

Abstract
Rheb1 is an immediate early gene that functions to activate mammalian target of rapamycin (mTor) selectively in complex 1 (mTORC1). We have demonstrated previously that Rheb1 is essential for myelination in the CNS using a Nestin-Cre driver line that deletes Rheb1 in all neural cell lineages, and recent studies using oligodendrocyte-specific CNP-Cre have suggested a preferential role for mTORC1 is myelination in the spinal cord. Here, we examine the role of Rheb1/mTORC1 in mouse oligodendrocyte lineage using separate Cre drivers for oligodendrocyte progenitor cells (OPCs) including Olig1-Cre and Olig2-Cre as well as differentiated and mature oligodendrocytes including CNP-Cre and Tmem10-Cre. Deletion of Rheb1 in OPCs impairs their differentiation to mature oligodendrocytes. This is accompanied by reduced OPC cell-cycle exit suggesting a requirement for Rheb1 in OPC differentiation. The effect of Rheb1 on OPC differentiation is mediated by mTor since Olig1-Cre deletion of mTor phenocopies Olig1-Cre Rheb1 deletion. Deletion of Rheb1 in mature oligodendrocytes, in contrast, does not disrupt developmental myelination or myelin maintenance. Loss of Rheb1 in OPCs or neural progenitors does not affect astrocyte formation in gray and white matter, as indicated by the pan-astrocyte marker Aldh1L1. We conclude that OPC-intrinsic mTORC1 activity mediated by Rheb1 is critical for differentiation of OPCs to mature oligodendrocytes, but that mature oligodendrocytes do not require Rheb1 to make myelin or maintain it in the adult brain. These studies reveal mechanisms that may be relevant for both developmental myelination and impaired remyelination in myelin disease.
Keywords: differentiation, mTORC1, myelination, OPC, Rheb1
Introduction
The myelination of axons by oligodendrocytes (OLs) in the brain is essential for rapid saltatory transduction of neuronal impulses along neural networks, and is regulated by both developmental (Emery, 2010) and activity-dependent mechanisms (Fields and Stevens-Graham, 2002). The formation of myelin in the brain requires mature, myelinating OLs, which provide essential signals for radial growth of axons, as well as metabolic support to axons to maintain their long-term structural integrity (Lee et al., 2012). OLs are derived from oligodendrocyte precursor cells (OPCs) that are identified by expression of PDGFRα and NG2 proteoglycan (Rivers et al., 2008). OPCs exit the cell cycle, becoming postmitotic OLs that further mature into late-stage, myelinating OLs. The maturation of OLs can be monitored by the expression of cellular markers for mature OLs including, adenomatous polyposis coli, also known as CC1, proteolipid protein (PLP) and Tmem10, aka Opalin, a newly discovered marker for myelinating OLs (Golan et al., 2008; Jiang et al., 2013).
Previous studies have shown that expression of constitutively active AKT in OLs increases mTORC1 activity and enhances myelination in the brain (Flores et al., 2008). This effect is reversed by the mTor inhibitor rapamycin (Narayanan et al., 2009) suggesting that the effect of AKT on myelination is mediated by mTor. Studies of cultured cells showed that rapamycin arrests the differentiation of OPCs at O4+ stage, suggesting a role of mTor in the early stage OPC differentiation (Tyler et al., 2009). However, another report showed that inhibition of mTORC1 in cultured OLs only affects the late-stage transition of premyelinating OLs to fully matured OLs (Guardiola-Diaz et al., 2012). In vivo ablation of mTor or Raptor (an essential component of mTORC1 complex) in OLs using CNP-Cre driver reveals a severe disruption in OL differentiation and myelination in spinal cord, but not in the brain, suggesting a region-dependent requirement of mTor or mTORC1 on OL and myelin formation in the CNS (Bercury et al., 2014; Wahl et al., 2014). Thus, the role for mTORC1 signaling in OPCs versus OLs and its contribution to myelination in the brain remains unclear.
To address this challenge, we have used four different Cre lines to target Rheb1/mTORC1 activity in OPCs (Olig1-Cre, Olig2-Cre) and OLs (CNP-Cre, Tmem10-Cre). We targeted Rheb1 since it is essential for mTORC1 (Y. Li et al., 2004; Zou et al., 2011) and its neuronal expression can be naturally regulated by activity (Yamagata et al., 1994). Rheb1 deletion in vivo in neural progenitor cells using Nestin-Cre driver, which results in reduction of mTORC1 in all types of neural cells, prevents OPC maturation and myelination (Zou et al., 2011). We report that OL-intrinsic signaling of Rheb1 and mTor is essential for the early stage OPC differentiation to OLs in the brain, but Rheb1 is not required for the survival of OLs or generation and maintenance of myelin.
Materials and Methods
Animals.
Cre lines include Olig1-Cre (Lu et al., 2002), Olig2-Cre (Schüller et al., 2008), CNP-Cre (Lappe-Siefke et al., 2003), Tmem10-Cre (lab made), or Nestin-Cre was crossed to mice carrying the floxed allele of Rheb1 (Rheb1f/f; Zou et al., 2011) or mTor (B6;129s4-Mtortm1.2Koz/J; The Jackson Laboratory) to generate Rheb1 or mTor conditional knock-out animals. The Tmem10-Cre mice were bred with tdTomato reporter mice (B6;129S6-Gt(ROSA)26tm9(CAG-tdTomato)Hze/J; The Jackson Laboratory) to validate Cre recombinase activity. Aldh1L1 knock-out mice were generated with a knockin/knock-out strategy by inserting eGFP-Cre cDNA into the locus right after the Aldh1L1 promoter in our lab. The insertion of eGFP-Cre cDNA disrupts the reading frame of Aldh1L1. All strains were on C57/BL/6 and 129s4 mixed backgrounds. Both males and females were used in all analyses. All mouse protocols were conducted in accordance with the guidelines set forth by Sichuan University and Johns Hopkins University.
Antibodies.
Phosphorylated-S6 (Ser240/244), total AKT, phosphorylated-AKT (Ser473), phosphorylated-4EBP (T37/46), and phosphorylated-histone3 (Ser10) antibodies were purchased from Cell Signaling Technology; Olig2, NG2, GFAP, MOG, and CNPase antibodies from Millipore; CC1 and MBP antibodies from Calbiochem; PDGFRα from Becton Dickinson; PLP, BrdU, and Ki67 from Abcam; and Iba-1 from Wako Chemicals. Rheb1 antibody was generated by immunizing New Zealand white rabbits with bacterial GST fusion protein (Zou et al., 2011). Tmem10 antibody was generated by immunizing New Zealand white rabbits with bacterial His-tagged fusion protein (Jiang et al., 2013). Aldh1L1 antibody was generated by immunizing New Zealand white rabbits with bacterial GST fusion protein (150 AA of mouse Aldh1L1 in C-terminal) in our lab.
Western blotting.
Mice were rapidly decapitated and brains were removed. The brain was dissected into cortical, hippocampus, and cerebellum regions. To make cell extracts, tissues were homogenized in lysis buffer (2% SDS with proteinase inhibitors and phosphatase Inhibitor). The protein concentration of each extract was measured using the BCA Protein Assay kit (Thermo Scientific Pierce). Equal amounts of proteins from each extract were loaded into SDS-PAGE gel and blotted with various antibodies, according to standard Western blotting procedures. Western blotting and densitometry was performed using the ECL system (Thermo Scientific Pierce) and ImageJ.
Immunohistochemistry, histology, and electron microscopy.
Tissues for immunohistochemistry and electron microscopy were prepared as described previously (Zou et al., 2011). For electron microscopy, ultrathin sections were obtained using Ultracut UCT (Leica) and stained with 2% uranyl acetate and lead citrate. Electron micrographs were taken with a Hitachi electron microscope.
BrdU labeling and in situ hybridization.
For OPC proliferation analysis, we injected mice intraperitoneally with BrdU (100 mg/kg; Sigma). Two hours later, the perfused brains were dissected out and brain cryosections were stained with anti-BrdU and anti-Olig2 antibodies to visualize proliferating OPCs. For cell-cycle exit experiment, mice were intraperitoneally injected with BrdU (100 mg/kg) and killed 24–36 h later. The cell-cycle exit index was measured as the percentage of the OPCs that exited the cell cycle (BrdU+/Ki67−) divided by total BrdU+ cells in the corpus callosum.
For in situ hybridization, brain sections were prepared the same way as with immunohistochemistry. Briefly, a probe for Plp was amplified using gene-specific PCR primers (Allen Institute for Brain Science, Seattle, WA) from mouse brain. The resulting fragment was cloned into pKSII transcription vectors. All the buffers were made with DEPC-treated water. In situ hybridization was then performed on sectioned tissue as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993). Brain sections were fixed in 4% buffered paraformaldehyde, treated with 0.5% acetic anhydride/1.5% triethanolamine, and equilibrated in PBS. After 1 h prehybridization at 65°C, DIG-UTP labeled Plp probe was added to each slide and hybridization was performed in a humidified box at 60°C for 16 h. Slides were washed to a final stringency of 0.2× SSC at 60°C. Afterward, anti-DIG-alkaline phosphatase antibody (1:5000 BMB; catalog #1093274) was applied in blocking solution with 10% normal goat serum and 0.1% Triton X-100 in PBS and kept at 4°C overnight. Slides were washed and equilibrated in Tris-buffered saline (with 0.1% Tween 20) and developed in BM-Purple AP substrate (with 0.5 mg/ml levamisole, σ) at room temperature in the dark. The reaction was terminated by washing in 1× PBS, 0.1% Triton X-100, and then rinsed with distilled water, air dried, and mounted with antifade media (Invitrogen).
RNA extraction and RT-PCR.
Total RNAs were extracted from tissues using TRIzol reagent (Invitrogen). RNA was subjected to reverse transcription with reverse transcriptase as Manufacturer's instructions (Fermentas). Quantitative real-time PCR was performed using the Bio-Rad CFX96 system, and the relative gene expression was normalized to internal control as gapdh. Primer sequences for SYBR Green probes of target genes are as follows: Mbp: ATCCAAGTACCTGGCCACAG and CCTGTCACCGCTAAAGAAGC; Cnp: TTCTGGAGATGAACCCAAGG and TCTCTTCACCACCTCCTGCT; Plp: CTGGCTGAGGGCTTCTACAC and GACTGACAGGTGGTCCAGGT; Mog: AAATGGCAAGGACCAAGATG and AGCAGGTGTAGCCTCCTTCA; Mobp: CATTTGCTTCCCATTCACCT and AGGATGCCTCCATTTCCTCT; Tmem10: TGAGCCCGTAGAGGAGACT and CATATATGTGCCCTTCTTGG; Mag: TGGGCCTACGAAACTGTACC and GCTCCGAGAAGGTGTACTGG; Gapdh: GGTGAAGGTCGGTGTGAACG and CTCGCTCCTGGAAGATGGTG.
Cells counts.
Three to four animals per genotype were used to examine the cellular marker expression for each time point. In the cortex, three to five nonadjacent sections were counted per animal. In the corpus callosum, images were acquired to include only the corpus callosum at the midline. Counts were statistically compared using Student's t test.
Statistical analysis.
Data represent the mean and SEM. Student's t test (one-tailed for Western blot and qRT-PCR, two-tailed for the others) was performed for all statistical significance analysis using GraphPad Prism software. *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Hypomyelination in brain of the Rheb1f/f;Olig1Cre+/−mouse
To examine the cell-intrinsic effect of Rheb1 on oligodendrogenesis, we crossed floxed Rheb1 mouse with Olig1-Cre deleter mouse (Lu et al., 2002) in which Cre activity is expressed at the stage of OPCs and throughout the entire OL lineage (Xin et al., 2005). The resulting mutant Rheb1f/f;Olig1Cre+/− mice exhibited normal postnatal growth in comparison with their control littermates (Rheb1f/+;Olig1Cre+/− or Rheb1+/+;Olig1Cre+/−). The success of the genetic deletion of Rheb1 was evidenced by undetectable phosphorylation of S6 in OL lineage cells (Olig2+) in the corpus callosum (Fig. 1A), decreased Rheb1 protein, and pronounced increase in pAKT S473 in the cortex of P28 Rheb1f/f;Olig1Cre+/− mice (Fig. 1F,G). Rheb1f/f;Olig1Cre+/− mice showed severe hypomyelination in the corpus callosum, cortex, and striatum of the brain at P14 and P28 as indicated by Black Gold and MBP staining (Fig. 1B,C). Electron microscopy further demonstrated reduced number of myelinated axons in the optic nerve of Rheb1f/f;Olig1Cre+/− mice (Fig. 1D). In P28 controls, 58% of axons were myelinated, whereas only 25% of axons were myelinated in Rheb1f/f;Olig1Cre+/− (Fig. 1D). In addition, the proportion of small-diameter axons (<1.0 μm) was increased by 37%, whereas the large-diameter axons, particularly in the range of 1.0–1.5 μm, decreased by 26% (Fig. 1D), suggesting that the axon diameter might be reduced in the Rheb1f/f;Olig1Cre+/− mice. The g-ratio of the remaining myelinated axons in Rheb1f/f;Olig1Cre+/− was comparable to that in controls (data not shown). Consistent with hypomyelination in the brain, the mRNA levels of myelin genes including myelin basic protein (Mbp), 2′3′ cyclic nucleotide 3′-phosphodiesterase (Cnp), myelin oligodendrocyte glycoprotein (Mog), and Tmem10, were reduced in the cortex of Rheb1f/f;Olig1Cre+/− at P28 (Fig. 1E). Corresponding myelin proteins were diminished in the brain from P28 (Fig. 1F,G) to 8 months (Fig. 1H,I), indicating severe myelination deficit in the brain.
Figure 1.

Genetic deletion of Rheb1 in OL-lineage cells by Olig1-Cre causes hypomyelination in CNS. A, Immunostaining of phospho-S6 (red) and Olig2 (green) shows mTORC1 signaling reduced in the corpus callosum of Rheb1f/f; Olig1Cre+/− mouse. Scale bar, 40 μm. B, Black Gold staining of P28 Rheb1f/f; Olig1Cre+/− and control littermates (Rheb1f/+; Olig1Cre+/− or Rheb1+/+; Olig1Cre+/−). Arrows indicate the Black Gold-positive areas in control and comparable areas in mutant brain. Scale bar, 200 μm. Insets show higher magnification of the boxed regions. Scale bar, 100 μm. C, Immunostaining with MBP antibody shows reduction of MBP+ fibers (arrows) in the cortex (CTX) and striatum (STR) of P14 and P28 Rheb1f/f; Olig1Cre+/− mice. Scale bar, 200 μm. D, EMs from the optic nerve (ON) of P28 mice. Unmyelinated axons are indicated by red asterisks. Scale bars: 1 μm; insets, 20 μm. Average percentage of myelinated axons in P28 control and Rheb1f/f;Olig1Cre+/− (KO; graph 1). The distribution of axonal size in optic nerve at P28 (graph 2). Independent specimens from optic nerve were evaluated. Data represent mean ± SEM, n = 3, **p = 0.0055. E, Expression of Mbp, Mog, Tmem10, and Cnp in the cortex of P28 control and Rheb1f/f; Olig1Cre+/− mice assayed by quantitative real-time PCR. All quantifications are expressed as percentage control ± SEM. Mbp, **p = 0.0086; Mog,*p = 0.0141; Tmem10, **p = 0.0020; Cnp, **p = 0.0016. F–I, Western blot analysis of protein expression in isolated cortex of control and Rheb1f/f; Olig1Cre+/− mice at P28 (F, G) and 8 months (H, I). All quantifications are expressed as percentage control ± SEM. G, MBP, **p = 0.0073; MOG,*p = 0.0124; Tmem10, **p = 0.0011; CNP, **p = 0.006; pAKT, *p = 0.0410; pS6, p = 0.0509; Rheb1, *p = 0.0260. (I) MBP, **p = 0.0256; MOG,*p = 0.0222; Tmem10, ***p = 0.0004; CNP, **p = 0.0036; pAKT, *p = 0.0361; pS6, p = 0.2693.
Reduced oligodendrogenesis in brain of the Rheb1f/f;Olig1Cre+/− mouse
To determine whether the observed hypomyelination was a consequence of impaired OPC differentiation, we analyzed expression of cellular markers for OPCs and differentiated OLs. The results showed that the total number of OL lineage cells indicated by Olig2+ cells was not significantly reduced (p = 0.1038); however, the number of differentiated OLs expressing markers CC1 and Olig2 (CC1+/Olig2+) cells was reduced by 54% compared with control (Fig. 2A,B). In addition, expression of mature OL markers such as PLP and Tmem10 was diminished in the cortex of mutant at P14 and P28 (Fig. 2C–F). Tmem10 immunofluorescence reveals that the numbers of mature OLs and myelinated fibers are reduced in the cortex of Rheb1f/f;Olig1Cre+/− mice (Fig. 2E,F). In contrast to the reduction of differentiated OLs, the number of OPCs with PDGFRα+ was significantly increased (Fig. 2G,H). These results indicate that Rheb1 contributes to the regulation of OPC differentiation to mature OLs in the brain.
Figure 2.

Rheb1 ablation in OL lineage cells affects differentiation of OPCs. A, B, Immunostaining of CC1+/Olig2+ cells (arrows) in the cortex (CTX) of P28 Rheb1f/f; Olig1Cre+/− and control mice (Rheb1f/+; Olig1Cre+/− or Rheb1+/+; Olig1Cre+/−). B, Quantification of total OL lineage cells (Olig2+, red) and differentiated OLs (CC1+/Olig2+, yellow) in the cortex; n = 3, p = 0.1038; *p = 0.0111. C, D, In situ hybridization of Plp on the brain of control and Rheb1f/f; Olig1Cre+/− mice at P28. D, Quantification of Plp+ cells (arrows indicate cells in C) in the cortex; n = 3, **p = 0.0052. E, F, Immunostaining shows the reduction in the number of mature OLs (Tmem10+, arrows) in the cortex of P14 mutant Rheb1f/f; Olig1Cre+/−. F, Quantification of Tmem10+ cells in the cortex; n = 3, *p = 0.0117. G, PDGFRα as marker of OPC expression in the cortex of control and mutant at P28. H, Quantification of PDGFRα+ cells in the cortex; n = 4, **p = 0.0012. All of the quantifications are presented as mean ± SEM. Scale bars: A, C, E, G, 50 μm; insets of A and E, 20 μm. CC, corpus callosum.
Reduced oligodendrogenesis and myelination in brain of the Rheb1f/f;Olig2Cre+/− mouse
The effect of Rheb1 deletion on OPC differentiation in the cortex and corpus callosum was reproduced in another Rheb1 conditional deletion mouse (Fig. 3) that used Olig2-Cre (Schüller et al., 2008) driver, which starts to express Cre activity in the earliest stage of OL lineage cells, like Olig1-Cre. The severe impairment in oligodendrogenesis and myelination in Rheb1f/f;Olig1Cre+/− and Rheb1f/f;Olig2Cre+/− mice indicate that Rheb1 plays a crucial role in the differentiation of OPCs in the brain.
Figure 3.

Deletion of Rheb1 by Olig2-Cre causes hypomyelination and deficit of OPC differentiation in CNS. A, Immunostaining with MBP antibody shows reduction of MBP-positive fibers in the cingulum bundles (Cing) and cortex (CTX) of P28 Rheb1f/f;Olig2Cre+/− mice. Scale bar, 200 μm. B, Western blot analysis of protein expression in isolated cortex, hippocampus (HIP), and cerebellum (CB) of control and Rheb1f/f;Olig2Cre+/− mice at P28. Bottom, Quantification of protein levels (n = 5, *p < 0.05,**p < 0.01,***p < 0.0001, mean ± SEM). C, D, Immunofluorescence of CC1 and Olig2 double-positive cells as a marker of differentiated OLs (arrows) in the cortex of P28 Rheb1f/f;Olig2Cre+/− and control (Rheb1f/+; Olig2Cre+/− or Rheb1+/+; Olig2Cre+/−). Scale bar, 50 μm. D, Quantifications of total Olig2+ and CC1+/Olig2+ cells in the cortex (CTX); n = 3, *p = 0.0112, mean ± SEM. E, F, Immunofluorescence of PDGFRα as marker of OPCs (arrows) in the cortex of P28 Rheb1f/f;Olig2Cre+/− and control. Scale bar, 50 μm. F, Quantification of PDGFRα+ cells in the cortex; n = 3, *p = 0.0175, mean ± SEM.
Normal myelination in brain of the Rheb1f/f;CNPCre+/− mouse
To further investigate the role of Rheb1 for developing OLs to differentiate, we generated CNP-Cre (Lappe-Siefke et al., 2003)-mediated Rheb1 knock-out mice. Unlike Olig1 or Olig2, CNP is expressed in a subpopulation of OPCs and its expression is strongly induced early during OL differentiation (Yu et al., 1994; Baumann and Pham-Dinh, 2001). Excision of Rheb1 by CNP-Cre appeared successful since the phosphorylation of S6 was almost undetectable in the corpus callosum, consistent with the expression of Cre recombinant protein in Olig2+ cells (data not shown). Rheb1f/f;CNPCre+/− mice had a comparable number of CC1+/Olig2+ cells in the P14 and P28 corpus callosum and cortex, and the total number of OL lineage cells was not different from controls (Rheb1f/+;CNPCre+/− or Rheb1+/+;CNPCre+/−) in the corpus callosum and cortex (Fig. 4A,C; data not shown). The number of Plp+ OLs in the P14 cortex appeared to be modestly reduced, but was not different from controls in the corpus callosum (Fig. 4B,D). These results suggest that the differentiation of OPCs in the brain of Rheb1f/f;CNPCre+/− mouse is normal. Further, expression of mRNA and protein encoded by most myelin genes were not different in the cortex of P14 and P28 Rheb1f/f;CNPCre+/− mice (Fig. 4E–H). However, we noted reduced Plp mRNA and protein level in the cortex of Rheb1f/f;CNPCre+/−, suggesting that certain components of myelin were affected by the loss of Rheb1 in Rheb1f/f;CNPCre+/− mice. MBP staining showed no dramatic changes of myelin structure in the corpus callosum and cortex of Rheb1f/f;CNPCre+/− mice (Fig. 4I). Consistent with this result, electron microscopy analysis of the optic nerve at P28 revealed no difference in the number of myelinated axons or myelin thickness between Rheb1f/f;CNPCre+/− versus control mice (Fig. 4J). All the above results indicate that myelination in the brain of Rheb1f/f;CNPCre+/− mouse is not grossly affected.
Figure 4.
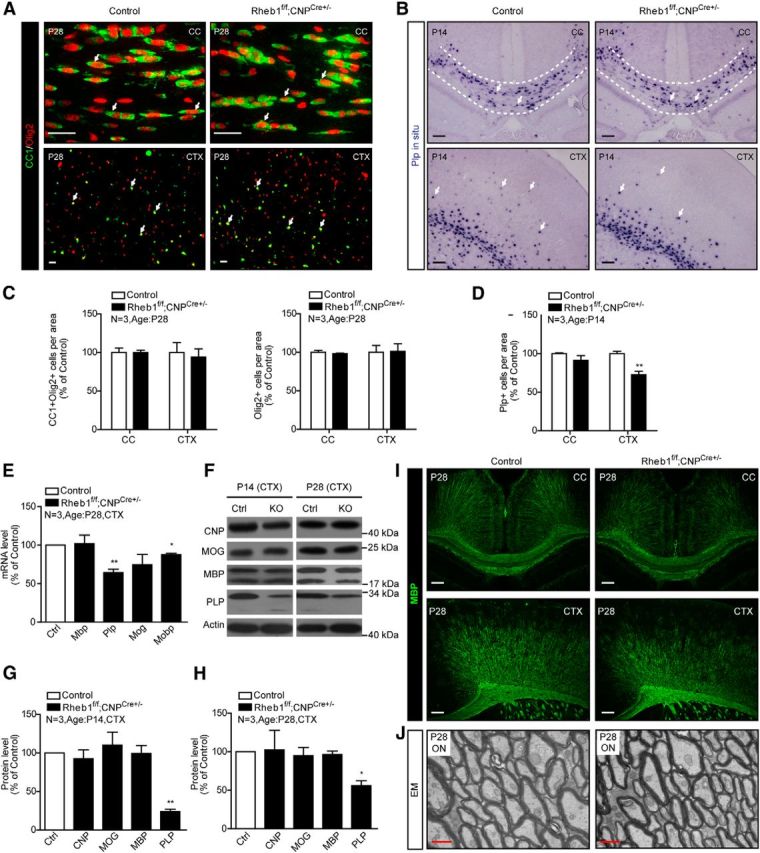
Loss of Rheb1 in Rheb1f/f; CNPCre+/− mice does not affect differentiation of OPCs but does affect certain components of myelin. A, Immunofluorescence of CC1+/Olig2+ cells (arrows) in the corpus callosum (CC) and cortex (CTX) of P28 control and Rheb1f/f;CNPCre+/− brains. Scale bar, 40 μm. B, In situ hybridization showing Plp+ cells (indicated by arrows) in brain of control (Rheb1f/+; CNPCre+/− or Rheb1+/+; CNPCre+/−) and Rheb1f/f;CNPCre+/− mice in the corpus callosum outlined by dashed lines and cortex at P14. Scale bar, 100 μm. C, Quantification of OL lineage cells (Olig2+) and differentiated OLs (CC1+/Olig2+) in the corpus callosum and cortex of P28 animals (mean ± SEM, n = 3). D, Quantification of Plp+ cells in the corpus callosum and cortex of Rheb1f/f;CNPCre+/− mice at P14 (mean ± SEM, n = 3,**p = 0.0061). E, mRNA expression of myelin genes in the cortex at P28 control and Rheb1f/f;CNPCre+/− mice (Mbp, p = 0.8836; Plp, **p = 0.0073; Mog, p = 0.2008; Mobp,*p = 0.0121; mean ± SEM, n = 3). F–H, Western blot analysis of myelin protein expression in the cortex of control and Rheb1f/f;CNPCre+/− mice at P14 (left panel of F, G) and P28 (right panel of F, H). All quantifications are expressed as percentage control ± SEM; **p = 0.0015, *p = 0.0201, n = 3. I, MBP staining reveals no dramatic reduction of MBP-positive fibers in the CC and CTX of Rheb1f/f;CNPCre+/− mice at P28. Scale bars: 100 μm. J, EMs from the optic nerve (ON) of control and Rheb1f/f;CNPCre+/− mice at P28. Scale bar, 500 nm.
Impairment in cell-cycle exit of brain OPCs by Rheb1 deletion
Because mTor plays a role in the regulation of cell cycle (Fingar et al., 2004), we wondered if the impairment in OPC differentiation in Rheb1f/f;Olig1Cre+/− mouse was a result of impairment in cell-cycle exit of OPCs. We first examined if the proliferating capacity of OPCs was affected by the loss of Rheb1. Toward this goal, we labeled early postnatal Rheb1f/f;Olig1Cre+/− and littermate control (Rheb1f/+;CNPCre+/− or Rheb1+/+;CNPCre+/−) mice (P4) with BrdU (100 mg/kg) for 2 h and found the number of proliferating OPCs (BrdU+/Olig2+) was normal compared with that of controls (Fig. 5A,B). This suggests that Rheb1 is not required for the proliferation of OPCs. To test the possibility that loss of Rheb1 impairs cell-cycle exit of OPCs, we measured the cell-cycle exit index (Sanada and Tsai, 2005). BrdU was injected into control and Rheb1f/f; Olig1Cre+/− mice at P4. After 24–36 h, brains were collected and analyzed by immunohistochemistry with anti-BrdU and anti-Ki67 (a marker for cells in active phases of the cell cycle—G1, S, G2, M) antibodies (Fig. 5C). The total number of Ki67+ cells was not altered (Fig. 5D). Because >70% of BrdU+ cells in the normal adult brain and >90% in the corpus callosum are OPCs (Horner et al., 2000; Dawson et al., 2003; data not shown), we counted the number of BrdU+/Ki67− cells in the corpus callosum to analyze the cell-cycle exit of OPCs. Results show that 85% of OPCs exited the cell cycle in the corpus callosum of controls, whereas only 65% of progenitors exited the cell cycle in the comparable region of Rheb1f/f;Olig1Cre+/− mice (Fig. 5D; *p = 0.0258). Rapamycin inhibits the G2/M progression in cell cycle of cultured OPCs (Min et al., 2012). Accordingly we colabeled mitotic marker phospho-histone3 (S10) [P-H3] and Olig2 of cycling OPCs in M-phase in P4 Rheb1f/f;Olig1Cre+/− mice, and found that the number of P-H3+/Olig2+ cells was reduced in the corpus callosum of Rheb1f/f;Olig1Cre+/− mice, compared with that in controls (Fig. 5E,F). The reduction in the number of P-H3+/Olig2+ cells in the corpus callosum is indicative of impaired G2/M progression in cell cycle. The above results suggest that Rheb1 contributes to the regulation of G2/M progression in cycling OPCs, such that deletion of Rheb1 in OPCs impairs their cycle exit and thus OPC differentiation.
Figure 5.
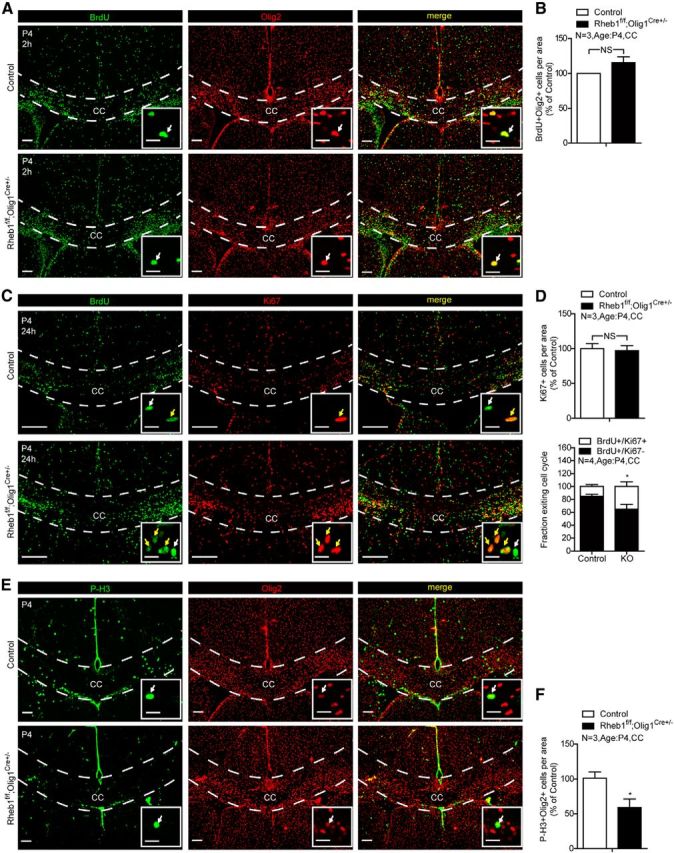
Deletion of Rheb1 in OPCs affects the cell-cycle exit. A, Rheb1f/f;Olig1Cre+/− or Rheb1f/+;Olig1Cre+/− (control) mice at P4 were labeled with BrdU (100 mg/kg) for 2 h. Proliferating OPCs were BrdU and Olig2 double-positive cells (arrows). Scale bars: 200 μm; insets, 20 μm. B, Quantification of BrdU+ and Olig2+ colabeled cells per area at P4 in the corpus callosum (CC; dashed line area of A) of control and mutant (n = 3, p = 0.2010, mean ± SEM). C, BrdU was injected into mice intraperitoneally at P4. Mice were killed after 24–36 h. OPCs labeled with BrdU but not Ki67 are those that exit the cell cycle (white arrows). OPCs labeled with both BrdU and Ki67 remain in the cell cycle (yellow arrows). Scale bars: 200 μm; insets, 20 μm. D, Quantification of total Ki67+ cells in the corpus callosum showed comparable number of OPCs in the cell cycle (top graph). The cell-cycle exit index is measured as the percentage of the OPCs in the corpus callosum (dashed line areas) that exited the cell cycle (BrdU+Ki67−) divided by total BrdU-positive (BrdU+) cells (bottom graph;*p = 0.0258, n = 4, mean ± SEM). E, F, Immunostaining of P4 control and Rheb1f/f;Olig1Cre+/− brains with phospho-histone3(S10) [P-H3] and Olig2 antibodies. F, Quantification of P-H3 and Olig2 double-positive cells shows reduction of M-phase OPCs in the corpus callosum of Rheb1f/f;Olig1Cre+/− mice (n = 3, *p = 0.0197, mean ± SEM).
Astrocyte formation in the brain not affected by Rheb1 deletion
Because NG2+ OPCs are shown to generate both OLs and gray mater astrocytes (Zhu et al., 2008), which can share a common progenitor in certain tissues (Rompani and Cepko, 2010), we examined if Rheb1 deletion reduces the formation of gray matter astrocytes, which may affect myelination in the cortex. Because GFAP antibody does not label gray matter astrocytes in the cortex, we generated Aldh1L1 antibody that recognizes both gray and white matter astrocytes in the brain; therefore, Aldh1L1 is a pan-astrocyte marker (Cahoy et al., 2008). The Aldh1L1 antibody readily detected a band of expected 99 kDa protein in multiple brain tissues, which was absent in Aldh1L1 knock-out tissues (Fig. 6A,B). Immunostaining shows that it labels both gray matter astrocytes in the cortex and white matter astrocytes in corpus callosum (Fig. 6C; data not shown). In white matter, it labels the same astrocytes as GFAP antibody labels (data not shown). Using Aldh1L1 antibody, we found that the number of gray matter astrocytes in the cortex was normal in Rheb1f/f;Olig1Cre+/− mouse (Fig. 6D,F). To further examine the effect of Rheb1 on astrocyte formation, we assayed the number of astrocytes in the brain of Rheb1f/f;NestinCre+/− mouse, where Rheb1 was deleted in neural progenitors that give rise to neurons and glia. The results indicate Rheb1 is not essential for astrocyte formation (Fig. 6E,F).
Figure 6.

Astrocyte formation is not affected by Rheb1 deletion. A, Western blotting analysis of Aldh1L1 expression in P28 cortex (CTX) and cerebellum (CB) of wild-type mouse. B, Western blots showing that Aldh1L1 immunoreactivity is absent in extracts of P28 Aldh1L1 knock-out brain. C, Immunostaining of wild-type mouse brain with Aldh1L1 (green) and GFAP (red) antibody. Scale bar, 200 μm. Insets show higher magnification of the boxed regions. Scale bar, 50 μm. D–F, Immunostaining of control (Rheb1f/+; Olig1Cre+/−) and Rheb1f/f;Olig1Cre+/− (D) and control (Rheb1f/+; NestinCre+/−) and Rheb1f/f;NestinCre+/− (E) staining of brain sections with Aldh1L1 antibody. Scale bar, 200 μm. F, Quantification of Aldh1L1+ cells in the cortex shows comparable number of astrocytes in mutants and controls. (Data represent mean ± SEM, n = 3.)
mTor deletion in OPCs phenocopies Rheb1 knock-out in the brain
mTor functions in two complexes—mTORC1 and mTORC2 (Sarbassov et al., 2005a, b; Jacinto et al., 2006). Our previous work confirmed that Rheb1 is required for mTORC1 activity in vivo (Zou et al., 2011). To examine if mTor mediates the effect of Rheb1 on OL differentiation and myelination in the brain, we generated mTor conditional knock-out mice by breeding mTorf/f (Gangloff et al., 2004) with the Olig1-Cre deleter mouse. mTorf/f;Olig1Cre+/− mice were viable and gained body weight and brain size similar to littermate controls (mTorf/+;Olig1Cre+/− or mTor+/+;Olig1Cre+/−). Downstream targets of mTORC1, such as pS6 and p4EBP, were reduced in the cortex and hippocampus of mTorf/f;Olig1Cre+/− mice (Fig. 7D,E). Similar to Rheb1f/f;Olig1Cre+/− mice, mTorf/f;Olig1Cre+/− mice showed reduced Black Gold-positive myelinated fibers in the cortex, corpus callosum, and striatum in the brain at P28 (Fig. 7A). MBP and Tmem10 staining similarly revealed lack of myelin in the cortical layers, cingulum bundles, and corpus callosum (Fig. 7B,C). Western blots and quantifications confirmed reduction of myelin proteins in the cortex and hippocampus of mTorf/f;Olig1Cre+/− mice (Fig. 7D,E). Consistent with the reduction in myelin proteins, mRNA expression of myelin genes was significantly reduced (Fig. 7F). These observations indicate mTorf/f;Olig1Cre+/− phenocopies Olig1-Cre-mediated Rheb1 deletion mice.
Figure 7.

Hypomyelination in CNS of mTorf/f;Olig1Cre+/− mice. A, Black Gold staining of P28 control (mTorf/+; Olig1Cre+/− or mTor+/+; Olig1Cre+/−) and mTorf/f;Olig1Cre+/− brains. Arrows indicate the Black Gold-positive areas in control and comparable areas in mutant brain. Scale bar, 200 μm. Insets show higher magnification of the boxed regions. Scale bar, 100 μm. B, Immunostaining of control and mTorf/f;Olig1Cre+/− brains with MBP antibody. Arrows indicate the MBP-positive areas in control and comparable areas in mutant brain. Scale bar, 50 μm. C, Immunostaining of control and mTorf/f;Olig1Cre+/− brains with Tmem10 antibody. Arrows indicate the Tmem10-positive fibers in the cingulum bundles (Cing; arrows indicate areas, top) and corpus callosum (CC; dashed line areas, bottom) of control and mutant. Scale bar, 50 μm. D, E, Western blot analysis of myelin protein expression in the cortex (CTX) and hippocampus (HIP) of control (mTorf/+; Olig1Cre+/− or mTor+/+; Olig1Cre+/−) and mTorf/f;Olig1Cre+/− mice at P21. All quantifications are expressed as percentage control ± SEM, n = 3, MBP, *p = 0.0270; MOG,*p = 0.0317; Tmem10, **p = 0.0051; CNP, *p = 0.0398; p4EBP, *p = 0.0330; pS6, *p = 0.0191. F, mRNA expression of myelin genes in the cortex of P21 mTorf/+; Olig1Cre+/− and mTorf/f;Olig1Cre+/− assayed by quantitative real-time PCR. Mbp, **p = 0.0058; Mog, **p = 0.0040; Tmem10, ***p = 0.0009; Cnp, *p = 0.0320; Mag, **p = 0.0059.
To determine whether the observed hypomyelination in mTorf/f;Olig1Cre+/− mice was also a consequence of impaired OPC differentiation, we analyzed the numbers of differentiated OLs (CC1+/Olig2+) and OPCs (PDGFRα+/NG2+) in mTorf/f;Olig1Cre+/− mice (Fig. 8A–E). Overall, the number of Olig2+ cells was comparable in the cortex and slightly reduced in the corpus callosum (*p = 0.0389) compared with control (mTorf/+;Olig1Cre+/−). There was a significant decrease in the number of OLs identified as CC1+/Olig2+ cells in the cortex and corpus callosum (Fig. 8A; quantified in B; **p = 0.0096, ***p = 0.0007), Conversely, the number of OPCs indicated by PDGFRα and NG2 was increased in the cortex of mTorf/f;Olig1Cre+/− mice (Fig. 8C,E; **p = 0.0068). These results confirm impaired OPC differentiation in mTorf/f;Olig1Cre+/− mice. Similar to Rheb1f/f;Olig1Cre+/− mice, the cell-cycle exit of OPCs was impaired in mTorf/f;Olig1Cre+/− mice; we observed ∼50% reduction of M-phase OPCs in the corpus callosum of mTorf/f;Olig1Cre+/− brain (Fig. 8D,F; *p = 0.0229). These data support the notion that Rheb1/mTor controls myelination in the brain by regulating the differentiation OPCs to become OLs in the brain.
Figure 8.

Deletion of mTor by Olig1-Cre affects differentiation of OPCs. A, Immunostaining of differentiated OLs (CC1+/Olig2+ cells; arrows) in the cortex (CTX) and corpus callosum (CC) of P28 control (mTorf/+; Olig1Cre+/− or mTor+/+; Olig1Cre+/−) and mTorf/f;Olig1Cre+/− mice. Scale bar, 40 μm. B, Quantification of total Olig2+ and CC1+/Olig2+ cells in the cortex and corpus callosum. (Data represent mean ± SEM, n = 3, *p = 0.0389, **p = 0.0096, ***p = 0.0007.) C, Immunostaining of OPCs (PDGFRα+/NG2+) in the cortex of P28 mTorf/f;Olig1Cre+/− brain. Arrows indicate the PDGFRα/NG2 double-positive cells. Scale bar, 20 μm. D, Immunostaining of P4 control and mTorf/f;Olig1Cre+/− brains with phospho-histone3 (S10) [PH3] and Olig2 antibodies shows reduction of M-phase OPCs in the corpus callosum of mTorf/f;Olig1Cre+/− mice. E, F, Quantification of PDGFRα+/NG2+ and P-H3+/Olig2+ cells. (Data represent mean ± SEM, n = 3, **p = 0.0068, *p = 0.0458.)
Myelin maintenance in the brain not affected by Rheb1 deletion in mature OLs
A previous study using CNP-Cre deletion of Raptor suggested that mTORC1 may be important for maintenance of myelin (Bercury et al., 2014). As noted, CNP-Cre is expressed early in OL differentiation, which leaves open the possibility that it impacts OL differentiation independently of myelin maintenance. To address this question, we generated a Tmem10-Cre knockin mouse in which Cre cDNA was inserted into the open-reading frame of Tmem10. Tmem10-Cre activity was expressed in fully matured OLs, not in OPCs, astrocytes, or microglia as assessed by visualizing tdTomato (tdT) reporter (Madisen et al., 2010) in the cortex of Rosa26tdTomato;Tmem10Cre+/− mice (Fig. 9A). The pattern of Tmem10-Cre-mediated tdT is consistent with the expression pattern of native Tmem10 protein (Golan et al., 2008). We found that Rheb1f/f;Tmem10Cre+/− mice have a comparable number of total Olig2+ and OL (CC1+/Olig2+) cells in the cortex and corpus callosum at P28 as controls (Rheb1f/+;Tmem10Cre+/− or Rheb1+/+;Tmem10Cre+/−; Fig. 9B,C). This indicates that Rheb1 is not required for survival of mature OLs. To determine whether the expression of myelin proteins and myelin maintenance were affected in Rheb1f/f;Tmem10Cre+/− mice, we examined myelin proteins by Western blotting and myelin structure by electron microscopy at different time points compared with control mice. Representative Western blots and quantification revealed comparable expression of myelin proteins at P28 and 6 months (Fig. 9D,E). MBP staining also revealed comparable patterns of myelin fibers in the cortex at P28 (Fig. 9F). No difference in myelin structure was observed in 5 month optic nerves between Rheb1f/f;Tmem10Cre+/− and control mice (Fig. 9G,H). These results indicate that Rheb1 expression in mature OLs is not required for the synthesis or maintenance of myelin.
Figure 9.

Rheb1 is not required for myelin maintenance. A, tdTomato reporter of Tmem10-Cre activity in the cortex (CTX) at P28 revealed tdTomato reporter only expressed in (CC1+) OLs, but not in (PDGFRα+) OPCs, (GFAP+) astrocytes, or (Iba-1+) microglia. Scale bar, 50 μm. B, Immunofluorescence of differentiated OLs (CC1+/Olig2+; arrows) in the cortex and corpus callosum (CC) of P28 Rheb1f/f;Tmem10Cre+/− and Rheb1f/+;Tmem10Cre+/− (control) mice. Scale bars: 20 μm. C, Quantification of CC1+/Olig2+ cells in the cortex and corpus callosum, mean ± SEM, n = 3. D, E, Western blot analysis of myelin protein expression in the cortex of control (Rheb1f/+;Tmem10Cre+/− or Rheb1+/+;Tmem10Cre+/−) and Rheb1f/f;Tmem10Cre+/− mice at P28 (left, D and E) and 6 months (right, D and E). All quantifications are expressed as percentage control ± SEM. n = 3, p > 0.1. F, Normal myelination in the cortex of P28 Rheb1f/f;Tmem10Cre+/− mice revealed by MBP immunostaining. Scale bars: 100 μm. G, EMs from the optic nerve (ON) of control and Rheb1f/f;Tmem10Cre+/− mice at 5 months. Scale bars: 1 μm. H, Average percentage of myelinated axons in the optic nerve of 5 month controls and Rheb1f/f;Tmem10Cre+/− mutants. Independent specimens from optic nerve were evaluated. Data represent mean ± SEM, n = 3.
Hypomyelination and reduced oligodendrogenesis in the spinal cord of Rheb1f/f;Olig1Cre+/− and Rheb1f/f;CNPCre+/− mice
Because of differential effects of CNP-Cre-mediated deletion of Raptor or mTor deletion on brain and spinal cord myelination (Bercury et al., 2014; Wahl et al., 2014), we examined if similar regional effect exists in Rheb1f/f;CNPCre+/− mice, in comparison with Olig1-Cre-mediated Rheb1 deletion. Black Gold staining of myelin was reduced in P28 spinal cords of both Rheb1f/f;Olig1Cre+/− and Rheb1f/f;CNPCre+/− mice. Myelinated fibers were modestly reduced in gray matter of Rheb1f/f;CNPCre+/− mice but absent in Rheb1f/f;Olig1Cre+/− mice (Fig. 10A,B). In addition, we found that the levels of myelin proteins were consistently reduced in the spinal cords of both Rheb1f/f;Olig1Cre+/− and Rheb1f/f;CNPCre+/− mice (Fig. 10C,D). At P14, the reduction in myelin proteins was comparable between these two Rheb1 KO lines. At P28, myelin proteins remained reduced by ∼50% in the spinal cord of Rheb1f/f;Olig1Cre+/− mice, compared with∼30% reduction in Rheb1f/f;CNPCre+/− mice. These results indicate hypomyelination in the spinal cord of both Rheb1f/f;Olig1Cre+/− and Rheb1f/f;CNPCre+/− mice, with a more severe reduction in Rheb1f/f;Olig1Cre+/−. Similar to the hypomyelination in the brain of Rheb1f/f;Olig1Cre+/− mice, hypomyelination in the spinal cord appears to be caused by impairment in oligodendrogenesis since the number of differentiated OLs (Olig2+/CC1+) was reduced in both P14 and P28 spinal cord of Rheb1f/f;Olig1Cre+/− mice (Fig. 10E). The total number of OL lineage cells (Olig2+) was slightly reduced at P14 and P28, but not statistically significant. In the Rheb1f/f;CNPCre+/− mouse, we found that differentiated OLs were reduced in P14 but recovered in P28 spinal cord (Fig. 10F). The total number of OLs was comparable to controls in both P14 and P28 spinal cord of Rheb1f/f;CNPCre+/−. These data indicated that deletion of Rheb1 in OPCs or OLs affects spinal cord OL differentiation and myelination. In addition, the effects of Rheb1 deletion from Olig1-Cre were more severe than CNP-Cre deletion in the spinal cord.
Figure 10.
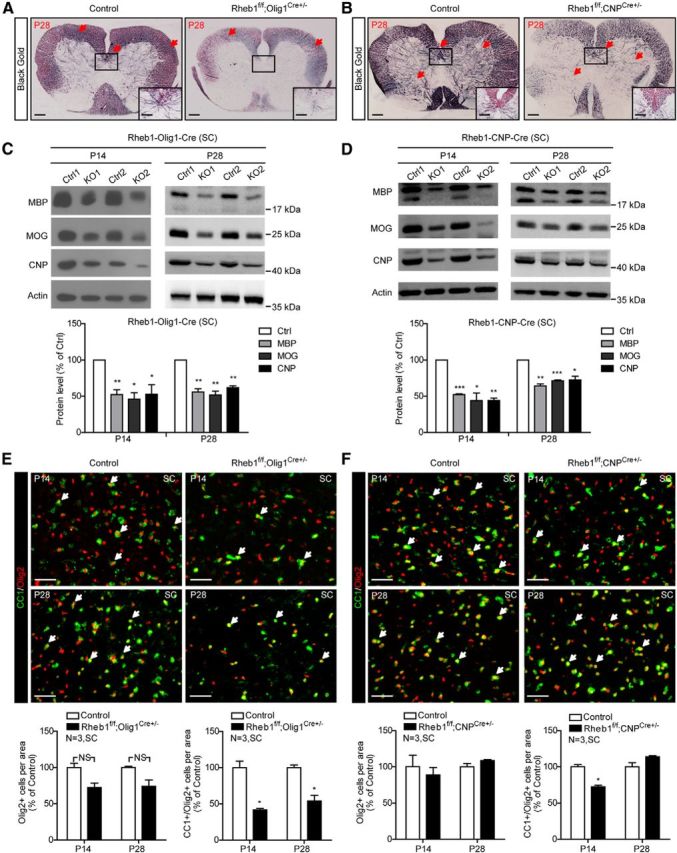
Myelin deficit in the spinal cord of Rheb1f/f;Olig1Cre+/− and Rheb1f/f;CNPCre+/− mice. A, B, Black Gold staining of P28 control (Rheb1f/+;Olig1Cre+/−) and Rheb1f/f; Olig1Cre+/− (A) and control (Rheb1f/+;CNPCre+/−) and Rheb1f/f; CNPCre+/− (B) mice. Arrows indicate the Black Gold-positive areas in control and comparable areas in mutant brain. Scale bar, 200 μm. Insets show higher magnification of the boxed regions. Scale bar, 100 μm. C, D, Western blot analysis of myelin protein expression in the spinal cord (SC) of Rheb1f/f; Olig1Cre+/− (C) and Rheb1f/f; CNPCre+/− mice (D) at P14 and P28. All quantifications are expressed as percentage control ± SEM. C, P14: MBP, **p = 0.0096; MOG,*p = 0.0132; CNP, *p = 0.0356; P28: MBP, **p = 0.0049; MOG, **p = 0.0055; CNP, **p = 0.0022. D, P14: MBP, ***p = 0.0003; MOG,*p = 0.0164; CNP, **p = 0.0020; P28: MBP, **p = 0.0029; MOG, ***p = 0.0004; CNP, *p = 0.0170. E, F, Immunostaining of CC1+/Olig2+ cells (arrows) in the spinal cord of Rheb1f/f;Olig1Cre+/− (E) and Rheb1f/f;CNPCre+/− (F) mutants at P14 and P28. Quantification of total Olig2+ (OL lineage cells) and CC1+/Olig2+ (differentiated OLs) in the spinal cord are shown (bottom). E, P14, *p = 0.0254; P28,*p = 0.0102; (F) P14, *p = 0.0271. Scale bar, 40 μm.
Discussion
Rheb1/mTor regulates early stage differentiation of OPCs in the brain and spinal cord
Our study reveals that Rheb1 and mTor function in OPCs to regulate early stage OPC differentiation in the brain and spinal cord. Rheb1 is not required in mature OLs to make or maintain myelin. Rheb1/mTor-dependent defects in CNS myelination appear to be caused by failure to generate sufficient numbers of mature OLs, which are the source of myelin. The observation that Rheb1/mTor plays an essential yet selective role in OPCs to control their maturation identifies a critical nodal point in myelin biology.
Our conclusions are based on comparisons of four Cre driver lines that span the OPC to mature OL lineage. Olig1-Cre and Olig2-Cre are the earliest, and Rheb1 excision in OPCs with these drivers markedly reduces the formation of OLs in the brain without disrupting OPC proliferation. The effect of Rheb1 on early stage OPC differentiation is mediated by mTor, because mTor deletion by Olig1-Cre leads to a similar degree of reduction in mature OLs and myelination in the brain as in Rheb1f/f;Olig1Cre+/− mouse. In contrast, CNP-Cre-mediated deletion of Rheb1 reduces OL formation and myelination selectively in the spinal cord, with minimal effects in the brain. This regional difference is consistent with reported results of CNP-Cre-mediated mTor and Raptor deletion (Bercury et al., 2014; Wahl et al., 2014). One possible reason for this regional effect might be related to differential expression of CNP-Cre in subpopulations of OPCs. In the spinal cord, ventrally derived OPCs are generated in early waves of oligodendrogenesis and account for 85% OPCs in the spinal cord (Yu et al., 1994). In the forebrain, ventrally derived OPCs and OLs are replaced by more dorsal precursors with each OPC pool contributing ∼50% (Kessaris et al., 2006; Richardson et al., 2006). Accordingly, differences between spinal cord and brain might be explained if floxed Rheb1 (or mTor/Raptor) were not be recombined as efficiently in the cortex/dorsal-derived OPCs. The differential action of these Cre drivers suggests that there is a narrow developmental time window in early stage differentiation of OPCs in brain during which Rheb1/mTor function is required.
Rheb1/mTORC1 is not required for myelin gene/protein expression and myelin maintenance
Tmem10-Cre is expressed in mature OLs and cleanly excludes a requirement for Rheb1 in myelination or myelin gene expression in these cells. Results from the Tmem10-Cre-mediated Rheb1 knock-out mouse indicate that mTor (TORC1)-dependent regulation of OPC differentiation is not through the control of myelin gene transcription or myelin protein synthesis. Regulated expression of myelin genes is an important aspect of myelin formation, and transcription of multiple myelin genes is upregulated in close correlation with OPC maturation. The finding that rapamycin elevates the expression of negative transcription factors of OL differentiation suggests that mTORC1 regulates OL differentiation through transcriptional control of early stage OPC differentiation (Wood et al., 2013). In support of this model, knockdown of mTORC1 activity by means of siRNA to Raptor reduces MBP protein in cultured OLs and suggests that mTORC1 may regulate OPC differentiation in a coordinated or closely hierarchical fashion with myelin protein synthesis (Tyler et al., 2009). These in vitro effects of mTORC1 on OL differentiation are consistent with the well documented role of mTORC1 in gene transcription (H. Li et al., 2006; Chaveroux et al., 2013) and protein synthesis (Bilanges et al., 2007; Thoreen et al., 2012) in other cell types. However, deletion of Rheb1 in mature OLs by Tmem10-Cre driver does not affect the number of mature OLs or expression of myelin gene mRNAs and proteins. The normal number of mature OLs and expression of mRNA and protein of myelin genes and myelination in the brain of Rheb1f/f;Tmem10Cre+/− mice as old as 6 months suggests that Rheb1/mTORC1 is not essential for the sustained expression of myelin genes, myelin turnover, or maintenance. This highlights the notion that Rheb1/mTORC1-dependent OPC differentiation to OL is a distinct process from control of myelin gene expression. The observation that Rheb1 is not required in mature OLs for myelin expression is surprising since mTORC1 plays an integrative role in energy metabolism and lipid synthesis (Cunningham et al., 2007; Peterson et al., 2011), both of which are required for myelin membrane synthesis (Stevens et al., 2002; Verheijen et al., 2009; Rinholm et al., 2011; Wake et al., 2011).
A recent study shows a modest increase in g-ratio of spinal cord axons of Raptor knock-out mouse, suggesting a modest impairment in myelin maintenance and more pronounced increase in g-ratio in Raptor/Rictor double knock-out mouse, one year after the deletion of corresponding genes in mature OLs (Lebrun-Julien et al., 2014). The enhanced impairment in myelin maintenance may suggest an involvement of mTORC2 activity in myelin maintenance. mTORC2 activity as indicated in pAKT (S473) was found to be increased in the brain of Rheb1 knock-out mouse, which may also be attributable to normal myelin maintenance.
Rheb1/mTORC1 regulates cell-cycle exit of OPCs
The effect of mTORC1 activity on OPC differentiation is likely through the control of cell-cycle exit, because reduced cell-cycle exit of OPCs is present both in Olig1-Cre-mediated Rheb1 and mTor knock-out mice. The reduced cell-cycle exit of OPCs seems to be due to impairment in G2/M progression. The effect of Rheb1 deletion on G2/M progression is consistent with previous report showing that inhibition of mTORC1 activity by rapamycin impairs growth factor-stimulated G2/M progression of cultured OPCs (Min et al., 2012). The notion that Rheb1/mTORC1 regulates OPC differentiation through its effect on cell-cycle exit is also supported by other studies. Magri et al. (2014) reported that the time course of cell-cycle exit coincides with the initiation of differentiation toward myelinating phenotypes, i.e., expression of myelin genes and the decline in the cell-cycle regulator–E2F1 transcription factor, while genetic ablation of the inhibitor gene coding for cyclin-dependent kinase p27Kip1 impairs the differentiation of OPCs to mature OLs (Casaccia-Bonnefil et al., 1997). Our results indicate that OPCs are capable of proliferation in absence of Rheb1/mTor since OPC numbers increase simultaneously with reduced OL generation. The remarkably restricted requirement for mTORC1 is consistent with previous observations. For example, Rheb1 deletion in neural progenitor cells using Nestin-Cre does not prevent formation of neurons, astrocytes, or OPCs (Zou et al., 2011), nor is it required for Schwann cell formation in the peripheral nervous system (data not shown). Similar findings are noted for non-neural cell types, such as T-lineage lymphocytes, where Rheb1/mTORC1 selectively affects the formation of a subtype of T-lymphocytes (Delgoffe et al., 2009). Thus, while mTORC1 can regulate many aspects of cellular function, it appears to be required for rather distinct aspects of cell biology that varies between cell types. This may rationalize the therapeutic value of modifiers of the mTor pathway.
In summary, our analysis of OL lineage Rheb1 and mTor knock-out mice indicates that OL-intrinsic mTORC1 activity regulated by Rheb1 plays a critical role in the formation of mature OLs, which in turn are response for generation of myelin. There is a narrow time window during which mTORC1 is essential for early stage differentiation of OPCs. Rheb1/mTORC1 is not critical for the expression of most myelin genes in differentiated OLs or essential for the maintenance of myelin in the brain. Our study clarifies the essential role of OPC-intrinsic Rheb1/mTORC1 activity in myelin program in the brain.
Footnotes
This work was supported by grants from the National Natural Science Foundation of China (31371484 to B.X.), National Basic Research Program of China (2009CB941400 to B.X.) and the National Institutes of Health (5P50DA000266-42 to P.F.W.), and by the National Basic Research Program of China (2010CB529900). We thank the Transgenic Facility of Johns Hopkins University School of Medicine for assistance with the generation of Tmem10 and Aldh1L1 knock-out/knockin mouse models, Klaus-Armin Nave for providing CNP-cre mice, and Guo Yang of the Electron Microcopy Facility of West China School of Preclinical and Forensic Medicine for assistance with the electron microscopy.
The authors declare no competing financial interests.
References
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL, Macklin WB. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J Neurosci. 2014;34:4466–4480. doi: 10.1523/JNEUROSCI.4314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilanges B, Argonza-Barrett R, Kolesnichenko M, Skinner C, Nair M, Chen M, Stokoe D. Tuberous sclerosis complex proteins 1 and 2 control serum-dependent translation in a TOP-dependent and -independent manner. Mol Cell Biol. 2007;27:5746–5764. doi: 10.1128/MCB.02136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Jr, Chao MV, Koff A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev. 1997;11:2335–2346. doi: 10.1101/gad.11.18.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaveroux C, Eichner LJ, Dufour CR, Shatnawi A, Khoutorsky A, Bourque G, Sonenberg N, Giguère V. Molecular and genetic crosstalks between mTOR and ERRalpha are key determinants of rapamycin-induced nonalcoholic fatty liver. Cell Metab. 2013;17:586–598. doi: 10.1016/j.cmet.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1 alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/S1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24:9508–9516. doi: 10.1128/MCB.24.21.9508-9516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan N, Adamsky K, Kartvelishvily E, Brockschnieder D, Möbius W, Spiegel I, Roth AD, Thomson CE, Rechavi G, Peles E. Identification of Tmem10/Opalin as an oligodendrocyte enriched gene using expression profiling combined with genetic cell ablation. Glia. 2008;56:1176–1186. doi: 10.1002/glia.20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiola-Diaz HM, Ishii A, Bansal R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia. 2012;60:476–486. doi: 10.1002/glia.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci. 2000;20:2218–2228. doi: 10.1523/JNEUROSCI.20-06-02218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jiang W, Yang W, Zhang J, Pang D, Gan L, Luo L, Fan Y, Liu Y, Chen M. Identification of Tmem10 as a novel late-stage oligodendrocytes marker for detecting hypomyelination. Int J Biol Sci. 2013;10:33–42. doi: 10.7150/ijbs.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F, Bachmann L, Norrmén C, Trötzmuller M, Köfeler H, Rüegg MA, Hall MN, Suter U. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci. 2014;34:8432–8448. doi: 10.1523/JNEUROSCI.1105-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tsang CK, Watkins M, Bertram PG, Zheng XF. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature. 2006;442:1058–1061. doi: 10.1038/nature05020. [DOI] [PubMed] [Google Scholar]
- Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29:32–38. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/S0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri L, Swiss VA, Jablonska B, Lei L, Pedre X, Walsh M, Zhang W, Gallo V, Canoll P, Casaccia P. E2F1 coregulates cell cycle genes and chromatin components during the transition of oligodendrocyte progenitors from proliferation to differentiation. J Neurosci. 2014;34:1481–1493. doi: 10.1523/JNEUROSCI.2840-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Singh S, Fitzgerald-Bocarsly P, Wood TL. Insulin-like growth factor I regulates G2/M progression through mammalian target of rapamycin signaling in oligodendrocyte progenitors. Glia. 2012;60:1684–1695. doi: 10.1002/glia.22387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci. 2006;7:11–18. doi: 10.1038/nrn1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci. 2011;31:538–548. doi: 10.1523/JNEUROSCI.3516-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompani SB, Cepko CL. A common progenitor for retinal astrocytes and oligodendrocytes. J Neurosci. 2010;30:4970–4980. doi: 10.1523/JNEUROSCI.3456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada K, Tsai LH. G protein betagamma subunits and AGS3 control spindle orientation and asymmetric cell fate of cerebral cortical progenitors. Cell. 2005;122:119–131. doi: 10.1016/j.cell.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005a;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005b;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855–868. doi: 10.1016/S0896-6273(02)01067-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, Wood TL. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29:6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen MH, Camargo N, Verdier V, Nadra K, de Preux Charles AS, Médard JJ, Luoma A, Crowther M, Inouye H, Shimano H, Chen S, Brouwers JF, Helms JB, Feltri ML, Wrabetz L, Kirschner D, Chrast R, Smit AB. SCAP is required for timely and proper myelin membrane synthesis. Proc Natl Acad Sci U S A. 2009;106:21383–21388. doi: 10.1073/pnas.0905633106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci. 2014;34:4453–4465. doi: 10.1523/JNEUROSCI.4311-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TL, Bercury KK, Cifelli SE, Mursch LE, Min J, Dai J, Macklin WB. mTOR: a link from the extracellular milieu to transcriptional regulation of oligodendrocyte development. ASN Neuro. 2013;5:e00108. doi: 10.1042/AN20120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Yue T, Ma Z, Wu FF, Gow A, Lu QR. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J Neurosci. 2005;25:1354–1365. doi: 10.1523/JNEUROSCI.3034-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, Worley PF. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem. 1994;269:16333–16339. [PubMed] [Google Scholar]
- Yu WP, Collarini EJ, Pringle NP, Richardson WD. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 1994;12:1353–1362. doi: 10.1016/0896-6273(94)90450-2. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhou L, Du XX, Ji Y, Xu J, Tian J, Jiang W, Zou Y, Yu S, Gan L, Luo M, Yang Q, Cui Y, Yang W, Xia X, Chen M, Zhao X, Shen Y, Chen PY, Worley PF, et al. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev Cell. 2011;20:97–108. doi: 10.1016/j.devcel.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]