

Abstract
The hippocampus is particularly sensitive to sleep loss. Although previous work has indicated that sleep deprivation impairs hippocampal cAMP signaling, it remains to be determined whether the cognitive deficits associated with sleep deprivation are caused by attenuated cAMP signaling in the hippocampus. Further, it is unclear which cell types are responsible for the memory impairments associated with sleep deprivation. Transgenic approaches lack the spatial resolution to manipulate specific signaling pathways selectively in the hippocampus, while pharmacological strategies are limited in terms of cell-type specificity. Therefore, we used a pharmacogenetic approach based on a virus-mediated expression of a Gαs-coupled Drosophila octopamine receptor selectively in mouse hippocampal excitatory neurons in vivo. With this approach, a systemic injection with the receptor ligand octopamine leads to increased cAMP levels in this specific set of hippocampal neurons. We assessed whether transiently increasing cAMP levels during sleep deprivation prevents memory consolidation deficits associated with sleep loss in an object–location task. Five hours of total sleep deprivation directly following training impaired the formation of object–location memories. Transiently increasing cAMP levels in hippocampal neurons during the course of sleep deprivation prevented these memory consolidation deficits. These findings demonstrate that attenuated cAMP signaling in hippocampal excitatory neurons is a critical component underlying the memory deficits in hippocampus-dependent learning tasks associated with sleep deprivation.
Keywords: hippocampus, learning, memory, pharmacogenetics, sleep, sleep disruption
Introduction
It is generally acknowledged that sleep promotes memory consolidation, while sleep deprivation attenuates this process, particularly for hippocampus-dependent memories (Meerlo et al., 2009; Havekes et al., 2012a; Abel et al., 2013; Rasch and Born, 2013). Studies in rodents have shown that 5–6 h of sleep deprivation directly after training impairs the consolidation of contextual fear memories (Graves et al., 2003; Vecsey et al., 2009; Hagewoud et al., 2010b, 2011) and hippocampus-dependent object–location memories (Florian et al., 2011; Binder et al., 2012; Prince et al., 2014). The negative impact of sleep deprivation on hippocampal function is emphasized by the observation that sleep-deprived animals preferentially use a behavioral strategy that relies on systems other than the hippocampus such as the striatum (Hagewoud et al., 2010c; Watts et al., 2012). In line with these behavioral observations are electrophysiological studies showing that sleep deprivation attenuates long-lasting forms of long-term potentiation (LTP), a cellular model of memory (Havekes et al., 2012a).
We found that 5 h of sleep deprivation attenuates hippocampal cAMP levels as a result of increased activity and expression of the cAMP-degrading phosphodiesterase 4 (PDE4) family (Vecsey et al., 2009), and cAMP signaling increases during sleep (Luo et al., 2013). Systemic injections with the nonspecific PDE4 inhibitor rolipram made memory consolidation resistant to sleep loss (Vecsey et al., 2009), indicating that hampered cAMP signaling contributes to the memory deficits associated with sleep deprivation. However, important gaps in our understanding of the mechanisms underlying sleep deprivation-induced memory impairments remain both at the systems and cellular level. First, it remains to be determined whether the cognitive deficits associated with sleep deprivation are caused by attenuated cAMP signaling specifically in the hippocampus. No studies have assessed whether transiently increasing cAMP levels in the hippocampus during sleep deprivation is sufficient to prevent the memory deficits associated with sleep loss. Second, it remains to be established which cell types are responsible for the cognitive impairments after sleep deprivation. This is of importance because various components of cAMP signaling pathways such as G-protein-coupled receptors are present in excitatory neurons, interneurons, and glial cells (Milner et al., 2000; Färber et al., 2005; Zsiros and Maccaferri, 2008).
Here, we used a pharmacogenetic approach combined with a viral strategy to transiently increase cAMP signaling in a brain region-specific and cell type-specific fashion. Such cell type specificity as well as temporal, and spatial resolution cannot be obtained with other pharmacological or transgenic strategies. We used adeno-associated viruses (AAVs) in combination with a CaMKIIα promoter fragment to express the Drosophila Gαs-coupled octopamine receptor (DmOctβ1R; CG6919) selectively in hippocampal excitatory neurons. The receptor could be activated at specific time points by giving mice a systemic injection of octopamine, leading to a transient increase in cAMP levels selectively in hippocampal excitatory neurons. Using this strategy, we found that transiently increasing cAMP levels in hippocampal excitatory neurons during the course of sleep deprivation is sufficient to prevent the deficits in memory consolidation that occurs with sleep loss.
Materials and Methods
Subjects.
Male C57BL/6J mice were obtained from The Jackson Laboratory at 6 weeks old, and were housed on a 12 h light/12 h dark schedule with lights on at 7:00 A.M. Food and water was available ad libitum. The animals underwent surgery at 8–12 weeks of age, were single housed for 5 d to recover, and then pair-housed with a littermate. Experiments were conducted according to National Institutes of Health guidelines for animal care and use and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
DNA manipulation and virus constructs.
The octopamine receptor gene construct (DmOctβ1R; Balfanz et al., 2005) was generated using Geneart (Millipore). A 0.4 kb CamKIIα promoter fragment was used to restrict expression to excitatory neurons. An HA peptide tag was included at the C-terminal end to monitor receptor expression using immunohistochemistry. Control mice were injected with an AAV to drive enhanced green fluorescent protein (EGFP) expression in excitatory neurons (pAAV9-CaMKIIα0.4-EGFP). The pAAV9-CaMKIIα0.4-DmOctβ1R-HA and pAAV9-CaMKIIα0.4-EGFP recombinants were constructed by standard methods and packaged by the University of Pennsylvania Vector Core. Viral titers ranged from 2.4 × 1012 to 1.86 × 1013 genome copy numbers/μl.
Viral surgeries.
The virus suspension (∼1 μl, depending on the titer) was injected bilaterally at −1.9 mm anteroposterior to, ±1.5 mm lateral/medial to, and 1.5 mm below bregma using a 33 ga beveled NanoFil needle (World Precision Instruments), a microsyringe pump (UMP3; WPI), and controller (Micro4; WPI).
Octopamine preparation.
The DmOctβ1Rs were stimulated by systemically injecting mice with the receptor ligand octopamine. Octopamine (Sigma) was prepared fresh on the day of the experiment in PBS, and mice received one or multiple intraperitoneal injections of octopamine (1 mg/kg). PBS without octopamine served as a vehicle solution.
Brain collection, immunohistochemistry, and biochemical analysis.
To validate the virally induced octopamine receptor expression, we used a separate set of mice to collect perfusion-fixed brains for immunohistohemical analysis. Transcardial perfusions and immunohistochemistry were conducted as described previously (Havekes et al., 2006; van der Borght et al., 2007; Isiegas et al., 2008). To detect DmOctβ1R-HA, we used a monoclonal antibody directed against the HA tag (Roche), biotinylated goat anti-rabbit secondary antibody (1:500; Jackson ImmunoResearch), and avidin–biotin–horseradish peroxidase complex (1:500; ABC Kit; Vector Laboratories). Tyramide Signal Amplification plus fluorescein (PerkinElmer) was used as a chromogen. Sections were imaged using a Leica SP8 confocal microscope.
Another set of mice was used to assess the potency of the virally mediated expression of octopamine receptors to increase cAMP levels. Mice were killed by cervical dislocation 30 min after a systemic injection with vehicle solution or octopamine. The hippocampus and prefrontal cortex were rapidly dissected on wet ice and immediately snap frozen on dry ice. Tissue was lysated, and cAMP content was measured using the ELISA-based assay kit (ENZO Life Sciences) following the manufacturer's instructions.
Object–place recognition task.
The object–place recognition task was conducted as described previously (Oliveira et al., 2010; Havekes et al., 2012b; Prince et al., 2014). Four weeks after surgery and before training, mice were handled for 2 min a day on 5 consecutive days during which animals also received a mock intraperitoneal injection. Training commenced at lights on (zeitgeber time 0) and totaled four 6 min training sessions with a 3 min intersession interval. The first session consisted of habituation to the arena in the absence of objects. In the next three sessions, the arena contained three distinct objects (a glass bottle, metal tower, and plastic cylinder). Directly after training, two of four groups of mice were sleep deprived for 5 h. After 24 h, mice were re-exposed to the context for 6 min, with one object moved to a novel spatial location. Object identities and locations were balanced between treatment groups. Behavior was video monitored and scored off-line by an experimenter that was blind to treatment. Object exploration was defined as facing and actively sniffing the objects within close proximity (i.e., within 1 cm) and/or touching them. The detection of spatial novelty was assessed by calculating the time spent exploring the relocated object relative to all objects during the test session.
Sleep deprivation.
Animals were sleep deprived for 5 h using the gentle stimulation method (Hagewoud et al., 2010a,b,c, 2011; Vecsey et al., 2009, 2013; Prince et al., 2014). Animals were kept awake by gentle tapping on the cage, gentle shaking the cage, or disturbing their bedding. This procedure is effective in keeping rodents awake for several hours, as established by electroencephalic recordings (Meerlo et al., 2001). Furthermore, this method was chosen because we and others have shown that the cognitive deficits associated with sleep deprivation are not due to elevated plasma corticosterone levels or the gentle stimulation method itself (Meerlo and Turek, 2001; Ruskin et al., 2006; van der Borght et al., 2006; Tiba et al., 2008; Hagewoud et al., 2010a; Vecsey et al., 2009). The role of glucocorticoids in synaptic plasticity and memory deficits associated with sleep deprivation has been extensively discussed previously (Havekes et al., 2012a).
Statistics.
Data analysis was performed using SPSS. Student's t tests were used to analyze the biochemical data. Two-way ANOVAs were used to analyze the behavioral data. A post hoc Dunnett's test was used for post hoc comparisons. Differences were considered statistically significant when p < 0.05. All data are plotted as the mean ± SEM.
Results
We aimed to express a Gαs-coupled Drosophila octopamine receptor (DmOctβ1R) or EGFP in excitatory hippocampal neurons of adult male C57BL/6J mice by using a CaMKIIα promoter fragment (Fig. 1A,B). Four weeks after injection of the AAVs into the hippocampus, DmOctβR1 protein expression was observed in excitatory neurons in areas CA1 and CA2/CA3, and in granular cells in the dentate gyrus (DG; Fig. 1C–L).
Figure 1.
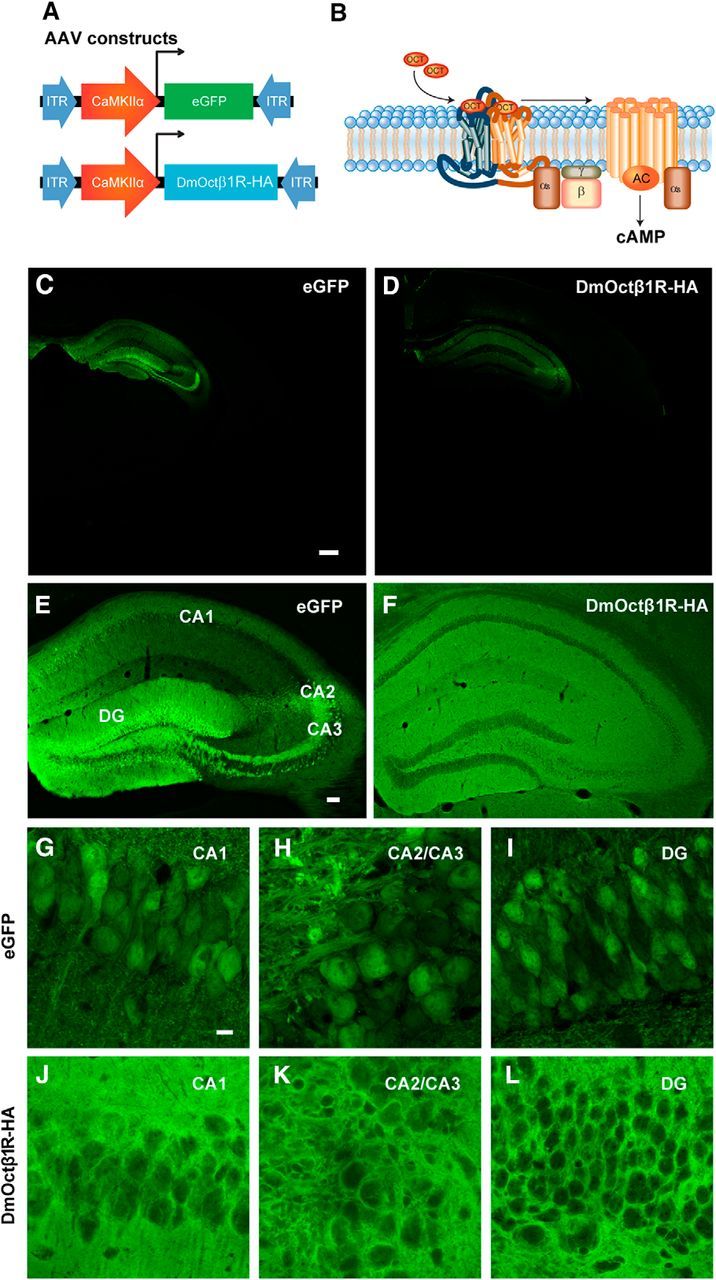
Viral expression of DmOctβR1 is restricted to excitatory neurons of the hippocampus. A, Mice were injected with pAAV9-CaMKIIα0.4-EGFP or pAAV9-CaMKIIα0.4-DmOctβ1R-HA to drive expression of EGFP or the Gαs-coupled DmOctβ1R in hippocampal excitatory neurons. B, Activation of the DmOctβ1R by its ligand octopamine (OCT) stimulates adenylyl cyclase activity, which synthesizes cAMP. C, D, Representative low-magnification images of EGFP and DmOctβ1R expression in coronal brain sections. Scale bar, 570 μm. E, F, Representative images EGFP and DmOctβ1R expression in all three major hippocampal subregions. Scale bar, 200 μm. G–L, Higher-magnification images of area CA1 (E, H), CA3 (F, I), and dentate gyrus (G, J). Scale bar, 20 μm. AC, Adenyl cyclase; ITR, inverted terminal repeats.
To examine whether expression of the receptor leads to increased levels of cAMP in the hippocampus, specifically upon stimulation with its natural ligand octopamine, mice expressing EGFP or DmOctβ1R received one systemic injection with PBS or octopamine (1 mg/kg). Thirty minutes after the injection, hippocampal tissue was harvested and cAMP content was measured. A systemic injection of octopamine led to a significant increase in hippocampal cAMP levels selectively in mice expressing the DmOctβ1R (t test, p = 0.034; Fig. 2A). Importantly, receptor expression by itself did not alter cAMP levels (Fig. 2A). Subsequently, we assessed whether the octopamine-induced increase in cAMP levels is restricted to the hippocampus. We found that hippocampal octopamine receptor expression and activation does not lead to an altered cAMP level in the prefrontal cortex (t test, p = 0.668; Fig. 2B). Together, these findings indicate that this pharmacogenetic approach can successfully increase cAMP levels selectively in excitatory neurons of the hippocampus.
Figure 2.
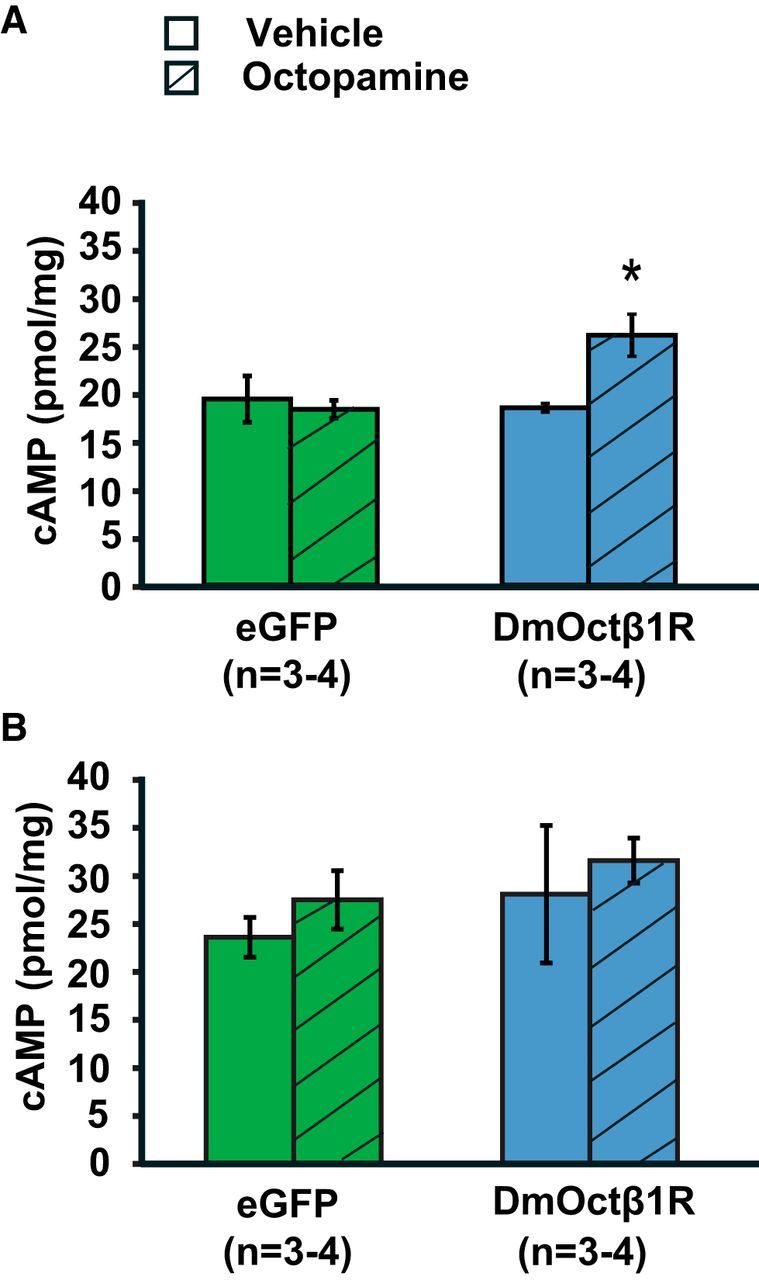
Activation of the DmOctβ1R in hippocampal neurons leads to increased cAMP levels selectively in the hippocampus. A, A systemic injection with octopamine (1 mg/kg) increased hippocampal cAMP levels 30 min after injection (*p = 0.034, t test) in mice expressing DmOctβ1R. B, Systemic injection with octopamine (1 mg/kg) did not alter cAMP levels in the prefrontal cortex 30 min after injection in mice expressing DmOctβ1R (p = 0.668, t test). All data are expressed as the mean ± SEM.
The main aim of this study was to test whether transiently increasing cAMP levels selectively in hippocampal excitatory neurons during sleep deprivation would be sufficient to prevent the memory deficits normally seen after sleep deprivation. We therefore trained mice expressing DmOctβ1R or EGFP in the object–place recognition task (Fig. 3A). Across the three training sessions with the objects, all mice gradually decreased the object exploration times (ANOVA effect of session F(2,70) = 58.723, p = 0.0001; Fig. 3B), and the level of exploration during training was not altered by the expression of DmOctβ1R (ANOVA effect of virus: F(1,35) = 1.640, p = 0.209; interaction effect: F(2,70) = 0.936, p = 0.397; Fig. 3B). Immediately after training, half of the group of DmOctβ1R- and EGFP-expressing mice were kept awake for 5 h while the remaining animals were left undisturbed. There are two specific time windows during which cAMP signaling contributes to hippocampus-dependent memory consolidation (Bourtchouladze et al., 1998; Graves et al., 2001). The first period is directly after training, and the second period is ∼3–4 h after training. Furthermore, octopamine can be traced back in tissue at least 1 h after application (Barron et al., 2007). To transiently increase cAMP levels during both periods, all mice received two systemic injections of octopamine (1 mg/kg), one immediately after training at the start of sleep deprivation, and another one 2.5 h after training (corresponding to 2.5 h into sleep deprivation). The next day, mice were tested for object–location memory. The analyses revealed that neither octopamine receptor expression nor sleep deprivation altered the total exploration time of the nondisplaced objects during the test session (EGFP: non-sleep-deprived animals, 6.98 ± 0.76; sleep-deprived animals, 8.26 ± 1.03; DmOctβ1R: non-sleep-deprived animals, 6.283 ± 0.56; sleep-deprived animals, 7.05 ± 1.00; ANOVA effect of sleep deprivation, F(1,39) = 1.397, p = 0.245; ANOVA: effect of virus, F(1,39) = 1.228, p = 0.275; interaction effect, F(1,39) = 0.089, p = 0.786). However, there were clear treatment effects on the preference for the displaced object. A two-way ANOVA revealed significant effects of sleep deprivation (F(1,35) = 6.603, p = 0.015) and virus treatment (F(1,35) = 20.256, p < 0.001), and an interaction effect between the two (F(1,35) = 4.565, p = 0.04). Mice expressing EGFP that had not been sleep deprived preferentially explored the displaced object, indicating that they identified the spatial change in object location and therefore had successfully consolidated the original object locations (Fig. 3C). In contrast, EGFP-expressing mice that were sleep deprived failed to detect that one object was displaced, indicating that sleep deprivation impaired the consolidation of object–place memories (EGFP non-sleep-deprived group vs EGFP sleep-deprived group, post hoc Dunnett's test, p < 0.05; Fig. 3C). However, mice expressing the DmOctβ1R and being injected with octopamine showed a strong preference for the displaced object regardless of sleep deprivation (DmOctβ1R groups vs EGFP sleep-deprived group, post hoc Dunnett's test, p < 0.05; Fig. 3C). These data show that transiently increasing cAMP levels in hippocampal excitatory neurons during sleep deprivation is sufficient to prevent memory deficits in the object–place recognition task.
Figure 3.

Increasing neuronal cAMP levels in hippocampal neurons prevents memory deficits caused by sleep deprivation. A, A diagram illustrating the training schedule and time points at which all animals received intraperitonal injections with octopamine. B, Viral expression of the Gαs-coupled DmOctβ1R in hippocampal neurons does not affect object exploration levels during training in the object–place recognition task (effect of virus: F(1,35) = 1.640, p = 0.209). All groups showed decreased total object exploration time during consecutive training sessions in a similar fashion (effect of session: F(2,70) = 58.723, p = 0.0001; interaction effect: F(2,70) = 0.936, p = 0.397). C, Mice expressing EGFP or DmOctβ1R were trained in the hippocampus-dependent object–place recognition task and sleep deprived for 5 h immediately after training [sleep-deprived (SD) animals] or were left undisturbed [non-sleep-deprived (NSD) animals]. Mice received systemic injections with octopamine directly after training and 2.5 h into sleep deprivation. Boosting cAMP signaling by activating the of DmOctβ1R prevents memory deficits caused by sleep deprivation (n = 9–10; effect of sleep deprivation: F(1,35) = 6.603, p = 0.015; effect of virus: F(1,35) = 20.256, p < 0.001; interaction effect: F(1,35) = 4.565, p = 0.04; EGFP SD group vs other groups, p < 0.05). Dotted line indicates chance performance. Error bars denote SEM. *p < 0.05. All data are expressed as the mean ± SEM.
Discussion
Previous work indicated that 5 h of total sleep deprivation reduced cAMP levels in hippocampal tissue and demonstrated that a general, nonspecific enhancement of cAMP through the systemic inhibition of PDE4 activity prevented impairments in contextual fear memory formation (Vecsey et al., 2009). However, it remained to be defined whether attenuation of cAMP levels in the hippocampus was ultimately causing the memory deficits associated with sleep loss. In addition, it was unclear which cell types in the hippocampus are vulnerable to sleep deprivation and thus are responsible for the cognitive deficits caused by sleep deprivation. To answer these two important questions, we used a pharmacogenetic approach and expressed the Gαs-coupled DmOctβR1 selectively in hippocampal excitatory neurons. Biochemical analyses revealed that hippocampal cAMP levels were elevated 30 min after giving animals a single systemic injection with octopamine. We found that activating the octopamine receptor in hippocampal excitatory neurons during the 5 h of sleep deprivation that started directly after training in the object–location memory task was sufficient to prevent sleep deprivation-induced memory deficits. Although we did not directly assess whether cAMP levels were elevated due to activation of the octopamine receptor during the course of sleep deprivation, our finding suggests that sleep deprivation directly impacts cAMP signaling in hippocampal excitatory neurons and that this misregulation of the cAMP pathway plays a central role in the cognitive deficits associated with sleep loss.
Various studies have emphasized the importance of the cAMP signaling pathway in long-lasting forms of hippocampal LTP and memory (Abel et al., 1997; Bernabeu et al., 1997). Local inhibition of cAMP-dependent protein kinase A (PKA) activity in the hippocampus is sufficient to impair the consolidation of long-term contextual fear memories (Bourtchouladze et al., 1998). In addition, activation of the exchange protein activated by cAMP (Epac) in the hippocampus enhances memory consolidation (Ma et al., 2009). Thus, the impairment of cAMP signaling caused by sleep loss could very well impair signaling mechanisms that require activation of PKA or Epac. Indeed, we and others previously observed that sleep deprivation reduces the phosphorylation of the cAMP response element-binding protein (CREB) in the hippocampus under basal conditions (Vecsey et al., 2009) and prevents the increases in CREB phosphorylation observed after high-frequency stimulation (Alhaider et al., 2011; Park et al., 2012), learning (Hagewoud et al., 2010c), or exercise (Zagaar et al., 2013). Because long-term memory formation critically depends on CREB-mediated gene transcription (Vecsey et al., 2007), one potential mechanism by which transiently increasing cAMP levels in the hippocampus during the course of sleep deprivation is the restoration of CREB-mediated gene transcription. Another substrate of the cAMP–PKA signaling pathway that is important for memory and synaptic plasticity is the GluA1 subunit of the AMPA receptor (AMPAR; Roche et al., 1996; Kim et al., 2011). Because sleep loss reduces hippocampal AMPAR function (Dubiela et al., 2013), including AMPAR GluA1 phosphorylation (Hagewoud et al., 2010a; Ravassard et al., 2009), our pharmacogenetic approach to increase cAMP levels during the course of sleep deprivation may restore AMPAR function, leading to proper consolidation of object–location memories regardless of sleep deprivation. In future studies, it would be interesting to determine whether boosting cAMP levels during the course of sleep deprivation restores hippocampal CREB and AMPAR function as well as the phosphorylation state of other PKA and Epac substrates.
The hippocampus consists of distinct subregions that each contribute differently to the memory consolidation process. Models of hippocampal function as well as studies testing these models using selective lesions of hippocampal subregions suggest that, particularly, area CA3 and the dentate gyrus play a critical role in detecting a mismatch between the actual environment and a previous experience, such as detecting the relocation of a familiar object to a novel location (Hasselmo, 2005; Lee et al., 2005). Because sleep deprivation impairs forms of hippocampal synaptic plasticity that depend on proper cAMP signaling in area CA3, such as LTP induced by forskolin treatment and theta-burst stimulation (Vecsey et al., 2009; Park et al., 2014), it would be of interest to restrict DmOctβ1R expression to CA3 excitatory neurons and to determine whether transiently increasing cAMP levels in this single subregion would be sufficient to prevent the memory deficits associated with sleep loss.
One potential concern with sleep deprivation studies is that the method used to deprive animals of sleep rather than the sleep loss itself may actually cause memory deficits due to interference (Robertson, 2012). The theory suggests that memory consolidation can be impaired by the stimuli themselves during the consolidation period. A recently published study assessed whether loss of sleep or interference as a result of the sleep deprivation method was the main cause of the cognitive deficits associated with sleep deprivation (Hagewoud et al., 2010b). Using a sleep deprivation protocol similar to the one used in the present study, this article describes how 6 h of sleep deprivation by manual stimulation during the first half of the light phase/sleep phase immediately following acquisition impairs hippocampus-dependent memory consolidation. In a follow-up experiment, the authors performed 6 h of sleep deprivation during the dark phase/active phase, again immediately after acquisition, and subjected animals to the exact same amount of stimulation over the course of 6 h that was needed to keep animals awake during the light phase in the previous experiment (in other words, animals received stimulation, whether they were already awake or not, to exactly match the number of stimulations per hour given during the previous experiment in the light phase). If the interference was the driving factor behind the cognitive deficits in the first experiment, then one would expect that applying the exact amount of stimulation during the dark phase would also lead to memory impairment. We showed, however, that this exact amount of stimulation when applied in the dark phase does not cause a memory deficit. Thus, while interference may be able to perturb memory consolidation, it does not seem to be a dominant factor in the cognitive deficits associated with sleep deprivation using the gentle stimulation method.
Millions of people regularly obtain insufficient sleep. Therefore, understanding the cellular and molecular pathways affected by sleep deprivation is of great social and clinical importance. In the present study, we used a mouse model to study memory, and, by applying a cell type-specific, brain region-specific, and time-specific pharmacogenetic approach, we demonstrated that selectively increasing cAMP levels in excitatory neurons of the hippocampus during sleep deprivation can prevent the memory deficits that normally occur due to sleep loss. In future work, the downstream targets of cAMP in the hippocampus that ultimately mediate the cognitive deficits associated with sleep loss will be addressed. Because the cAMP signaling pathway is differentially regulated during wakefulness and sleep (Luo et al., 2013) and is negatively impacted by sleep loss (Vecsey et al., 2009), in future studies it will also be of importance to define whether different mechanisms are involved in sleep-related as opposed to wake-related memory consolidation.
Footnotes
This research was supported by the Netherlands Organization for Scientific Research NWO-Rubicon Grant 825.07.029 (to R.H.), a University Research Foundation grant (to R.H. and T.A.), National Institutes of Health Grants 1R01MH086415 (to T.A.), 5K12GM081529 [to J.C.T. (Principal Investigator, Y. Paterson], and 2T32NS7413–16 [to S.L.F. (Principal Investigator, M. B. Robinson)]. We thank other Abel laboratory members for their help with these experiments and for their comments on the manuscript.
The authors declare no competing financial interests.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/S0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013;23:R774–R788. doi: 10.1016/j.cub.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhaider IA, Aleisa AM, Tran TT, Alkadhi KA. Sleep deprivation prevents stimulation-induced increases of levels of P-CREB and BDNF: protection by caffeine. Mol Cell Neurosci. 2011;46:742–751. doi: 10.1016/j.mcn.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Balfanz S, Strünker T, Frings S, Baumann A. A family of octopamine [corrected] receptors that specifically induce cyclic AMP production or Ca2+ release in Drosophila melanogaster. J Neurochem. 2005;93:440–451. doi: 10.1111/j.1471-4159.2005.03034.x. [DOI] [PubMed] [Google Scholar]
- Barron AB, Maleszka J, Vander Meer RK, Robinson GE, Maleszka R. Comparing injection, feeding and topical application methods for treatment of honeybees with octopamine. J Insect Physiol. 2007;53:187–194. doi: 10.1016/j.jinsphys.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M, Izquierdo I, Medina JH. Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci U S A. 1997;94:7041–7046. doi: 10.1073/pnas.94.13.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder S, Baier PC, Mölle M, Inostroza M, Born J, Marshall L. Sleep enhances memory consolidation in the hippocampus-dependent object-place recognition task in rats. Neurobiol Learn Mem. 2012;97:213–219. doi: 10.1016/j.nlm.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem. 1998;5:365–374. [PMC free article] [PubMed] [Google Scholar]
- Dubiela FP, Queiroz CM, Moreira KD, Nobrega JN, Sita LV, Tufik S, Hipolide DC. AMPA receptors mediate passive avoidance deficits induced by sleep deprivation. Behav Brain Res. 2013;257:189–196. doi: 10.1016/j.bbr.2013.09.037. [DOI] [PubMed] [Google Scholar]
- Färber K, Pannasch U, Kettenmann H. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci. 2005;29:128–138. doi: 10.1016/j.mcn.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Florian C, Vecsey CG, Halassa MM, Haydon PG, Abel T. Astrocyte-derived adenosine and A1 receptor activity contribute to sleep loss-induced deficits in hippocampal synaptic plasticity and memory in mice. J Neurosci. 2011;31:6956–6962. doi: 10.1523/JNEUROSCI.5761-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves LA, Heller EA, Pack AI, Abel T. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem. 2003;10:168–176. doi: 10.1101/lm.48803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves L, Pack A, Abel T. Sleep and memory: a molecular perspective. Trends Neurosci. 2001;24:237–243. doi: 10.1016/S0166-2236(00)01744-6. [DOI] [PubMed] [Google Scholar]
- Hagewoud R, Havekes R, Novati A, Keijser JN, Van der Zee EA, Meerlo P. Sleep deprivation impairs spatial working memory and reduces hippocampal AMPA receptor phosphorylation. J Sleep Res. 2010a;19:280–288. doi: 10.1111/j.1365-2869.2009.00799.x. [DOI] [PubMed] [Google Scholar]
- Hagewoud R, Whitcomb SN, Heeringa AN, Havekes R, Koolhaas JM, Meerlo P. A time for learning and a time for sleep: the effect of sleep deprivation on contextual fear conditioning at different times of the day. Sleep. 2010b;33:1315–1322. doi: 10.1093/sleep/33.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagewoud R, Havekes R, Tiba PA, Novati A, Hogenelst K, Weinreder P, Van der Zee EA, Meerlo P. Coping with sleep deprivation: shifts in regional brain activity and learning strategy. Sleep. 2010c;33:1465–1473. doi: 10.1093/sleep/33.11.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagewoud R, Bultsma LJ, Barf RP, Koolhaas JM, Meerlo P. Sleep deprivation impairs contextual fear conditioning and attenuates subsequent behavioural, endocrine and neuronal responses. J Sleep Res. 2011;20:259–266. doi: 10.1111/j.1365-2869.2010.00895.x. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. The role of hippocampal regions CA3 and CA1 in matching entorhinal input with retrieval of associations between objects and context: theoretical comment on Lee et al (2005) Behav Neurosci. 2005;119:342–345. doi: 10.1037/0735-7044.119.1.342. [DOI] [PubMed] [Google Scholar]
- Havekes R, Nijholt IM, Luiten PG, Van der Zee EA. Differential involvement of hippocampal calcineurin during learning and reversal learning in a Y-maze task. Learn Mem. 2006;13:753–759. doi: 10.1101/lm.323606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Vecsey CG, Abel T. The impact of sleep deprivation on neuronal and glial signaling pathways important for memory and synaptic plasticity. Cell Signal. 2012a;24:1251–1260. doi: 10.1016/j.cellsig.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Canton DA, Park AJ, Huang T, Nie T, Day JP, Guercio LA, Grimes Q, Luczak V, Gelman IH, Baillie GS, Scott JD, Abel T. Gravin orchestrates protein kinase A and β2-adrenergic receptor signaling critical for synaptic plasticity and memory. J Neurosci. 2012b;32:18137–18149. doi: 10.1523/JNEUROSCI.3612-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isiegas C, McDonough C, Huang T, Havekes R, Fabian S, Wu LJ, Xu H, Zhao MG, Kim JI, Lee YS, Lee HR, Ko HG, Lee N, Choi SL, Lee JS, Son H, Zhuo M, Kaang BK, Abel T. A novel conditional genetic system reveals that increasing neuronal cAMP enhances memory and retrieval. J Neurosci. 2008;28:6220–6230. doi: 10.1523/JNEUROSCI.2935-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Park AJ, Havekes R, Chay A, Guercio LA, Oliveira RF, Abel T, Blackwell KT. Colocalization of protein kinase A with adenylyl cyclase enhances protein kinase A activity during induction of long-lasting long-term-potentiation. PLoS Comput Biol. 2011;7:e1002084. doi: 10.1371/journal.pcbi.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Hunsaker MR, Kesner RP. The role of hippocampal subregions in detecting spatial novelty. Behav Neurosci. 2005;119:145–153. doi: 10.1037/0735-7044.119.1.145. [DOI] [PubMed] [Google Scholar]
- Luo J, Phan TX, Yang Y, Garelick MG, Storm DR. Increases in cAMP, MAPK activity, and CREB phosphorylation during REM sleep: implications for REM sleep and memory consolidation. J Neurosci. 2013;33:6460–6468. doi: 10.1523/JNEUROSCI.5018-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N, Abel T, Hernandez PJ. Exchange protein activated by cAMP enhances long-term memory formation independent of protein kinase A. Learn Mem. 2009;16:367–370. doi: 10.1101/lm.1231009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerlo P, Turek FW. Effects of social stimuli on sleep in mice: non-rapid-eye-movement (NREM) sleep is promoted by aggressive interaction but not by sexual interaction. Brain Res. 2001;907:84–92. doi: 10.1016/S0006-8993(01)02603-8. [DOI] [PubMed] [Google Scholar]
- Meerlo P, de Bruin EA, Strijkstra AM, Daan S. A social conflict increases EEG slow-wave activity during subsequent sleep. Physiol Behav. 2001;73:331–335. doi: 10.1016/S0031-9384(01)00451-6. [DOI] [PubMed] [Google Scholar]
- Meerlo P, Mistlberger RE, Jacobs BL, Heller HC, McGinty D. New neurons in the adult brain: the role of sleep and consequences of sleep loss. Sleep Med Rev. 2009;13:187–194. doi: 10.1016/j.smrv.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Shah P, Pierce JP. beta-adrenergic receptors primarily are located on the dendrites of granule cells and interneurons but also are found on astrocytes and a few presynaptic profiles in the rat dentate gyrus. Synapse. 2000;36:178–193. doi: 10.1002/(SICI)1098-2396(20000601)36:3<178::AID-SYN3>3.0.CO%3B2-6. [DOI] [PubMed] [Google Scholar]
- Oliveira AM, Hawk JD, Abel T, Havekes R. Post-training reversible inactivation of the hippocampus enhances novel object recognition memory. Learn Mem. 2010;17:155–160. doi: 10.1101/lm.1625310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park AJ, Havekes R, Choi JH, Luczak V, Nie T, Huang T, Abel T. A presynaptic role for PKA in synaptic tagging and memory. Neurobiol Learn Mem. 2014;114:101–112. doi: 10.1016/j.nlm.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Kang WS, Paik JW, Kim JW. Effect of valproic acid through regulation of NMDA receptor-ERK signaling in sleep deprivation rats. J Mol Neurosci. 2012;47:554–558. doi: 10.1007/s12031-011-9673-5. [DOI] [PubMed] [Google Scholar]
- Prince TM, Wimmer M, Choi J, Havekes R, Aton S, Abel T. Sleep deprivation during a specific 3-hour time window post-training impairs hippocampal synaptic plasticity and memory. Neurobiol Learn Mem. 2014;109:122–130. doi: 10.1016/j.nlm.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch B, Born J. About sleep's role in memory. Physiol Rev. 2013;93:681–766. doi: 10.1152/physrev.00032.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravassard P, Pachoud B, Comte JC, Mejia-Perez C, Scoté-Blachon C, Gay N, Claustrat B, Touret M, Luppi PH, Salin PA. Paradoxical (REM) sleep deprivation causes a large and rapidly reversible decrease in long-term potentiation, synaptic transmission, glutamate receptor protein levels, and ERK/MAPK activation in the dorsal hippocampus. Sleep. 2009;32:227–240. doi: 10.1093/sleep/32.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson EM. New insights in human memory interference and consolidation. Curr Biol. 2012;22:R66–R71. doi: 10.1016/j.cub.2011.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/S0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Ruskin DN, Dunn KE, Billiot I, Bazan NG, LaHoste GJ. Eliminating the adrenal stress response does not affect sleep deprivation-induced acquisition deficits in the water maze. Life Sci. 2006;78:2833–2838. doi: 10.1016/j.lfs.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Tiba PA, Oliveira MG, Rossi VC, Tufik S, Suchecki D. Glucocorticoids are not responsible for paradoxical sleep deprivation-induced memory impairments. Sleep. 2008;31:505–515. doi: 10.1093/sleep/31.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Borght K, Ferrari F, Klauke K, Roman V, Havekes R, Sgoifo A, van der Zee EA, Meerlo P. Hippocampal cell proliferation across the day: increase by running wheel activity, but no effect of sleep and wakefulness. Behav Brain Res. 2006;167:36–41. doi: 10.1016/j.bbr.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Van der Borght K, Havekes R, Bos T, Eggen BJ, Van der Zee EA. Exercise improves memory acquisition and retrieval in the Y-maze task: relationship with hippocampal neurogenesis. Behav Neurosci. 2007;121:324–334. doi: 10.1037/0735-7044.121.2.324. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, Huang T, Brown KM, Li XY, Descalzi G, Kim SS, Chen T, Shang YZ, Zhuo M, Houslay MD, Abel T. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature. 2009;461:1122–1125. doi: 10.1038/nature08488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, Wimmer ME, Havekes R, Park AJ, Perron IJ, Meerlo P, Abel T. Daily acclimation handling does not affect hippocampal long-term potentiation or cause chronic sleep deprivation in mice. Sleep. 2013;36:601–607. doi: 10.5665/sleep.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts A, Gritton HJ, Sweigart J, Poe GR. Antidepressant suppression of non-REM sleep spindles and REM sleep impairs hippocampus-dependent learning while augmenting striatum-dependent learning. J Neurosci. 2012;32:13411–13420. doi: 10.1523/JNEUROSCI.0170-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagaar M, Dao A, Levine A, Alhaider I, Alkadhi K. Regular exercise prevents sleep deprivation associated impairment of long-term memory and synaptic plasticity in the CA1 area of the hippocampus. Sleep. 2013;36:751–761. doi: 10.5665/sleep.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsiros V, Maccaferri G. Noradrenergic modulation of electrical coupling in GABAergic networks of the hippocampus. J Neurosci. 2008;28:1804–1815. doi: 10.1523/JNEUROSCI.4616-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]