

Abstract
Ampakines are allosteric modulators of AMPA receptors that facilitate hippocampal long-term potentiation (LTP) and learning, and have been considered for the treatment of cognition and memory deficits. Here, we show that the ampakine CX546 raises the amplitude and slows the decay time of excitatory postsynaptic currents (EPSCs) at cerebellar parallel fiber (PF) to Purkinje cell synapses, thus resembling CX546 effects described at hippocampal synapses. Using the fluorescent calcium indicator dye Oregon Green BAPTA-2 and an ultra-high-speed CCD camera, we also monitored calcium transients in Purkinje cell dendrites. In the presence of CX546 in the bath, PF-evoked calcium transients were enhanced and prolonged, suggesting that CX546 not only enhances synaptic transmission, but also boosts dendritic calcium signaling at cerebellar synapses. In contrast to previous observations in the hippocampus, however, CX546 applied during cerebellar recordings facilitates long-term depression (LTD) rather than LTP at PF synapses. These findings show that ampakines selectively modify the LTP–LTD balance depending on the brain area and type of synapse, and may provide tools for the targeted regulation of synaptic memories.
At glutamatergic synapses onto hippocampal and neocortical pyramidal cells, respectively, coincident pre- and postsynaptic activity elicits long-term potentiation (LTP), which typically requires the activation of N-methyl-d-aspartate (NMDA) receptors (Bliss and Collingridge 1993). Long-term depression (LTD), in contrast, may or may not require NMDA receptor activation, but results from activity patterns that activate the postsynaptic neuron in a less efficient or less well-timed manner (Artola and Singer 1993; Kirkwood et al. 1993). It has been suggested that LTP requires a larger calcium signal amplitude for its induction than LTD (Bienenstock et al. 1982; Bear et al. 1987; Hansel et al. 1997). It seems likely that other calcium signaling parameters are crucial as well (Neveu and Zucker 1996; Nevian and Sakmann 2006), but the amplitude of calcium signaling might be the one parameter that most faithfully reflects synaptic input strength during the induction phase. Efforts to develop memory-enhancing drugs have thus focused on the strategy that the magnitude and induction probability of LTP might be enhanced by drugs that boost synaptic transmission and calcium signaling. Ampakines, which are allosteric modulators of AMPA receptors that enhance transmission by slowing both desensitization and deactivation of AMPA receptors (Lynch 2002), have indeed been shown to facilitate LTP induction (Stäubli et al. 1994; Arai et al. 2004). These observations demonstrate that a selective manipulation of synaptic activation strength can affect the LTP induction probability. Moreover, ampakines positively affect memory encoding and recall in humans (Ingvar et al. 1997; Lynch et al. 1997; Lynch and Gall 2006) and reduce effects of sleep deprivation on cognitive performance (Porrino et al. 2005). These data show that enhancing synaptic transmission using drugs that interfere with AMPA receptor function can positively affect synaptic plasticity as well as cognitive performance and learning.
To elucidate the applicability of this strategy to other types of glutamatergic synapses, we examined the effects of the ampakine CX546 on synaptic plasticity at cerebellar parallel fiber (PF) to Purkinje cell synapses. At these synapses LTD and LTP can be induced (Lev-Ram et al. 2002; Coesmans et al. 2004), but they provide an exceptional case regarding LTP/LTD induction rules in that a higher calcium threshold needs to be reached for LTD than for LTP induction (Coesmans et al. 2004) and the molecular induction pathways—cerebellar LTD is PKC/CaMKII-dependent, while LTP depends on phosphatase activation—provide a mirror image of their hippocampal counterparts (Jörntell and Hansel 2006). LTD at PF synapses is facilitated by coactivation of the climbing fiber (CF) input, which helps to reach the higher threshold for LTD induction by providing large, widespread calcium transients (Ross and Werman 1987). This complex interaction involving a qualitatively different heterosynaptic input makes it difficult to predict whether ampakine-mediated enhancement of AMPA receptor signaling at PF synapses can substitute for CF stimulation and facilitate LTD induction under conditions that otherwise trigger LTP, because CF coactivity not only leads to enhanced calcium signaling (Wang et al. 2000), but also causes the release of corticotropin-releasing factor (CRF) that, too, facilitates LTD induction (Miyata et al. 1999). Here, we thus address the question whether ampakines have an impact on cerebellar plasticity at all, and if so, which direction of synaptic gain change they promote.
Results
To examine the effects of CX546 on PF synaptic transmission, we performed whole-cell patch-clamp recordings from Purkinje cells in P18–25 rat cerebellar slices (200–250 μm). At concentrations of 200- and 300-μm, bath-applied CX546 enhanced both the peak amplitude (200 μM: 162.4 ± 34.1% of baseline ± SEM; n = 4; P < 0.05; 300 μM: 160.9 ± 17.1%; n = 5; P < 0.05; t = 30 min; Fig. 1A) and the area of PF-EPSCs (200 μM: 262.4 ± 53.2%; n = 4; P < 0.05; 300 μM: 374.5 ± 43.8%; n = 5; P < 0.05; t = 30 min; Fig. 1A,B). The observed slowed decay time of PF-EPSCs is in line with the note that CX546 acts by destabilizing the desensitized receptor conformation (Nagarajan et al. 2001). There was no change in the paired-pulse facilitation (PPF) ratio of peak EPSC amplitudes (EPSC 2/1; 200 μM: 92.8 ± 6.8% of baseline recorded before CX546 wash-in; n = 4; P > 0.05; 300 μM: 102.9 ± 4.2%; n = 5; P > 0.05; t = 30 min; Supplemental Fig. 1), confirming that CX546 acted postsynaptically. These results show that CX546 enhances and prolongs EPSCs at cerebellar PF synapses, thus boosting basic synaptic transmission the same way as previously observed at hippocampal synapses (Nagarajan et al. 2001; Arai et al. 2002).
Figure 1.

The ampakine CX546 enhances and prolongs PF-EPSCs. (A,B) At concentrations of 200 μM (left; n = 4) and 300 μM (right; n = 5), respectively, CX546 increases the peak amplitude and slows the decay time of PF-EPSCs. (A) Typical traces recorded upon wash-in of 200 μM CX546 (left) and 300 μM CX546 (right), respectively. (B, Top) Time graphs showing changes in the EPSC1 integral when CX546 is bath-applied (bar), bottom: time graphs showing the integral of EPSC2. Error bars are mean ± SEM.
The ultimate goal of this study was to examine how ampakines affect cerebellar synaptic plasticity, which, like hippocampal synaptic plasticity, is controlled by dendritic calcium signaling (Jörntell and Hansel 2006; Hansel and Bear 2008). To examine whether bath application of CX546 enhances calcium transients in Purkinje cell dendrites, we performed fluorometric calcium imaging experiments using an ultra-high-speed CCD camera (NeuroCCD-SMQ; RedShirtImaging), and the fluorescent calcium indicator dye Oregon Green BAPTA-2 (200 μM). PF stimulation evoked dendritic calcium transients that were localized to specific branches (see also Miyakawa et al. 1992; Denk et al. 1995; Eilers et al. 1995). This spatial restriction is illustrated in Figure 2: in this example, PF stimulation that is sufficiently strong to evoke PF-EPSCs of ∼300 pA (600 pA for EPSC2) elicits a calcium transient in a small region of interest (ROI) located on a dendritic branch (region 1; Fig. 2), but not in a ROI on a neighboring branch (region 2; Fig. 2). In the following recordings, we routinely monitored calcium signals from those ROIs that showed the most pronounced calcium transients.
Figure 2.
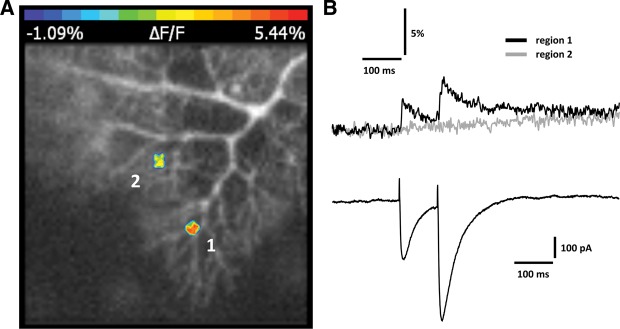
PF stimulation elicits spatially restricted calcium transients. (A) Original image showing two regions of interest (ROI) indicating spatial restriction of calcium transient hotspots (false-color coding). Calcium transients are reported as normalized fluorescence changes (ΔF/F). (B) PF-EPSCs (bottom) and associated calcium transients (top) recorded from regions 1 and 2 as shown in A. Fluorescence measurements were performed using an ultra-high-speed CCD camera (NeuroCCD-SMQ; RedShirtImaging) and the fluorescent calcium indicator Oregon Green BAPTA-2 (200 μM).
To assess the effect of CX546 bath-application on PF-evoked calcium signaling, we monitored calcium transients during a 5-min baseline period, washed-in CX546 (200 μM) for 5 min, and recorded calcium signals in the presence of CX546 for at least five more minutes. During the wash-in period, data acquisition was discontinued to limit the amount of light exposure and phototoxicity, and thus to allow for long-term recordings of calcium transients under stable baseline fluorescence conditions (Weber et al. 2003; Yasuda et al. 2003). In the presence of CX546, the calcium transients were enhanced (203.5 ± 37.9%; n = 5, t = 13–15 min; P < 0.01; Fig. 3; Supplemental Table 1). These calculations are based on the area under the curve (100-msec time window) of the calcium transient evoked by the second stimulus to take advantage of the more pronounced and more stable calcium signals. There was no change in the paired-pulse ratio of the calcium transients (peak of calcium transient 2/1; 117.8 ± 10.7%; n = 5; t = 13–15 min; P > 0.05). In the absence of CX546, the calcium transients remained stable (99.6 ± 12.0%; n = 6; P > 0.05; t = 13–15 min; Fig. 3C; Supplemental Fig. 2). These results show that CX546 not only enhances and prolongs PF-EPSCs, but also boosts dendritic calcium transients.
Figure 3.

CX546 enhances PF-evoked calcium transients. (A) Original image with false-color coded ROI. (B) PF-EPSCs (bottom) and calcium transients (top) before (gray line) and after wash-in of CX546 (black line). (C) Time graph showing the area under the curve of the calcium transients under control conditions (closed dots; n = 6) and when CX546 was applied to the bath (open dots; n = 5). In these recordings, data acquisition was limited to three sweeps per minute to reduce the amount of light exposure. The traces shown are averages over three subsequent sweeps. During wash-in of CX546, data acquisition was discontinued (5 min). Error bars are mean ± SEM.
Ampakines have received attention as potential memory enhancers following the discovery that they facilitate the induction of hippocampal LTP (Stäubli et al. 1994; Lynch 2002). This LTP-enhancing effect has also been demonstrated specifically for CX546 (Arai et al. 2004). Because cerebellar LTP and LTD are governed by induction rules that differ from those described in hippocampal and neocortical circuits (Jörntell and Hansel 2006; Hansel and Bear 2008), we tested whether enhancing AMPA receptor-mediated PF-EPSCs by CX546 bath application (200 and 300 μM, data were pooled) affects PF synaptic plasticity. Under control conditions, PF stimulation at 1 Hz for 5 min elicited a weak, but significant potentiation (120.1 ± 5.7%; t = 20–30 min; n = 8; P < 0.05/t = 30–40 min: 111.8 ± 7.0%; n = 8; P < 0.05; Fig. 4). In contrast, in the presence of CX546 in the bath, application of the same PF stimulation protocol caused LTD (78.7 ± 11.6%; t = 20–30 min; n = 9; P > 0.05/t = 30–40 min: 68.3 ± 10.2%; n = 9; P < 0.05; Fig. 4). The change in the PF-EPSC amplitude in the presence of CX546 was significantly different from that observed under control conditions (Mann–Whitney U-test; t = 30–40 min; P < 0.05). Thus, while ampakines facilitate LTP induction at hippocampal synapses (Stäubli et al. 1994; Arai et al. 2004), at cerebellar PF synapses we observed the opposite effect: application of a weak LTP induction protocol results in the induction of LTD instead. To examine whether the switch in polarity might be caused by the CX546-mediated amplification of calcium transients described above, we applied the PF stimulation protocol with CX546 present in the bath, and a lower Ca2+/Mg2+ ratio in the ACSF (from 2 mM Ca2+/2 mM Mg2+ to 1 mM Ca2+/3 mM Mg2+), in effect reducing voltage-gated calcium influx. In these experiments, the stimulus strength was enhanced to compensate for the accompanying reduction in release and keep baseline EPSC levels in the same amplitude range (100–200 pA) used in all LTP/LTD experiments shown here. Under these conditions, application of the PF stimulation protocol induced LTP (110.9 ± 9.0%; t = 20–30 min; n = 5; P > 0.05/t = 30–40 min: 127.8 ± 10.7%; n = 5; P < 0.05; Fig. 4). This change in PF-EPSC amplitudes was significantly different from the LTD seen in the presence of CX546 alone (t = 30–40 min; P < 0.01). These findings are in line with the hypothesis that CX546 enhances the probability for LTD induction by strengthening AMPA receptor signaling and subsequent calcium influx through voltage-gated calcium channels.
Figure 4.

CX546 enhances the probability for LTD induction. (A) Typical traces obtained in the absence (A1) and in the presence of CX546 (A2) as well as in the presence of CX546 when [Ca2+]o was lowered to 1 mM (A3). (B) Time graph showing changes in the amplitude of EPSC1. Under control conditions, PF stimulation at 1 Hz for 5 min causes a potentiation (black dots; n = 8). In the presence of CX546, application of the same PF stimulation protocol causes LTD instead (white dots; n = 9). When CX546 is present in ACSF that also contains a lowered Ca2+/Mg2+ ratio (1:3) LTP is preserved (gray dots; n = 5). Error bars are mean ± SEM.
Discussion
In hippocampal and neocortical circuits, ampakines enhance synaptic transmission and promote the induction of LTP (Lynch 2002). Our study shows that in cerebellar circuits, the basic mode of action (slowing desensitization/deactivation of AMPA receptors) is the same, but the consequences of enhanced transmission are fundamentally different, as CX546 promotes LTD rather than LTP induction. At both types of synapses, the change in induction probabilities is likely due to an increase in synaptically evoked calcium signaling. Cerebellar Purkinje cells only weakly express GluA1 subunits (Baude et al. 1994) and plasticity is dominated by membrane trafficking of calcium-impermeable GluA2-containing AMPA receptors instead (Chung et al. 2003). Thus, the observed amplification of calcium transients is likely a consequence of strengthened activation of voltage-gated calcium channels, which results from enhanced AMPA receptor-mediated depolarization. Our finding that lowering the Ca2+/Mg2+ ratio in the ACSF prevents LTD induction when CX546 is applied in the bath supports this notion. The data presented here are also in line with our previous report that the polarity of gain change at PF synapses can be bidirectionally controlled by chelating calcium (BAPTA) and calcium uncaging, respectively (Coesmans et al. 2004). Ultimately, it is the inverse calcium thresholds for LTD and LTP induction that might explain why ampakines promote LTP in the hippocampus, but LTD in the cerebellum. Note that across different brain areas there is no difference in the way ampakines themselves affect transmission. It rather seems that at both hippocampal and cerebellar synapses ampakines—and high-calcium conditions in general—trigger kinase-dominated pathways that involve the activation of CaMKII (see Jörntell and Hansel 2006). It is the AMPA receptor subunit composition (dominance of GluA2 subunits in Purkinje cell synaptic transmission and plasticity; Baude et al. 1994; Chung et al. 2003) that determines which receptor trafficking rules apply.
At PF synapses, LTD induction is promoted by CF coactivity, but LTD can also be elicited in the absence of CF stimulation if calcium sources are recruited downstream from synaptic activation (Finch and Augustine 1998), or PF inputs are strongly stimulated (Hartell 1996; Han et al. 2007). Our finding that enhancement of AMPA receptor function by CX546 has the same effect and allows for LTD induction without CF coactivation shows that the strength of synaptic activation and local calcium transients ultimately determines the polarity of synaptic gain change at these synapses. This observation does not exclude the possibility that regulation of other factors that are critical for LTD induction, such as CRF signaling, may similarly control bidirectional PF plasticity. As LTD—in concert with other types of synaptic and nonsynaptic plasticity—potentially contributes to cerebellar memory formation (Hansel et al. 2001; Ito 2001; Gao et al. 2012; see also Schonewille et al. 2011) it will be interesting to find out whether ampakines affect cerebellum-dependent forms of motor learning in a similar way as hippocampus-dependent learning. The difference between hippocampal and cerebellar synaptic plasticity points toward a general problem when attempting to develop drugs for the treatment of memory deficits: LTP may not simply equal learning and memory formation across all types of synapses. Rather, LTP and LTD coexist at most types of synapses and play very specific roles in information storage (Malenka and Bear 2004; Gao et al. 2012). These specific roles need to be considered when interfering with the balance of LTP and LTD mechanisms using ampakines or other memory-enhancing drugs.
Can ampakines be used to compensate for LTD deficits? At cerebellar PF synapses, both LTD and LTP may be relevant for the fine-adjustment of motor output (Jörntell and Hansel 2006; Schonewille et al. 2010). LTD is induced upon PF and CF coactivation and thus results when PF activity coincides with error signals conveyed by the CF input (Ito et al. 1982; Ito and Kano 1982). LTP, in contrast, is induced by PF stimulation alone (Lev-Ram et al. 2002; Coesmans et al. 2004) and provides a reversal mechanism for LTD (formally, LTD can also provide a reversal mechanism for LTP; Jörntell and Hansel 2006). As demonstrated here, ampakines can shift the balance of cerebellar LTD/LTP toward a higher LTD induction probability and might thus be useful candidate drugs for the treatment of cerebellar dysfunctions that are caused by deficits in LTD induction/maintenance. A type of ataxia that might be related to a blockade of cerebellar LTD is paraneoplastic cerebellar ataxia, in which autoantibodies against the metabotropic glutamate receptor mGluR1 prevent the induction of LTD (Sillevis Smitt et al. 2000; Coesmans et al. 2003). Whether LTD deficits contribute to other forms of ataxia as well remains to be investigated (for discussion see Rinaldo and Hansel 2010; De Zeeuw et al. 2011).
While our findings demonstrate that cerebellar plasticity can be selectively regulated by CX546, it also needs to be pointed out that there are potential problems and risks when using ampakines for clinical treatment. First, proper learning requires intact cellular mechanisms for information storage and deletion. Saturation of a plasticity mechanism may prevent or impair further learning. Second, it is currently not well understood whether “uncoupling” of cerebellar LTD from the instructive CF signal would cause subsequent problems, because of the resulting inability of the CF input to convey error signals that are invaluable cues in a behavioral motor learning context. Third, it needs to be taken into consideration that excessive AMPA receptor activation can promote excitotoxicity (Yamada 1998).
In summary, our study shows that ampakines enhance PF synaptic transmission and dendritic calcium signaling, but promote LTD, rather than LTP induction. Thus, these findings challenge the view that enhanced synaptic activity generally equals a higher probability for LTP induction. Rather, different brain circuits and different types of synapses use synaptic plasticity mechanisms, such as LTP and LTD, in ways that are uniquely adapted. As a consequence, therapeutic treatments based on drugs interfering with synaptic learning processes need to be tailored to the specific types of memory deficits.
Materials and Methods
Slice preparation
Sagittal slices of the cerebellar vermis (200–250 μm) were prepared from P18–25 Sprague-Dawley rats in ice-cold artificial cerebrospinal fluid (ACSF), and were kept at room temperature for a maximum of 6 h in ACSF containing (in mM): 124 NaCl, 5 KCl, 1.25 Na2HPO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 d-glucose bubbled with 95% O2 and 5% CO2. Slices were continuously perfused with ACSF throughout recording. In one set of recordings, the ACSF was modified to contain 3 mM MgSO4 and 1 mM CaCl2 (Fig. 4). All drugs were purchased from Sigma, except for Oregon Green BAPTA-2 (Invitrogen).
Electrophysiology
Whole-cell patch-clamp recordings were performed at room temperature using an EPC-10 amplifier (HEKA Electronics). Currents were filtered at 3 kHz, digitized at 8 kHz, and acquired using PULSE software (HEKA). Patch pipettes (2.5–5.0 MΩ) were filled with a solution containing (in mM): 9 KCl, 10 KOH, 120 K-gluconate, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, and 17.5 sucrose (pH 7.25–7.35). Purkinje cells were voltage-clamped at holding potentials in the range of −65 to −70 mV. Picrotoxin (100 μM) was added to the ACSF to block GABAA receptors. To evoke EPSCs, parallel fibers were activated using glass electrodes that were filled with ACSF. These electrodes were placed in the upper one-third of the molecular layer to reduce the risk of unintentional climbing fiber stimulation. In all experiments, test responses were elicited using paired-pulse stimulation (interval 50–100 msec) at a frequency of 0.05 Hz with stimulus currents in the range of 0.5–2 μA. The paired-pulse facilitation (PPF) ratio was determined to confirm the postsynaptic origin of alterations. The PPF ratio was calculated as EPSC2/EPSC1. In all LTP/LTD experiments shown in Figure. 4, EPSC1 amplitudes recorded during the baseline period were adjusted to a range of 100–200 pA. The protocol for inducing LTP consisted of parallel fiber stimulation at 1 Hz for 5 min. For tetanization, the recording mode was switched from voltage- to current-clamp. Series and input resistances were monitored throughout the experiment by applying a hyperpolarizing voltage step (−10 mV) at the end of each sweep. Recordings were excluded from the study if the series or the input resistance varied by >15% over the course of the experiments. All values are shown as % of baseline (calculated from the 10 min of baseline recording) ± SEM. For statistical analysis, we used the Student t-test (paired/unpaired) and the Mann–Whitney U-test, when appropriate.
Calcium imaging
Microfluorometric recordings were performed using a NeuroCCD-SMQ camera (80 × 80 pixels) and NeuroPlex software (both RedShirtImaging) for data acquisition. Fluorescence was sampled at 2 kHz for sweep durations of 750 msec. Fluorescence was excited using a 100 W Xenon arc lamp (Cairn Research Ltd). Recordings started >30 min after patch formation to allow for the calcium indicator dye (Oregon Green BAPTA-2; 200 μM) to diffuse into the distal dendrites. The stimulus electrode was placed under visual guidance to activate fine dendritic branchlets. Fluorescence changes were normalized to resting levels and expressed as the ratio ΔF/F = (F(t) − F)/F, where F(t) is the fluorescence value at time t, and F is the averaged fluorescence obtained during the baseline period preceding the stimulus application.
Supplementary Material
Acknowledgments
We thank members of the Hansel laboratory for invaluable comments on the manuscript. This work was supported by grants from the Hersenstichting Nederland, the Netherlands Organization for Scientific Research (NWO-ALW and NWO-MW), and a National Institutes of Health (NIH) grant NS62771 to C.H.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.035220.114.
References
- Arai AC, Xia YF, Rogers G, Lynch G, Kessler M 2002. Benzamide-type AMPA receptor modulators form two subfamilies with distinct modes of action. J Pharmacol Exp Ther 303: 1075–1085. [DOI] [PubMed] [Google Scholar]
- Arai AC, Xia YF, Susuki E 2004. Modulation of AMPA receptor kinetics differentially influences synaptic plasticity in the hippocampus. Neuroscience 123: 1011–1024. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W 1993. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci 16: 480–487. [DOI] [PubMed] [Google Scholar]
- Baude A, Molnar E, Latawiec D, McIlhinney RAJ, Somogyi P 1994. Synaptic and nonsynaptic localization of the GluR1 subunit of the AMPA-type excitatory amino acid receptor in the rat cerebellum. J Neurosci 14: 2830–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF 1987. A physiological basis for a theory of synaptic modification. Science 237: 42–48. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW 1982. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci 2: 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ 2003. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 300: 1751–1755. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Smitt PA, Linden DJ, Shigemoto R, Hirano T, Yamakawa Y, van Alphen AM, Luo C, van der Geest JN, Kros JM, et al. 2003. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol 53: 325–336. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C 2004. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44: 691–700. [DOI] [PubMed] [Google Scholar]
- Denk W, Sugimori M, Llinas R 1995. Two types of calcium response limited to single spines in cerebellar Purkinje cells. Proc Natl Acad Sci 92: 8279–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK 2011. Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci 12: 327–344. [DOI] [PubMed] [Google Scholar]
- Eilers J, Augustine GJ, Konnerth A 1995. Subthreshold synaptic Ca2+ signaling in fine dendrites and spines of cerebellar Purkinje neurons. Nature 373: 155–158. [DOI] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ 1998. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature 396: 753–756. [DOI] [PubMed] [Google Scholar]
- Gao Z, van Beugen BJ, De Zeeuw CI 2012. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci 13: 619–635. [DOI] [PubMed] [Google Scholar]
- Han VZ, Zhang Y, Bell CC, Hansel C 2007. Synaptic plasticity and calcium signaling in Purkinje cells of the central cerebellar lobes of mormyrid fish. J Neurosci 27: 13499–13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Bear MF 2008. LTD-synaptic depression and memory storage. In Molecular mechanisms of memory, ed. Sweatt JD, Vol. 4of Learning and memory: a comprehensive reference (ed. Byrne J), pp. 327–366 Elsevier, Oxford. [Google Scholar]
- Hansel C, Artola A, Singer W 1997. Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. Eur J Neurosci 9: 2309–2322. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E 2001. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4: 467–475. [DOI] [PubMed] [Google Scholar]
- Hartell NA 1996. Strong activation of parallel fibers produces localized calcium transients and a form of LTD that spreads to distant synapses. Neuron 16: 601–610. [DOI] [PubMed] [Google Scholar]
- Ingvar M, Ambros-Ingerson J, Davis M, Granger R, Kessler M, Rogers GA, Schehr RS, Lynch G 1997. Enhancement by an ampakine of memory encoding in humans. Exp Neurol 146: 553–559. [DOI] [PubMed] [Google Scholar]
- Ito M 2001. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev 81: 1143–1195. [DOI] [PubMed] [Google Scholar]
- Ito M, Kano M 1982. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett 33: 253–258. [DOI] [PubMed] [Google Scholar]
- Ito M, Sakurai M, Tongroach P 1982. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J Physiol 324: 113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H, Hansel C 2006. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron 52: 227–238. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF 1993. Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science 260: 1518–1521. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Wong ST, Storm DR, Tsien RY 2002. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci 99: 8389–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G 2002. Memory enhancement: the search for mechanism-based drugs. Nat Neurosci 5: 1035–1038. [DOI] [PubMed] [Google Scholar]
- Lynch G, Gall CM 2006. Ampakines and the threefold path to cognitive enhancement. Trends Neurosci 29: 554–562. [DOI] [PubMed] [Google Scholar]
- Lynch G, Granger R, Ambros-Ingerson J, Davis CM, Kessler M, Schehr R 1997. Evidence that a positive modulator of AMPA-type glutamate receptors improves delayed recall in aged humans. Exp Neurol 145: 89–92. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF 2004. LTP and LTD: an embarrassment of riches. Neuron 44: 5–21. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Lev-Ram V, Lasser-Ross N, Ross WN 1992. Calcium transients evoked by climbing fiber and parallel fiber synaptic inputs in guinea pig cerebellar Purkinje neurons. J Neurophysiol 68: 1178–1189. [DOI] [PubMed] [Google Scholar]
- Miyata M, Okada D, Hashimoto K, Kano M, Ito M 1999. Corticotropin-releasing factor plays a permissive role in cerebellar long-term depression. Neuron 22: 763–775. [DOI] [PubMed] [Google Scholar]
- Nagarajan N, Quast C, Boxall AR, Shahid M, Rosenmund C 2001. Mechanism and impact of allosteric AMPA receptor modulation by the ampakine CX546. Neuropharmacology 41: 650–663. [DOI] [PubMed] [Google Scholar]
- Neveu D, Zucker RS 1996. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron 16: 619–629. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B 2006. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci 26: 11001–11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrino LJ, Daunais JB, Rogers GA, Hampson RE, Deadwyler SA 2005. Facilitation of task performance and removal of the effects of sleep deprivation by an ampakine (CX717) in nonhuman primates. PLoS Biol 3: 1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo L, Hansel C 2010. Ataxias and cerebellar dysfunction: involvement of synaptic plasticity deficits? Funct Neurol 25: 135–139. [PMC free article] [PubMed] [Google Scholar]
- Ross WN, Werman R 1987. Mapping calcium transients in the dendrites of Purkinje cells from the guinea-pig cerebellum in vitro. J Physiol 389: 319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, et al. 2010. Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron 67: 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, De Jeu MT, Steinberg JP, Takamiya K, Hoebeek FE, et al. 2011. Reevaluating the role of LTD in cerebellar motor learning. Neuron 70: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillevis Smitt P, Kinoshita A, De Leeuw B, Moll W, Coesmans M, Jaarsma D, Henzen-Logmans S, Vecht C, De Zeeuw C, Sekiyama N, et al. 2000. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 342: 21–27. [DOI] [PubMed] [Google Scholar]
- Stäubli U, Perez Y, Xu FB, Rogers G, Ingvar M, Stone-Elander S, Lynch G 1994. Centrally active modulators of glutamate receptors facilitate the induction of long-term potentiation in vivo. Proc Natl Acad Sci 91: 11158–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SSH, Denk W, Häusser M 2000. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neuroscience 3: 1266–1273. [DOI] [PubMed] [Google Scholar]
- Weber JT, De Zeeuw CI, Linden DJ, Hansel C 2003. Long-term depression of climbing fiber-evoked calcium transients in Purkinje cell dendrites. Proc Natl Acad Sci 100: 2878–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KA 1998. Modulating excitatory synaptic neurotransmission: potential treatment for neurological disease? Neurobiol Dis 5: 67–80. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K 2003. Plasticity of calcium channels in dendritic spines. Nat Neurosci 6: 948–955. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.