MC modulators, including depolarization (32.5 mm KCl), release from internal stores (20 μm carbachol), and saturating activation of purinergic (100 μm ATP) and glutamate receptors (100 μm glutamate; p < 0.0001, Tukey test; Fig. 2C).
Figure 2.

TRPV4 activation massively elevates Müller cell calcium levels. A, GSK101 dose-dependently increases calcium levels above spontaneous calcium spikes. B, The amplitude of Müller cell responses to GSK101 was quantified using fura-5F. n = 14, 12, and 4, respectively. C, 25 nm GSK101 increased calcium levels far more than other known modulators of Müller cell calcium homeostasis. n = 33, 26, 25, 19, and 19, respectively. D, According to the sigmoidal fit, the EC50 of GSK101 was ∼16 nm. n = 12. E, GSK101 induced a current with an IV plot characteristic of heteromeric TRPV4. Light gray trace represents voltage ramp-induced transmembrane current under resting conditions. Gray trace represents GSK101-induced current. Black trace represents difference current after subtraction. n = 10. F, TRPV4 opening by GSK101 resulted in a large inward conductance that coincided with an elevation in calcium. NS, Not significant (p > 0.05). ****p < 0.0001.
To examine the mechanism underlying the GSK101 response, we recorded the transmembrane current in voltage-clamped Müller cells. GSK101 (25 nm) induced an increase in conductance (p < 0.0212) averaging 1378 ± 522 pS (n = 9/10 cells). Consistent with imaging data, ITRPV4 showed little inactivation during 3–10 min stimulation with the agonist, showing a current–voltage relation typical of nonselective cation channels with a reversal at −4.5 ± 5.5 mV (Fig. 2E,F). This observation was reinforced by concurrent [Ca2+]i and whole-cell recordings that indicated substantial overlap between [Ca2+]i and ITRPV4 (Fig. 2F). GSK101-induced [Ca2+]MC increases were abolished in Ca2+-free saline (p < 0.0001, Fig. 3A,B), insensitive to the voltage-operated Ca2+ channel blocker Cd2+ (100 μm; p < 0.01; Fig. 3C,D) and inhibited by the nonselective TRP channel blocker Ruthenium Red (10 μm; p < 0.0001; Fig. 3E,F) and the selective TRPV4 antagonist HC-06 (p < 0.0001, Tukey test; Fig. 3G,H). The TRPV1 antagonist capsazepine (10 μm) had no effect on GSK101-induced [Ca2+]i elevations in Müller cells (data not shown).
Figure 3.
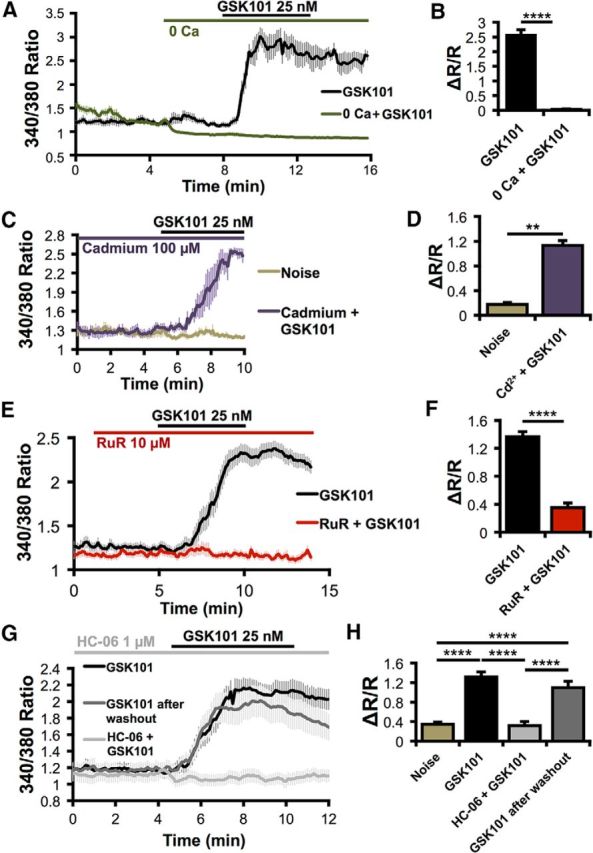
TRPV4 mediates responses to GSK101 in Müller cells. A, B, External calcium is required for GSK101-induced calcium elevations. n = 16. C, D, Responses to GSK101 persist in the presence of the voltage-gated channel blocker cadmium (Cd2+). n = 4. E, F, The nonselective TRP channel antagonist Ruthenium Red (RuR) blocks responses to GSK101. n = 21 for GSK101; n = 15 for GSK101 + RuR. G, H, GSK101 responses are also blocked by the selective TRPV4 antagonist HC-06. This inhibition is reversible following a washout. n = 30, except noise = 40. **p < 0.01. ****p < 0.0001.
The kinetics of TRPV4 activation differ in neurons and glia
In contrast to the inactivation of TRPV4 agonist-induced [Ca2+]i responses in RGCs, signals in Müller cells were markedly more sustained (Fig. 4). Concurrent recording from two cells identified post hoc by their immunoreactivity for GS and the RGC marker TuJ1 showed that GSK101 evokes a fast inactivating [Ca2+]i increase in the neuron and a delayed response in the glial cell (Fig. 4A). The dichotomy in the neuronal versus glial response kinetics stands out when TRPV4-mediated signals are normalized for peak [Ca2+]i (Fig. 4B) or when raw traces are statistically analyzed (Fig. 4C–F). GSK101 elevated [Ca2+]i to a peak 215 ± 15% and 217 ± 16% greater than the baseline in Müller glia and RGCs, respectively (Fig. 4C). Four minutes after the peak, these [Ca2+]i levels declined to 179 ± 11% and 116 ± 5% of the baseline during continued agonist stimulation (p < 0.05 for Müller glia and p < 0.0001 for RGCs; two-way repeated-measures ANOVA). Müller cells exhibited a slower response onset (95.3 ± 18.4 s vs 26.9 ± 3.6 s; p < 0.001, two-way ANOVA; Fig. 4D) and time-to-peak [Ca2+]i elevations (236.9 ± 17.7 s vs 93.0 ± 4.4 s; p < 0.0001, two-way ANOVA). The slope of the GSK101 response diverged for Müller glia (0.0132 ± 0.0027 ratio/s) and RGCs (0.0319 ± 0.0052 ratio/s; p < 0.01; Fig. 4E). The time constant of inactivation following the peak was larger in Müller glia than RGCs (τ = 346.2 ± 31.6 s vs 123.0 ± 12.3 s; p < 0.0001; Fig. 4F). Thus, glial TRPV4 is characterized by distinct modulation and/or gating compared with its neuronal counterpart. Nonetheless, exposure to Ca2+-free saline during initial response (Ryskamp et al., 2011; Fig. 3A) or the postpeak plateau phase of the agonist response (Fig. 4A) facilitated the recovery to baseline, indicating that the transduction and sustained components in both cell types are mediated primarily by plasma membrane Ca2+ entry rather than secondary release from internal stores.
Figure 4.

TRPV4 response kinetics differ in RGCs and Müller glia. A, Representative raw traces of calibrated GSK101-induced responses in a concurrently recorded RGC and Muller cell show a short-latency transient [Ca2+]i elevation in the RGC and a sustained [Ca2+]i response in the glial cell. Ca2+-free saline suppressed the sustained response component and decreased the [Ca2+]i baseline in the RGC. After the recording, the two cells were immunolabeled for TuJ1 and GS, respectively. B, Normalized fura-5F fluorescence from a GSK101-stimulated cohort of RGCs and glia (n = 25 Müller glia and 51 RGCs). Responses to GSK101 are fast and transient, whereas Müller cells exhibit delayed and sustained TRPV4 activation. C, GSK101 significantly increased calcium levels in both cell types; however, RGC calcium levels returned closer to baseline levels 4 min after the response peak in the continued presence of GSK101. D, The latency to the base (n = 17 Müller glia and 19 RGCs), 50% amplitude, and peak response (n = 21 Müller glia and 24 RGCs) was longer in Müller cells. E, F, The rate of TRPV4 activation (E; n = 20 Müller glia and 19 RGCs) and inactivation (F; n = 20 for each) was faster in RGCs. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Spatiotemporal TRPV4 activation in Müller cells involves transcellular Ca2+ waves
Müller glial Ca2+ signals evoked by agonists or membrane stretch typically took the form of Ca2+ waves propagating from the endfoot or the distal end toward the perikaryon (Fig. 5A–H). Ca2+ store depletion by cyclopiazonic acid (CPA), a reversible antagonist of sarco/endoplasmic Ca2+-ATPases, combined with stimulation of Ca2+ release by carbachol, reduced the amplitude of the GSK101 response by 34.5 ± 11.6% (p < 0.05; Fig. 5I). CPA also suppressed Ca2+ wave propagation (Fig. 5K–Q), suggesting that Ca2+-induced Ca2+ release amplifies the TRPV4 response.
Figure 5.
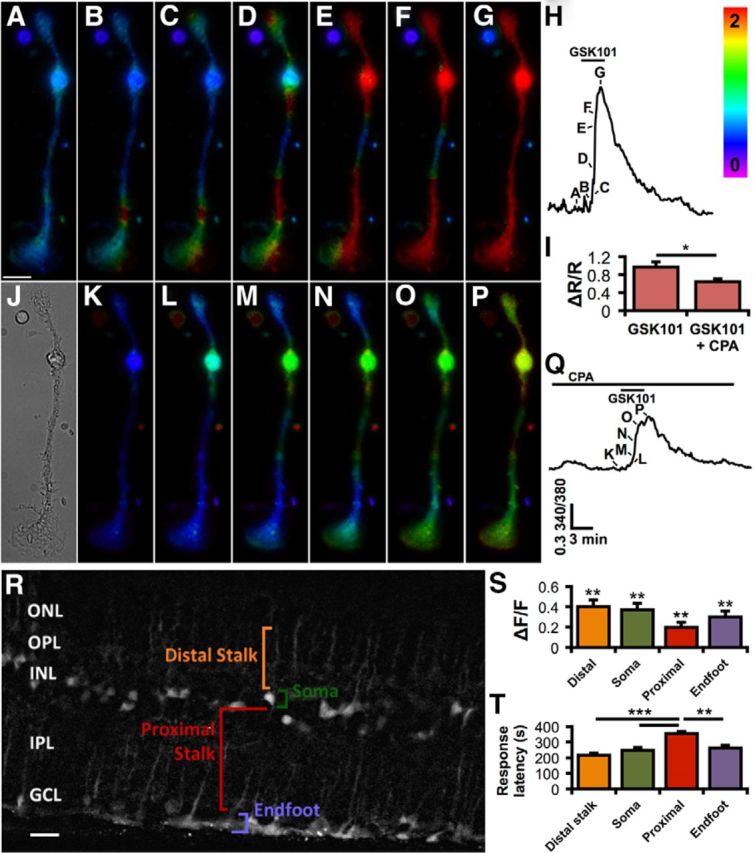
TRPV4 activation triggers Ca2+-induced Ca2+ release and calcium waves. A–H, Upon applying 25 nm GSK101 (B–G), calcium typically first flows into Müller glial endfeet (bottom) or their apical process (top). Calcium then elevates as a wave, filling the cytosol globally. Scale bar, 10 μm. The timing of the images (A–G) is indicated in the corresponding calcium trace (H). The rainbow scale specifies the 340/380 ratio level. I, TRPV4 stimulation by GSK101 further elevates Ca2+ via Ca2+ release (stores depleted by bathing cells with 5 μm CPA while stimulating release with applications of 20 μm carbachol). n = 19. J, A bright-field image of the Müller cell. K–P, Store depletion by CPA attenuates the response to GSK101 (L–P) and the resulting wave. The corresponding trace (Q) indicates timing of images (K, L). R, OBG-1 selectively loaded Müller glia in a retina slice. Four of the main Müller cell compartments are labeled for one cell. This image is the median composite of 10 temporally adjacent images separated by 3 s in time during the response to GSK101. The contrast between the Müller cells and the background was enhanced with the application of the rolling ball background subtraction function in ImageJ (10.0 pixel radius). ONL, Outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar, 10 μm. S, Responses to 10 μm GSK were significantly above the baseline for all cellular compartments. T, The distal stalks and endfeet were the primary transduction sites, as indicated by shorter response latencies (time to ½ peak). n = 13 for distal stalk, 23 for somata, 16 for proximal stalk, and 13 for endfeet. *p < 0.05. **p < 0.01. ***p < 0.001.
To follow glial TRPV4 activation in the retinal slice (Fig. 5R), glia were loaded with Oregon Green BAPTA-1 (OGB-1) (Kurth-Nelson et al., 2009). GSK101 induced Ca2+ waves that originated within focal points in the distal end of Müller glia and/or the endfoot. The agonist elevated [Ca2+]i in 12 of 14 (86%) distal stalks, 26 of 30 (87%) somata, 21 of 26 (81%) proximal stalks, and 20 of 28 (71%) endfeet. Peak response amplitudes for these regions in ΔF/F were as follows: distal stalk 0.4005 ± 0.0662, soma 0.3707 ± 0.0643, proximal stalk 0.1980 ± 0.0551, and endfoot 0.2988 ± 0.0642 (Fig. 5S). Somatic Ca2+ signals remained elevated after [Ca2+]i levels within apical and distal regions returned to the baseline. The response amplitudes for all Müller cell domains were greater than Ro (p < 0.01 in all cases; Dunnett's test). Response latencies were (in seconds): distal stalk 214.8 ± 15.2, soma 246.2 ± 18.1, proximal stalk 353.0 ± 14.3, and endfoot 262.6 ± 14.9 (p < 0.001–0.0001; Bonferroni test; Fig. 5T). Thus, TRPV4 channels can initiate and contribute to propagation of transretinal Ca2+ waves, representing a plausible candidate trigger mechanism for the regenerative phenomena reported previously (Newman and Zahs, 1998).
Differential TRPV4 channel activation mediates neuronal and glial responses to swelling
Astroglial swelling can compromise neuronal function through edema and excitotoxicity (Pasantes-Morales and Cruz-Rangel, 2010; Reichenbach and Bringmann, 2010; Thrane et al., 2011). Given that TRPV4 is activated by swelling (Strotmann et al., 2000; Becker et al., 2005; Benfenati et al., 2011), we examined the relationship between hypotonicity and volume regulation in Müller cells. HTS dose-dependently and reversibly increased the cell area and volume of Müller glia and RGCs (Fig. 6). Although both cell types express TRPV4, Müller cells were able to withstand larger hypotonic challenges than concomitantly recorded RGCs (Fig. 6B), possibly because of the greater elasticity of the glial membrane (Lu et al., 2006). As depicted in Figure 6, Müller cell swelling was associated with increases in [Ca2+]i with an EC50 of 34.15 ± 3.31% HTS (Fig. 6C,D). The [Ca2+]i response in Müller cells and RGCs was abolished by the removal of extracellular Ca2+ (Fig. 6E; Ryskamp et al., 2011) and was inhibited by HC-06 (1 μm) (p < 0.05; Fig. 6F,G). Consistent with optical imaging data, 50% HTS elicited inward currents (91.34 ± 17.66 pA) that were antagonized by HC-06 and were absent from TRPV4−/− cells (Fig. 6H,I). Thus, TRPV4 channels play a central role in the hypotonicity-induced Ca2+ signals of Müller glia.
Figure 6.

Differential TRPV4 channel activation mediates neuronal and glial responses to swelling. A, Changes in the volume of calcein-loaded Müller cells were apparent when switching from isotonic (Ai) to hypotonic saline (Aii). Aiii, The volume changes were approximated by measuring cell area. Black represents isotonicity; gray represents hypotonicity. Scale bar, 10 μm. B, Müller glia and RGCs swelled and shrunk as a function of tonicity. C, HTS elevated Müller cell calcium as they swelled in a representative experiment. D, Swelling-evoked calcium elevations were dose-dependent (95% confidence band around the sigmoidal fit). n = 13–20. E, Swelling-evoked [Ca2+]MC elevations required external calcium. F, PLA2 and TRPV4 contribute substantially to the hypoosmotic response of Müller glia. n = 10, 13, and 11, respectively. G, The PLA2 antagonist pBPB has no effect on RGC responses to cell swelling, even though these responses are mediated largely by TRPV4. n = 23, 16, and 9, respectively. H, I, The 50% HTS evoked inward currents in Müller glia (n = 7) that were absent in the presence of HC-06 (n = 8) or in TRPV4−/− cells (n = 7). J, Müller glia responsiveness to 35% HTS (n = 10 experiments) is reduced by the TRP channel antagonist Ruthenium Red (RuR 10 μm; n = 5) or 1 μm HC-06 (n = 6), but not the TRPV1 antagonist capsazepine (CPZ 10 μm; n = 3). K, RGC responsiveness to HTS (n = 6) is impaired by Ruthenium Red (n = 6) and HC-06 (n = 6), but not TRPV1 inhibition (n = 3). NS, Not significant (p > 0.05). *p < 0.05. **p < 0.01. ***p < 0.001.
TRPV4 activation might be secondary to stretch-induced stimulation of PLA2 (Watanabe et al., 2003a). Consistent with this, PLA2 inhibition with 4-bromophenacyl bromide (pBPB; 100 μm) blocked HTS-induced glial TRPV4 signals (p < 0.001; Fig. 6F). pBPB (n = 39) did not affect GSK101 responses (n = 23) (p > 0.05), suggesting that cell swelling and GSK101 activate Müller TRPV4 through different mechanisms. Surprisingly, swelling-induced [Ca2+]i increases in RGCs were unaffected by pBPB (p > 0.05; Fig. 6G). Thus, the Müller glial TRPV4 response to swelling requires the “canonical” PLA2 signaling pathway, whereas in neurons TRPV4 may either be directly force sensitive (Loukin et al., 2010) or reliant on a novel, indirect force transduction cascade.
Ruthenium Red reduced the percentage of Müller glia responding to 35% HTS with [Ca2+]i elevations from 59 ± 7% to 17 ± 5% (p < 0.01, Dunnett's test; Fig. 6J). HC-06 also reduced the percentage of HTS-responding Müller glia to 29 ± 8% (p < 0.05), whereas capsazepine, a competitive inhibitor of TRPV1 channels, had no effect at 5 μm (p > 0.05). Likewise, HTS-evoked [Ca2+]i responses of RGCs were diminished by Ruthenium Red and HC-06, but not capsazepine (Dunnett's test; Fig. 6K). The comparable efficacy of Ruthenium Red and HC-06 suggests that TRPV4 is the primary TRP channel contributing to volume sensing in Müller glia and RGCs. As neuronal and glial responses to HTS and GSK101 were almost completely absent in TRPV4−/− cells (Fig. 7A–D), the residual responsiveness in the presence of TRPV4 blockers could be ascribed to incomplete blockade by HC-06.
Figure 7.

TRPV4 mediates RGC and Müller cell responses to HTS and GSK101. A, The percentage of RGCs responding to 50% HTS and 25 nm GSK101 is reduced by TRPV4 ablation (n = 4–9 experiments, two-way ANOVA). B, Calcium elevations induced by HTS and GSK are smaller in TRPV1−/− RGCs (N = 12–74 cells, two-way ANOVA). C, The percentage of Müller cells responding to 50% HTS and 25 nm GSK101 is reduced by TRPV4 ablation (n = 3–8 experiments, two-way ANOVA). D, Calcium elevations induced by HTS and GSK are smaller in TRPV1−/− Müller glia (n = 14–66 cells, two-way ANOVA). *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
TRPV4 gating in Müller cells, but not RGCs, requires activation of PLA2
The prevailing model of TRPV4 gating involves PLA2 activation and biosynthesis of epoxyeicosatrienoic acids, which act as endogenous activators of TRPV4 (Watanabe et al., 2003a; Nilius et al., 2004; Jang et al., 2012) (Fig. 8A). To test the function of this canonical transduction pathway, we stimulated retinal cells with AA and its downstream metabolite 5,6-EET and tested the role of the crucial intermediary enzyme, CYP450. AA (100 μm) elevated Müller glial [Ca2+]i (ΔR/R = 0.39 ± 0.3; Fig. 8B,C) above spontaneous activity (noise; ΔR/R = 0.16 ± 0.03; p < 0.0001). Both the CYP450 antagonist clotrimazole (CLT; 10 μm) and HC-06 strongly suppressed these responses (p < 0.001; Fig. 8B–E), suggesting that AA-induced [Ca2+]MC responses are mediated by TRPV4 and the PLA2 signaling pathway. Although AA (10 μm) also induced [Ca2+]i elevations in RGCs (Fig. 8F,G), these responses were not antagonized by HC-06 (p > 0.05), despite the effective blockade of GSK101 responses (p < 0.05). 5,6-EET, a major astroglial epoxide metabolite of AA, was proposed as the final activator of TRPV4 (Nilius et al., 2004). 5,6-EET (5 μm) induced [Ca2+]i increases (ΔR/R = 0.83 ± 0.2 in Müller glia and 0.47 ± 0.08 in RGCs) that were inhibited by HC-06 (ΔR/R = 0.40 ± 0.07 in Müller glia and 0.28 ± 0.03 in RGCs; p < 0.05; Fig. 8H–J). We therefore conclude that glial but not neuronal TRPV4 activation in the retina involves an intermediary PLA2-CYP450 step.
Figure 8.
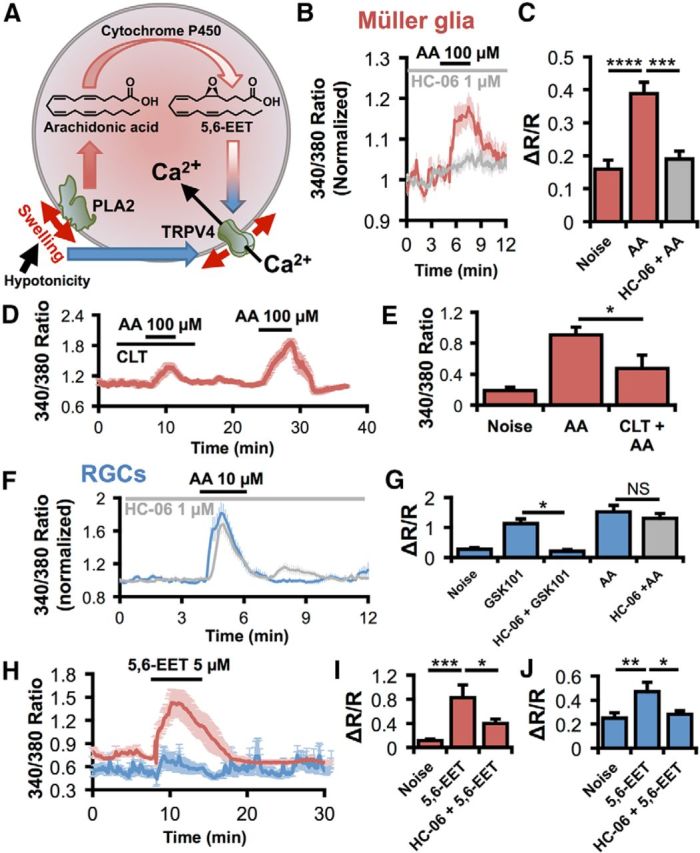
TRPV4 gating in Müller cells, but not RGCs, requires activation of PLA2. A, Working model for TRPV4 activation by cell swelling. In hypotonicity, both RGCs and Müller glia swell. This stretches the membrane and activates PLA2 at least in Müller cells. PLA2 synthesizes AA, which is metabolized by CYP450 into 5,6-EET, an endogenous agonist for TRPV4. 5,6-EET can activate TRPV4 in both cell types, but its potency is significantly more pronounced in Müller glia. Although required in Müller cells, this indirect force transduction pathway does not contribute to swelling responses in RGCs. Thus, RGCs either use a novel force transduction pathway (solid blue arrow) or direct gating of TRPV4 by membrane stretch (red arrows). B, C, AA elevates [Ca2+]MC by activating TRPV4. n = 34, 19, and 15, respectively. D, E, The CYP450 antagonist clotrimazole suppresses AA-induced [Ca2+]MC elevations. F, G, Although HC-06 (n = 16) completely inhibits TRPV4 activation by GSK101 (n = 10) in RGCs, the TRPV4 antagonist has no effect on AA responses (n = 46). H, Average traces from a representative experiment show that the 5,6-EET response is larger in Müller cells (red) than RGCs (blue). I, J, 5,6-EET activates TRPV4 in both Müller glia (I; n = 14) and RGCs (J; n = 54). NS, Not significant (p > 0.05). *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Calcium influx via TRPV4 exacerbates swelling in Müller glia and RGCs
Astroglial swelling is a major problem in traumatic brain injury and retinal diseases, such as diabetic retinopathy and glaucoma (Staub et al., 1994; Pannicke et al., 2006; Sofroniew, 2009; Pasantes-Morales and Cruz-Rangel, 2010; Pinar-Sueiro et al., 2011). We exposed HTS-stimulated retinal cells to HC-06 and compared the swelling response in wild-type cells with TRPV4−/− cells. As shown in Figure 9A, HTS-induced increases in cross-sectional area were counteracted by HC-06 (reduced by 46.9 ± 9.4%; p < 0.01; two-way ANOVA; Dunnett's test) and markedly reduced in TRPV4−/− Müller cells (reduced by 47.3 ± 6.1%; p < 0.01), indicating that swelling is facilitated by TRPV4 activation itself. HC-06 did not cause a further reduction in HTS-stimulated swelling in TRPV4−/− Müller glia, consistent with the central role for TRPV4 in the swelling response. Moreover, HTS-induced swelling was more pronounced in heterologously expressing HEK293:TRPV4 overexpressors compared with control cells (p < 0.001 in 35% HTS at each time, two-way ANOVA; Fig. 9B). Although the mechanism through which TRPV4 channels contribute to the HTS-induced swelling in recombinant cells, neurons, and glia remains to be determined, the effect is likely to be mediated by Ca2+ influx, as also indicated by the suppression of the HTS-induced increase retinal cell area by BAPTA-AM (200 μm; reduced by 45.7 ± 7.2% for RGCs; p < 0.0001; reduced by 33.2 ± 6.3% for Müller cells; p < 0.05; Fig. 9A).
Figure 9.

TRPV4 contributes to HTS-induced swelling. A, The maximal extent of Müller cell (red) and RGC (blue) swelling in 35% HTS was reduced by chelating intracellular calcium with BAPTA-AM or by inhibiting TRPV4 and thereby limiting calcium entry. TRPV4-mediated swelling was PLA2-dependent in Müller glia, but not RGCs. Swelling was reduced in cells from TRPV4−/− mice. HC-06 and pBPB had no additional effect on the reduction of swelling. For Müller glia, n = 96, 14, 11, 8, 13, 18, and 14. For RGCs, n = 145, 31, 36, 24, 21, 9, and 22. B, 35% HTS increased the cross-sectional area of calcein-loaded, WT (n = 51) and TRPV4-overexpressing HEK293 cells (n = 64); however, the extent of swelling was increased by the activation of TRPV4. NS, Not significant (p > 0.05). *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Given that Müller glial swelling was proposed to involve PLA2 (Pannicke et al., 2006; Reichenbach and Bringmann, 2010), we exposed wild-type and TRPV4−/− Müller cells to hypotonic saline in the presence of pBPB to test the hypothesis that this proposed obligatory activator of TRPV4 (Watanabe et al., 2003a; Nilius et al., 2004) is involved in the swelling response. The PLA2 antagonist reduced cell swelling in wild-type Müller cells (reduced by 66.5 ± 12.2%; p < 0.001) but was not effective in TRPV4−/− Müller glia (Fig. 9A). Moreover, pBPB did not suppress HTS-induced swelling in RGCs (reduced by 2.9 ± 9.2%; p < 0.05). Thus, hypotonicity-induced increases in cell volume are likely augmented by reciprocal stimulation between TRPV4-mediated Ca2+ influx and PLA2 activation in retinal glial cells but not neurons.
We next tested whether CYP450, the downstream enzyme in the signaling cascade proposed to govern HTS-mediated TRPV4 activation (Watanabe et al., 2003b; Nilius et al., 2004), contributes to swelling-induced Ca2+ responses in TRPV4-expressing retinal neurons and glia. Cells were stimulated with 35% and/or 50% HTS in the presence of the CYP450 antagonist 17-octadecynoid acid (17-ODYA; 10 μm). Consistent with the involvement of CYP450 in the transduction of glial swelling, the antagonist suppressed Müller glial hypotonic swelling in 35% HTS (p < 0.01) but had little effect on RGCs (p > 0.05) (Fig. 10A,B). Similar results were observed using both HTS stimuli as well as the other antagonist, clotrimazole (data not shown). The effect of 17-ODYA on swelling was substantiated at the level of Ca2+ homeostasis where the drug antagonized 50% HTS-evoked [Ca2+]i increases in Müller cells (p < 0.0001; n = 5), but had no effect on RGCs (p > 0.05; n = 10) (Fig. 10A).
Figure 10.

CYP450 metabolism regulates swelling-mediated [Ca2+]i responses in Müller glia but not RGCs. A, 140 mOsm HTS induced [Ca2+]i elevations in both cell types; however, only HTS-evoked responses in Müller glia were suppressed by 17-ODYA. B, Normalized cell area measurements in 190 mOsm saline. The CYP450 antagonist suppresses HTS-induced swelling in Müller glia but not in RGCs. NS, Not significant (p > 0.05). **p < 0.01. ****p < 0.0001.
TRPV4 activation or deletion is sufficient to trigger reactive gliosis
Reactive astrogliosis is an early indicator of CNS stress induced by mechanical stress and neuroinflammation (Tezel et al., 2003; Inman and Horner, 2007; Sofroniew, 2009; Lindqvist et al., 2010; Reichenbach and Bringmann, 2010). In control retinas, the gliotic marker GFAP was confined to astrocytes within the inner limiting membrane, whereas TRPV4−/− retinas exhibited a moderate level of GFAP-ir (Fig. 11A,B). After intravitreal injection of GSK101, a marked upregulation in GFAP-ir was observed, especially at the endfeet and proximal processes within the inner plexiform layer (p < 0.01; two-way ANOVA, Dunnett's test; Fig. 11A,B).
Figure 11.

TRPV4 regulates GFAP expression in Müller glia. A, Two weeks after a single intravitreal injection of a vehicle or 2 μl of 75 μm GSK101, mouse eyes were harvested and processed for IHC. Only astrocytes on the vitreal surface expressed GFAP in control eyes, whereas Müller glia also expressed GFAP in the GSK101-injected eyes of WT mice. TRPV4−/− Müller glia were reactive in both conditions, but GSK101 did not further increase gliosis. B, The GFAP optical density was significantly greater in the GSK101-injected eyes. n = 3 for each. Scale bar, 20 μm. OLM, Outer limiting membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; ILM, inner limiting membrane; a.u., arbitrary units. *p < 0.05. **p < 0.01.
Discussion
This study provides new insights into retinal physiology by identifying the Müller glial osmosensor and demonstrating a mechanistic framework that governs the relationship between glial osmosensing, Ca2+ homeostasis, acid metabolism, and swelling. The differential modulation of neuronal and glial TRPV4 channels has broader implications for our understanding of volume (dys)regulation and inflammatory signaling in the healthy and diseased CNS.
At EC50, GSK101-evoked [Ca2+]i signals far surpassed Müller responses to other known effectors of Ca2+ signaling, including depolarization, purinergic signaling, and Ca2+-induced Ca2+ release. The close match between TRPV4-mediated currents and [Ca2+]i suggests that both the initial and the sustained phases of the TRPV4 response are mainly mediated by plasma membrane influx. The I-V relationship underlying glial ITRPV4 did not exhibit the outward rectification typical of homomeric TRPV4 (Watanabe et al., 2003b; Loukin et al., 2010) but rather resembled the stretch-sensitive voltage-independent cation current observed in cultured Müller cells (Puro, 1991). It is possible that the conductance is linearized by heteromerization of TRPV4 with auxiliary TRPC1 or TRPP2 subunits (Da Silva et al., 2008; Köttgen et al., 2008; Ma et al., 2011) and/or modulatory influence of AQPs (Becker et al., 2005; Benfenati et al., 2011). Release from internal stores provided a minor contribution to the overall [Ca2+]i; however, as in brain astrocytes (Butenko et al., 2012; Dunn et al., 2013) and possibly mechanically stimulated retinal glia (Newman and Zahs, 1998), it fostered the propagation of TRPV4-initiated Ca2+ waves from the endfoot toward the distal retina.
The mechanism of TRPV4 gating has been controversial. The obligatory role of PLA2 has been questioned because TRPV4 is stretch-activated in excised patches of membrane, which are devoid of eicosanoids (Loukin et al., 2010, 2011) and the channel is force-sensitive in yeast, which do not express PLA2 (Loukin et al., 2009). Nonetheless, our conclusion that TRPV4 function in Müller glia requires AA and/or its metabolite 5,6-EET, is based on the following observations: (1) AA and 5,6-EET-induced [Ca2+]MC elevations are sensitive to TRPV4 antagonists; (2) AA potentiates Müller cell swelling, whereas PLA2 antagonists suppress TRPV4-evoked and HTS-induced [Ca2+]MC elevations; (3) PLA2 antagonists did not further suppress HTS-evoked swelling in TRPV4−/− Müller cells; (4) The lengthy activation lag time in Müller cells is consistent with activation of intermediary steps; (5) HTS-evoked inward current and [Ca2+]i increases were not observed in TRPV4−/− Müller cells or in the presence of the selective TRPV4 antagonist HC-06; and (6) CYP450 blockers antagonized HTS-evoked signals in Müller glia but not RGCs. By linking the proinflammatory AA pathway to the osmosensitive TRPV4 mechanism, these findings provide a molecular context for the reported effects of AA on glial swelling in cellular and tissue models of ischemia, traumatic brain injury, and diabetic edema and its role in reactive gliosis (Pannicke et al., 2006; Sofroniew, 2009; Nedergaard et al., 2010; Pasantes-Morales and Cruz-Rangel, 2010; Reichenbach and Bringmann, 2010). The simplest model to account for our results is that cell swelling, generated by hypotonic shock, induces TRPV4-dependent influx of cations and water into RGCs and Müller glia. In the glia, this ionic flux is augmented by PLA2 activation and possibly by the activation of the AQP4-mediated cell swelling.
Data from TRPV4−/− and HC-06-exposed retinal cells and HEK293 overexpressors show that TRPV4 activation contributes to cell swelling. This is in contrast to observations in salivary gland cells and keratinocytes where TRPV4/Ca2+ contributed to RVD (Becker et al., 2005; Liu et al., 2006), and cultured astrocytes in which RVD required AQP4, whereas hypotonic sensing was mediated by TRPV4 (Benfenati et al., 2011). AQP4 channels are strongly expressed in Müller endfeet and proximal processes (Nagelhus et al., 1998) and could modulate TRPV4 activation and glial Ca2+ homeostasis by boosting the rate of cell swelling. Another potential consequence of swelling-induced Ca2+ signaling is glial activation, which has been previously correlated with increased mechanical stress, arachidonic acid release, and elevated intraocular pressure (Inman and Horner, 2007; Reichenbach and Bringmann, 2010; Huang et al., 2011). We found that exposure to the TRPV4 agonist was sufficient to induce reactive gliosis, an effect that could have resulted directly from Ca2+ influx or as a secondary response to excitotoxic ATP release mediated by TRPV4-evoked degeneration of RGCs (Ryskamp et al., 2011; Križaj et al., 2014). Interestingly, unstimulated TRPV4−/− retinas also displayed mild gliosis, suggesting that basal TRPV4 activity in healthy retinas restrains glial reactivity.
Excitation mapping and HTS confirmed previous evidence of functional TRPV4 expression in RGCs (Ryskamp et al., 2011); however, the fast, inactivating [Ca2+]i responses in RGCs were easily distinguishable from lagging, sustained glial signals. Given the relative insensitivity of [Ca2+]RGC to 5,6-EET and the inability of pBPB, 17-ODYA, and clotrimazole to block HTS-induced [Ca2+]i elevations in RGCs, we conclude that, as reported for gastrointestinal neurons (Lechner et al., 2011), retinal neuronal responses are largely independent of the canonical transduction mechanism. This further suggests that TRPV4 might differentially contribute to volume sensing and AA-mediated proinflammatory signaling in retinal neurons versus glia. The origin of AA-mediated [Ca2+]i increases in RGCs is unclear but could involve modulation of other TRP channels (TRPV1–3, TRPA1), arachidonate-gated Ca (ARC) channels composed of Orai1/3 subunits, and/or auxiliary effects mediated through epoxygenase, lipoxygenase, or cyclooxygenase pathways downstream from AA (Meves, 2008; Ryskamp et al., 2014).
In conclusion, we report that TRPV4 is expressed in every Müller cell and that its activation is essential for the normal glial [Ca2+] response to hypotonic stress, whereas both its absence and overactivation are associated with reactive gliosis. Given that Müller glia perform key housekeeping, osmoregulatory, and mechanosensory functions in the retina, our findings implicate TRPV4 in both normal visual function and injury responses caused by excessive swelling and/or release of proinflammatory metabolites. Although Müller cells are not classically excitable, Ca2+ waves induced by endfoot TRPV4 channels might translate the effects of mechanical stress into interglial, gliovascular, and neuroglial signals (Newman and Zahs, 1998; Kurth-Nelson et al., 2009; Butenko et al., 2012; Dunn et al., 2013). Our data also reinforce the importance of TRPV4 signaling in the homeostatic response to mechanical stress. Because cell swelling, synaptic reorganization, and dendritic remodeling represent early RGC responses to elevated intraocular pressure (Pinar-Sueiro et al., 2011; Della Santina et al., 2013; Feng et al., 2013), TRPV4 targeting might mitigate the neuronal pressure-induced phenotype while inhibiting the inflammatory response associated with concomitant glial activation (Inman and Horner, 2007; Pinar-Sueiro et al., 2011; Ryskamp et al., 2011; Della Santina et al., 2013). Parenthetically, ablation of the closely related vanilloid channel TRPV1 was reported to have a protective effect on RGC survival in a mouse model of glaucoma (Ward et al., 2014). Given that retinal astroglial TRPV1 responses are markedly more sustained compared with the typical Ca2+-dependent inactivation observed in RGCs and other neurons (Koplas et al., 1997; Ho et al., 2014; Jo et al., 2014), the differences in response kinetics might represent a more general feature of Ca2+ homeostasis in neurons versus glia. Together, these studies reinforce the notion that vanilloid TRP isoforms contribute to the panoply of cell type-specific responses to stimulus modalities in the vertebrate retina that include pressure, osmotic gradients, temperature, and mechanical injury. Our findings provide novel insights into differential neuronal and astroglial Ca2+ responses to swelling and inflammation together with a mechanistic context for TRPV4 signaling in the retina but also suggest that targeting TRPV4 channels might offer a rational new tool for mitigating swelling-mediated neuronal damage and gliosis in patients with traumatic ocular/brain injury, edema, ischemia, and glaucoma.
Footnotes
This work was supported by Howard Hughes Medical Institute Med into Grad program to D.A.R., National Institutes of Health Grant T32DC008553 to D.A.R., Grant EY010542 to W.B.T., and Grants EY022076 and P30EY014800 Vision Core to D.K., Department of Defense W81XWH-12-1-0244, Glaucoma Research Foundation, State of Utah Technology Commercialization & Innovation Program, and Research to Prevent Blindness (Moran Eye Institute, University of Nebraska Medical Center). We thank Dr. Greg Hageman for the fixed human retinal tissue, Dr. Wolfgang Liedtke for TRPV4−/− mice, Dr. Chris Reilly for TRPV4-overexpressing HEK293 cells, and Wei Xing and Brian Argyle for technical support.
The authors declare no competing financial interests.
References
- Becker D, Blase C, Bereiter-Hahn J, Jendrach M. TRPV4 exhibits a functional role in cell-volume regulation. J Cell Sci. 2005;118:2435–2440. doi: 10.1242/jcs.02372. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108:2563–2568. doi: 10.1073/pnas.1012867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenko O, Dzamba D, Benesova J, Honsa P, Benfenati V, Rusnakova V, Ferroni S, Anderova M. The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS One. 2012;7:e39959. doi: 10.1371/journal.pone.0039959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da T, Verkman AS. Aquaporin-4 gene disruption in mice protects against impaired retinal function and cell death after ischemia. Invest Ophthalmol Vis Sci. 2004;45:4477–4483. doi: 10.1167/iovs.04-0940. [DOI] [PubMed] [Google Scholar]
- Da Silva N, Herron CE, Stevens K, Jollimore CA, Barnes S, Kelly ME. Metabotropic receptor-activated calcium increases and store-operated calcium influx in mouse Müller cells. Invest Ophthalmol Vis Sci. 2008;49:3065–3073. doi: 10.1167/iovs.07-1118. [DOI] [PubMed] [Google Scholar]
- Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong RO. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci. 2013;33:17444–17457. doi: 10.1523/JNEUROSCI.5461-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. Control of Müller glial cell proliferation and activation following retinal injury. Nat Neurosci. 2000;3:873–880. doi: 10.1038/78774. [DOI] [PubMed] [Google Scholar]
- Feng L, Zhao Y, Yoshida M, Chen H, Yang JF, Kim TS, Cang J, Troy JB, Liu X. Sustained ocular hypertension induces dendritic degeneration of mouse retinal ganglion cells that depends on cell type and location. Invest Ophthalmol Vis Sci. 2013;54:1106–1117. doi: 10.1167/iovs.12-10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández JM, Di Giusto G, Kalstein M, Melamud L, Rivarola V, Ford P, Capurro C. Cell volume regulation in cultured human retinal Müller cells is associated with changes in transmembrane potential. PLoS One. 2013;8:e57268. doi: 10.1371/journal.pone.0057268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 2000;26:395–404. doi: 10.1016/S0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Ho KW, Lambert WS, Calkins DJ. Activation of the TRPV1 cation channel contributes to stress-induced astrocyte migration. Glia. 2014;62:1435–1451. doi: 10.1002/glia.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Huang W, Xing W, Ryskamp DA, Punzo C, Krizaj D. Localization and phenotype-specific expression of ryanodine calcium release channels in C57BL6 and DBA/2J mouse strains. Exp Eye Res. 2011;93:700–709. doi: 10.1016/j.exer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. 2007;55:942–953. doi: 10.1002/glia.20516. [DOI] [PubMed] [Google Scholar]
- Jang Y, Jung J, Kim H, Oh J, Jeon JH, Jung S, Kim KT, Cho H, Yang DJ, Kim SM, Kim IB, Song MR, Oh U. Axonal neuropathy-associated TRPV4 regulates neurotrophic factor-derived axonal growth. J Biol Chem. 2012;287:6014–6024. doi: 10.1074/jbc.M111.316315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo AO, Ryskamp DA, Redmon S, Krizaj D. Nonretrograde endocannabinoid signaling modulates retinal ganglon cell calcium homeostasis through the TRPV1 cation channel. Soc Neurosci Abstr. 2014;55 E- 3021. [Google Scholar]
- Keirstead SA, Miller RF. Calcium waves in dissociated retinal glial (Müller) cells are evoked by release of calcium from intracellular stores. Glia. 1995;14:14–22. doi: 10.1002/glia.440140104. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, Shestopalov VI. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. 2014;39:105–119. doi: 10.3109/02713683.2013.836541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert-Keil C, Bisping F, Krüger J, Brinkmeier H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics. 2006;7:159. doi: 10.1186/1471-2164-7-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth-Nelson ZL, Mishra A, Newman EA. Spontaneous glial calcium waves in the retina develop over early adulthood. J Neurosci. 2009;29:11339–11346. doi: 10.1523/JNEUROSCI.2493-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Markworth S, Poole K, Smith ES, Lapatsina L, Frahm S, May M, Pischke S, Suzuki M, Ibañez-Tallon I, Luft FC, Jordan J, Lewin GR. The molecular and cellular identity of peripheral osmoreceptors. Neuron. 2011;69:332–344. doi: 10.1016/j.neuron.2010.12.028. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM. Abnormal osmotic regulation in TRPV4−/− mice. Proc Natl Acad Sci U S A. 2003;100:13698–13703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist N, Liu Q, Zajadacz J, Franze K, Reichenbach A. Retinal glial (Müller) cells: sensing and responding to tissue stretch. Invest Ophthalmol Vis Sci. 2010;51:1683–1690. doi: 10.1167/iovs.09-4159. [DOI] [PubMed] [Google Scholar]
- Liu X, Bandyopadhyay B, Nakamoto T, Singh B, Liedtke W, Melvin JE, Ambudkar I. A role for AQP5 in activation of TRPV4 by hypotonicity: concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery. J Biol Chem. 2006;281:15485–15495. doi: 10.1074/jbc.M600549200. [DOI] [PubMed] [Google Scholar]
- Loukin SH, Su Z, Kung C. Hypotonic shocks activate rat TRPV4 in yeast in the absence of polyunsaturated fatty acids. FEBS Lett. 2009;583:754–758. doi: 10.1016/j.febslet.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukin S, Zhou X, Su Z, Saimi Y, Kung C. Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J Biol Chem. 2010;285:27176–27181. doi: 10.1074/jbc.M110.143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PLoS One. 2011;6:e19533. doi: 10.1371/journal.pone.0019533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YB, Franze K, Seifert G, Steinhäuser C, Kirchhoff F, Wolburg H, Guck J, Janmey P, Wei EQ, Käs J, Reichenbach A. Viscoelastic properties of individual glial cells and neurons in the CNS. Proc Natl Acad Sci U S A. 2006;103:17759–17764. doi: 10.1073/pnas.0606150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Cheng KT, Wong CO, O'Neil RG, Birnbaumer L, Ambudkar IS, Yao X. Heteromeric TRPV4–C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium. 2011;50:502–559. doi: 10.1016/j.ceca.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE. Mapping glutamatergic drive in the vertebrate retina with a channel-permeant organic cation. J Comp Neurol. 1999;407:47–64. doi: 10.1002/(SICI)1096-9861(19990428)407:1<47::AID-CNE4>3.0.CO%3B2-0. [DOI] [PubMed] [Google Scholar]
- Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2010;2:435–442. doi: 10.1039/c0ib00034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meves H. Arachidonic acid and ion channels: an update. Br J Pharmacol. 2008;155:4–16. doi: 10.1038/bjp.2008.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar T, Barabas P, Birnbaumer L, Punzo C, Kefalov V, Križaj D. Store-operated channels regulate intracellular calcium in mammalian rods. J Physiol. 2012;590:3465–3481. doi: 10.1113/jphysiol.2012.234641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP. Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci. 1998;18:2506–2519. doi: 10.1523/JNEUROSCI.18-07-02506.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Rodríguez JJ, Verkhratsky A. Glial calcium and diseases of the nervous system. Cell Calcium. 2010;47:140–149. doi: 10.1016/j.ceca.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013;14:152–163. doi: 10.1038/embor.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;286:C195–C205. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Wurm A, Uckermann O, vom Hagen F, Reichenbach A, Wiedemann P, Hammes HP, Bringmann A. Diabetes alters osmotic swelling characteristics and membrane conductance of glial cells in rat retina. Diabetes. 2006;55:633–639. doi: 10.2337/diabetes.55.03.06.db05-1349. [DOI] [PubMed] [Google Scholar]
- Pasantes-Morales H, Cruz-Rangel S. Brain volume regulation: osmolytes and aquaporin perspectives. Neuroscience. 2010;168:871–884. doi: 10.1016/j.neuroscience.2009.11.074. [DOI] [PubMed] [Google Scholar]
- Pinar-Sueiro S, Urcola H, Rivas MA, Vecino E. Prevention of retinal ganglion cell swelling by systemic brimonidine in a rat experimental glaucoma model. Clin Exp Ophthalmol. 2011;39:799–807. doi: 10.1111/j.1442-9071.2011.02573.x. [DOI] [PubMed] [Google Scholar]
- Puro DG. Stretch-activated channels in human retinal Müller cells. Glia. 1991;4:456–460. doi: 10.1002/glia.440040505. [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Bringmann A. Müller cells in the healthy and diseased retina. New York: Springer; 2010. [DOI] [PubMed] [Google Scholar]
- Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, Lee SH, Chauhan S, Xing W, Rentería RC, Liedtke W, Križaj D. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci. 2011;31:7089–7101. doi: 10.1523/JNEUROSCI.0359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Redmon S, Jo AO, Križaj D. TRPV1 and endocannabinoids: emerging molecular signals that modulate mammalian vision. Cells. 2014;3:914–938. doi: 10.3390/cells3030914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub F, Winkler A, Peters J, Kempski O, Kachel V, Baethmann A. Swelling, acidosis, and irreversible damage of glial cells from exposure to arachidonic acid in vitro. J Cereb Blood Flow Metab. 1994;14:1030–1039. doi: 10.1038/jcbfm.1994.135. [DOI] [PubMed] [Google Scholar]
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- Szikra T, Barabas P, Bartoletti TM, Huang W, Akopian A, Thoreson WB, Križaj D. Calcium homeostasis and cone signaling are regulated by interactions between calcium stores and plasma membrane ion channels. PLoS One. 2009;4:e6723. doi: 10.1371/journal.pone.0006723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci. 2003;44:3025–3033. doi: 10.1167/iovs.02-1136. [DOI] [PubMed] [Google Scholar]
- Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Rangroo Thrane V, Enger R, Haj-Yasein NN, Skare Ø, Holen T, Klungland A, Ottersen OP, Nedergaard M, Nagelhus EA. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108:846–851. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Fu Y, Garcia-Elias A, Fernández-Fernández JM, Vicente R, Kramer PL, Klein RF, Hitzemann R, Orwoll ES, Wilmot B, McWeeney S, Valverde MA, Cohen DM. A loss-of-function nonsynonymous polymorphism in the osmoregulatory TRPV4 gene is associated with human hyponatremia. Proc Natl Acad Sci U S A. 2009;106:14034–14039. doi: 10.1073/pnas.0904084106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS, Ruiz-Ederra J, Levin MH. Functions of aquaporins in the eye. Prog Retin Eye Res. 2008;27:420–433. doi: 10.1016/j.preteyeres.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A. 2004;101:396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NJ, Ho KW, Lambert WS, Weitlauf C, Calkins DJ. Absence of transient receptor potential vanilloid-1 accelerates stress-induced axonopathy in the optic projection. J Neurosci. 2014;34:3161–3170. doi: 10.1523/JNEUROSCI.4089-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003a;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Janssens A, Wondergem R, Droogmans G, Nilius B. Modulation of TRPV4 gating by intra-and extracellular Ca2+ Cell Calcium. 2003b;33:489–495. doi: 10.1016/S0143-4160(03)00064-2. [DOI] [PubMed] [Google Scholar]