

Abstract
The female hormone progesterone (P4) promotes the expansion of stem-like cancer cells in estrogen receptor (ER) and progesterone receptor (PR) positive breast tumors. The expanded tumor cells lose expression of ER and PR, express the tumor-initiating marker CD44, the progenitor marker cytokeratin 5 (CK5), and are more resistant to standard endocrine and chemotherapies. The mechanisms underlying this hormone-stimulated reprogramming have remained largely unknown. In the present study, we investigated the role of microRNAs in progestin-mediated expansion of this dedifferentiated tumor cell population. We demonstrate that P4 rapidly downregulates miR-29 family members, particularly in the CD44+ cell population. Downregulation of miR-29 members potentiates the expansion of CK5+ and CD44+ cells in response to progestins, and results in increased stem-like properties in vitro and in vivo. We demonstrate that miR-29 directly targets Krüppel-like factor 4 (KLF4), a transcription factor required for the reprogramming of differentiated cells to pluripotent stem cells, and for the maintenance of breast cancer stem cells. These results reveal a novel mechanism whereby progestins increase the stem cell-like population in hormone-responsive breast cancers, by decreasing miR-29 to augment PR-mediated upregulation of KLF4. Elucidating the mechanisms whereby hormones mediate the expansion of stem-like cells furthers our understanding of the progression of hormone responsive breast cancers.
Keywords: breast cancer, microRNA, progesterone, KLF4, cancer stem cells
INTRODUCTION
The role of progesterone (P4) in the mammary gland is complex since it mediates both proliferation and differentiation. Progesterone receptor (PR) is indispensable for normal mammary ductal side branching and lobulo-alveolar development (1), yet progestins also play a deleterious role during breast cancer development and progression (2,3). In rodent models, P4 accelerates carcinogen-induced mammary tumorigenesis and PR is critical for this transformation (4). In women, inclusion of progestins in postmenopausal hormone therapy regimens increases the risk of breast cancer (5–7). Progestins increase the number of murine mammary stem cells (8,9), normal human breast progenitor cells (10), and stem-like cells in human breast tumor xenografts (11). Thus, progestin mediated expansion of stem-cell populations may explain its detrimental effects in breast carcinogenesis.
The mechanisms by which progestins increase the stem-cell compartment in the normal and malignant breast, may include both paracrine and autocrine signals. In the normal mammary gland, P4 acts in a paracrine manner to expand the basal/stem cell populations, and in rodents this is likely mediated through RANKL (12). In human breast cancer cells, evidence points to an autocrine mechanism by which progestins directly convert estrogen receptor (ER) and PR positive cells to receptor negative cells (ER−PR−) that acquire expression of the tumor initiating marker CD44 and cytokeratin 5 (CK5) (11). CK5+ cells in the normal human breast represent a bi-potent progenitor fraction capable of generating myoepithelial (smooth muscle actin SMA+) or luminal epithelial (CK8/18+) lineages (13). Furthermore, the expression of significant amounts of CK5/CK6 in primary human breast cancers correlates with high grade and worse prognosis (14). Accordingly, CK5+ cells are more prevalent in basal-like tumors than luminal ER+ tumors, but a subpopulation of ER−PR−CK5+ cells exists in many ER+ tumors (15) and this population is contained within the stem-like CD44+ fraction (11). These ER−PR−CK5+CD44+ cells are relatively resistant to endocrine and chemotherapies compared to the bulk ER+PR+CK5− tumor cells (16) and may be the reason that such tumors can recur many years post-treatment. However, virtually nothing is known about the mechanisms by which progestins reprogram ER+PR+CK5− cells into ER−PR−CK5+ cells.
Non-coding regulatory RNAs termed microRNAs (miRNAs) control various processes important for breast cancer progression and metastasis. Evidence suggests that miRNAs contribute to preserving the “stemness” of embryonic stem cells (ESC), because ESC deficient in miRNA processing cannot be maintained (17). MiRNAs have been implicated in the maintenance of the cancer stem cell (CSC) phenotype via their ability to affect multiple pathways including those regulating cell proliferation (18), cell death (19–21), cell-cell communication, and cell adhesion (22). MiRNAs can be regulated by hormones and target hormone receptors (reviewed in 23,24) and play a key role in the acquisition of resistance to hormonal-therapy (25–27). Importantly, forced expression of certain miRNAs can revert human somatic (normal skin keratinocytes and hair melanocytes) and cancer cells (cancerous melanoma Colo-829 and prostate cancer PC3 cells) into pluripotent stem cells (28,29). We recently discovered that progestins regulate multiple miRNAs in breast cancer cells (30). Thus, we hypothesized that P4-regulated miRNAs might play a role in the expansion of CK5+CD44+ cells in ER+PR+ tumors.
Here we demonstrate that P4 downregulates miR-29 family members, particularly in the CD44+ population expanded by progestins. Downregulation of miR-29 members potentiates the expansion of CK5+ and CD44+ cells in response to P4, resulting in increased mammosphere formation in vitro and increased tumor initiating capability in vivo. We find that miR-29 directly targets Krüppel-like factor 4 (KLF4, EZF, GKLF4), a transcription factor necessary for the reprogramming of differentiated cells into pluripotent stem cells and the maintenance of breast CSCs (31–33). Thus, both miR-29 downregulation and KLF4 upregulation contribute to maximal expansion of CK5+ cells in response to progestins in ER+PR+ breast cancer cells. These results provide a mechanism by which hormonal control of a potent reprogramming transcription factor mediates the expansion of a population of cells with stem-like properties in ER+PR+ breast tumors.
RESULTS
Natural and synthetic progestins induce the expansion of CK5+ cells in ER+PR+ breast cancer cells
To determine the extent of CK5+ cell expansion, we treated T47D and BT474 breast cancer cell lines with progestins and measured the relative amount of CK5+ cells. T47D cells express PR in the absence of estrogen stimulation, while BT474 represent the majority of ER+ breast cancer cell lines that require pre-treatment with estrogens to induce PR expression. The natural hormone P4, and the synthetic progestins medroxyprogesterone acetate (MPA) and R5020, equally promote the expansion of the CK5+ population from <1% to ~20% in T47D cells (Figure 1A, top). BT474 cells were pretreated for 48 h with 10 nM 17β-estradiol (E2) to induce PR expression, and subsequently treated with either vehicle or 100 nM progestins for 24 h. The CK5+ population increased from <1% to ~5% compared to vehicle treated BT474 cells (Figure 1A, bottom). Time course analyses show that CK5 mRNA levels increase during the first 24 h after progestin stimulation in both cell lines (Figure 1B, Supplementary Figure 1A). The degree to which progestins increase CK5 mRNA levels and the number of CK5+ cells varies in different ER+ cell lines (Figure 1B, right; Supplementary Figure 1B).
Figure 1. Promotion of CK5+ cells in ER+PR+ breast cancer cells by progestins.

A. Top: Treatment of T47D cells with 100 nM MPA, R5020 or P4 for 24h results in up to 20% increase in CK5+ cells compared to ethanol (OH) treated cells. Bottom: BT474 cells pretreated with estradiol for 48h to upregulate PR and then treated with the same progestins for 24h results in ~10% increase in CK5+ cells. CK5 expression was determined by immunohistochemistry and percentage of CK5+ cells was measured in 3 fields and at least 300 cells. DAPI shows nuclei. B. Left: By quantitative RT-PCR CK5 mRNA levels increase during the first 12h following P4 (T47D cells) or MPA (BT474 cells) treatment. Data represent fold change in CK5 mRNA normalized to β-actin mRNA levels relative to levels of control 0h-time point. Bars are mean ± SEM of biological triplicates. Asterisks represent statistical significance compared to 0h. Right: Western blots shows PR (PRA, PRB isoforms) and CK5 expression in T47D and BT474 cells after 24h treatment with P4. α-tubulin was used as loading control. CK5 antibody may recognize CK6. Since both CK5/6 are basal markers (55), both are P4-regulated, and are usually co-expressed(11), we refer to it as CK5 thereafter. C. By qRT-PCR, miR-29a b and c are decreased in T47D and BT474 cells at 6, and 18h after treatment with 100 nM P4 as compared to ethanol-treated (OH) cells. Data represent miRNA levels normalized to RNU6 relative to miR-29a levels in control cells. Bars = mean± SEM. In all experiments, BT474 cells were pre-treated with 10nM 17-β-estradiol for 48h to induce PR expression prior to progestin stimulation. D. CD44hi cells express significantly less miR-29abc than CD44lo cells. T47D cells were treated with vehicle or 100nM P4 for 24h. Top: Cells were immunolabeled and CD44+ (CD44hi) and CD44− (CD44lo) cells sorted from P4-treated triplicate samples. Bottom: miRNA expression relative to miR-29a expression in CD44lo cells (n=3). Bars =mean± SEM. For all panels, *P<0.05; **P<0.01; ***P<0.0001 ANOVA and Bonferoni post-hoc t-tests. Concentrations of progestins used in this study are within physiological range (i.e P4 ranges from 5.4 to 85.86nM during luteal phase in humans according to NIH clinical test guidelines).
Since most CK5+ cells are also CD44+ (11,34), we also determined if P4 would increase the CD44+ population in breast cancer cells. T47D cells were treated with vehicle or P4 and the CD44 content analyzed by flow cytometry. P4 increased the percentage of CD44+ cells from 3.8±2.3% to 22.75±15.18% (P<0.001) (Supplementary Figure 1C). This level is comparable to the P4-mediated increase in CK5+ cells.
Progesterone decreases the expression of miR-29abc in ER+PR+ breast cancer cell lines
Progestins regulate a cohort of microRNAs in breast cancer cells including potent downregulation of miR-29 family members (30). MiR-29a, b and c are lost in aggressive non-small cell lung cancers and melanomas (35–37); thus, we sought to determine whether loss of miR-29 plays a role in the P4-mediated expansion of more de-differentiated breast cancer cells. Time course experiments demonstrated that treatment of T47D cells with 100 nM P4 decreases the expression levels of mature miR-29 family members as early as 6 h after P4 treatment (Figure 1C). Likewise, BT474 cells treated for 48 h with E2 followed by stimulation with P4 showed a similar decrease in miR-29 levels (Figure 1C, Supplementary Figure 2). MiR-29 levels return to basal levels by 24 h in T47D cells (27).
In the previous experiments, miRNA levels were measured in a mixed population (the minority CD44+CK5+ plus the majority CD44−CK5− cells). To determine the expression of each miR-29 member specifically in the CD44+ population, we treated T47D cells with vehicle or P4 for 24 h and isolated the CD44hi and CD44lo populations by FACS (Figure 1D, top). Quantitative RT-PCR showed that expression of mature miR-29a, b, and c were all significantly reduced in CD44hi compared to CD44lo cells (Figure 1D, bottom).
Human miR-29 family members are encoded by the miR-29b-1/miR-29a cluster and the miR-29b-2/miR-29c cluster (38). Since miR-29a was relatively more abundant than miR-29b or miR-29c in both cell lines analyzed, we sought to determine the mechanism of P4-mediated downregulation of the miR-29a/b1 locus. P4 downregulated pri-miR-29a as early as 1h post-treatment (Figure 2A). The miR-29a/b1 promoter is repressed by c-Myc in stem cells (39), and progestins rapidly upregulate c-Myc (40) (Figure 2B), suggesting that P4-induced downregulation of miR-29a could be mediated by c-Myc. Consistent with this hypothesis, siMYC prevented the early upregulation of c-Myc in response to P4 (Figure 2B) and abrogated the ability of P4 to downregulate miR-29a in T47D (Figure 2C) and BT474 cells (Supplementary Figure 3A). siMYC also abolished P4-mediated downregulation of pri-miR-29a (Figure 2C, Supplementary Figure 3A). Uppregulation of c-Myc and downregulation of miR-29a by P4 were abrogated by the PR antagonist RU486 (Figure 2D, Supplementary Figure 3B). Furthermore, P4 did not affect levels of miRNA-processing enzyme Dicer in T47D cells (Supplementary Figure 3C), suggesting that miR-29a downregulation is not the result of decreased global miRNA processing. Taken together, these data suggest that the PR-dependent repression of miR-29a is transcriptional and likely mediated by c-Myc.
Figure 2. P4 induces transcriptional downregulation of miR-29a via c-Myc.
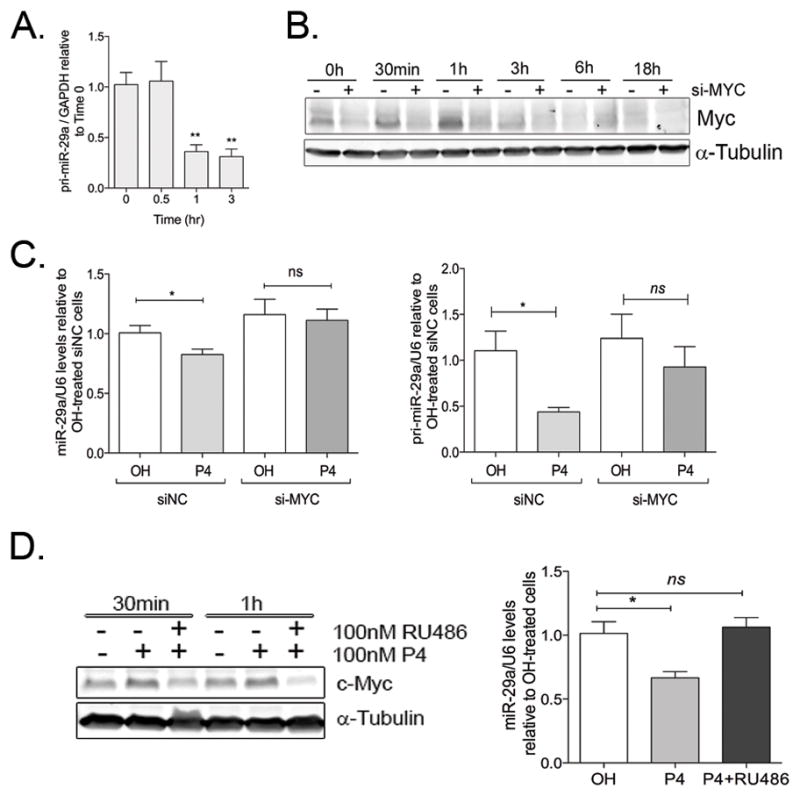
A. P4 decreases pri-miR-29a levels as early as 1h after treatment. T47D cells were treated for 0.5, 1 or 3h with 100nM P4 and pri-miR-29a levels analyzed by qRT-PCR. Data represents pri-miR-29a levels normalized to GAPDH, relative to pri-miR-29a levels at time 0. **P<0.01 compared to 0h. B. si-MYC prevents P4-induced upregulation of c-Myc in T47D cells. T47D cells were plated at 60% confluence, attached overnight and then transfected with either 20nM siNC (−) or siMYC (+). After 24h, cells were treated with 100nM P4 for the indicated times. Total cell lysates were used to measure c-Myc expression by western blot. α-tubulin is used as a loading control. C. c-Myc inhibition abolishes downregulation of mature miR-29 (left) and pri-miR-29a (right) 18h after P4 treatment. T47D cells were transfected with either 20nM siNC or 20nM siMYC and 24h later, treated with vehicle (OH) or 100nM P4 for 18h. Data represents miR-29a or pri-miR-29a levels normalized to RNU6B, relative to siNC-OH-treated cells. *P<0.05. D. P4 inhibitor RU486 prevents P4-mediated upregulation of c-Myc and repression of miR-29a. Left: T47D cells were treated for 18h with either vehicle (OH), 100nM P4 (P4) or 100nM P4 in combination with 100nM RU486 for the indicated times, and c-Myc expression analyzed by western blot. Right. qRT-PCR shows miR-29a levels normalized to RNU6B and relative to OH-treated cells. Bars represent mean± SEM from biological replicates (n=5). *P<0.05.
miR-29 downregulation potentiates the progestin mediated increase in CK5+ and CD44+ cells and results in increased tumor initiating ability
To test the dependency of P4-mediated induction of CD44+ and CK5+ cells on miR-29 function, we generated miR-29-inhibited stable cells (designated as 29aZIP, 29bZIP or 29cZIP) by transduction with lentiviral vectors expressing sequences complementary to each mature miRNA. Inhibition of miR-29a led to a small basal increase in the percentage of CD44+ cells in the absence of P4, but did not affect the basal percent of CK5+ cells (Figure 3A,B). However, inhibition of miR-29a, compared to SCR-ZIP control, significantly augmented the P4-mediated increase in CD44+ and CK5+ cells as analyzed by flow cytometry (Figure 3A) and immunocytochemistry (Figure 3B). In agreement with the finding that miR-29a is the most abundant member of this family in breast cancer cells and likely to represent the major functional player, inhibition of miR-29b and c did not significantly increase the CD44+ population (Figure 3A) or the CK5+ population (data not shown) in either the absence or presence of P4.
Figure 3. Stable inhibition of miR-29 increases the CK5+ and CD44+ populations in response to P4.
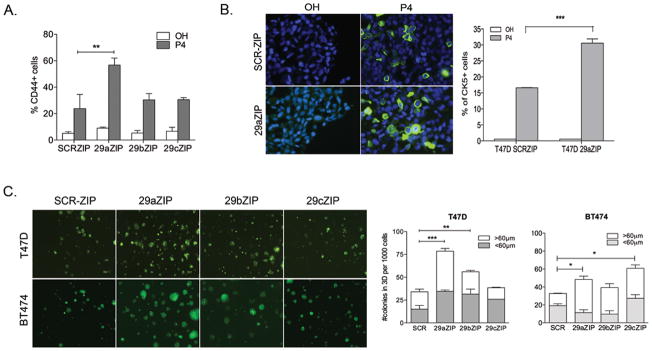
ER+PR+ breast cancer cells stably expressing inhibitors of miR-29a, b or c (29aZIP, 29bZIP or 29cZIP lentiviral vectors) were compared to SCR-control cells for expression of basal markers and stem-like properties. A. T47D 29aZIP cells express more CD44+ cells in response to P4 compared to SCR-ZIP cells. Cells were plated at 60% confluence, attached overnight and then treated with ethanol (OH) or 100nM P4 for 24h. Cells were labeled and CD44 expression measured by flow cytometry. Data represents percentage of CD44+ cells from at least 3 independent experiments. Bars = mean± SEM. B. T47D 29aZIP cells express more CK5+ cells in response to P4 compared to SCRZIP cells. Left: CK5 expression (green) was determined by immunofluorescence in vehicle or P4 treated T47D SCRZIP and 29aZIP cells. Representative image shows DAPI (blue) and CK5+ expression (20X objective). Right: Percentage of CK5+ cells (green) was measured in 3 fields and at least 300 cells per condition in biological triplicates. Bars = mean± SEM. C. T47D and BT474 cells expressing 29aZIPs form significantly more colonies in 3D-culture compared to SCRZIP control cells. 1000 cells were plated in matrigel and colonies counted 14 days later. Colonies and their diameter were digitally measured and a the number of colonies > or <60μm diameter (representing colonies of at least 10 cells) were counted. Bars represent mean± SEM from biological triplicates. For all panels, *P<0.05; **P<0.01; ***P<0.0001 ANOVA followed by Bonferoni post-hoc t-tests.
To test whether miR-29 has a role in self-renewal of stem-like cells in ER+ breast cancer cells, we cultured T47D and BT474 cells expressing either SCR-ZIP, 29aZIP, 29bZIP and 29cZIP in three-dimensional (3-D) Matrigel colony forming assays (Figure 3C). SCR-ZIP control cells formed colonies at significantly lower frequencies than 29aZIP cells in both T47D and BT474 cell lines (Figure 3C, right). In addition, 29aZIP cells formed more colonies that were larger than 60μm in diameter as compared to those in the SCR-ZIP controls (Figure 3C, right). 29bZIP and 29cZIP cells also formed significantly more colonies than the SCR-ZIP cells, but to a lesser extent than 29aZIP cells.
We next evaluated the ability of SCR-ZIP and 29aZIP cells to initiate tumors by injecting cells pretreated for 24 h with vehicle (OH) or 100 nM P4 in vitro into the 4th mammary gland of female nude (nu/nu) mice at dilutions ranging from 103–105. All mice were supplemented with estradiol (necessary for tumor growth in vivo). Table 1 summarizes limiting dilution experiments. 29aZIP cells were able to form tumors in mice more efficiently when compared to the SCRZIP cells in both the vehicle and P4 treated groups (Table 1). However, this was most pronounced in the P4 treated groups, where 29aZIP cells form palpable tumors in 9/10 animals while SCR-ZIP cells grew in 4/10 mice. Collectively, these data indicate that suppression of miR-29a potentiates tumor-initiating ability.
Table 1.
Tumor initiating ability of 29aZIP compared to SCR-ZIP T47D cells
| Number of cells injected per mammary fat pad | Number of tumors* | |
|---|---|---|
| SCR-ZIP OH | 29aZIP OH | |
| 1 x 105 | 10/10 | 10/10 |
| 1 x 104 | 9/10 | 10/10 |
| 1 x 103 | 6/10 | 8/10 |
| Tumor Initiating Frequency | (1/3596) | (1/621) |
| (95% CI) | (1/11484-1/2882) | (1/1342-1/288) |
| P value | 0.000987 | |
|
| ||
| SCR-ZIP P4 | 29aZIP P4 | |
| 1 x 105 | 7/10 | 9/10 |
| 1 x 104 | 7/10 | 10/10 |
| 1 x 103 | 4/10 | 9/10 |
| Tumor Initiating Frequency | 1/5753 | (1/434) |
| (95% CI) | (1/7348-1/3596) | (1/974-1/194) |
| P value | 0.0000024 | |
Limiting dilution analysis of the tumor initiating frequency of T47D-29aZIPs as compared to SCR-ZIP control cells. Mice were implanted with E2-releasing pellets and tumor formation followed for 4 weeks. SCRZIP and 29aZIP cells were injected in the contralateral 4th mammary fat pads of 4-week old nu/nu mice. Cells were pretreated with either vehicle (OH) or 100nM P4 for 24h prior to implantation. OH-pretreated or P4-pretreated cells were implanted in two independent groups (n=10 per group). CI, confidence interval.
Shown as number of tumors per number of injected fat pads. Data was analyzed using ELDA software (53).
Inhibition of miR-29a promotes tumor growth in vivo
To investigate the role of miR-29a inhibition in tumor growth in vivo, we injected T47D SCRZIP and T47D 29aZIPs (1x 106 cells) in the contralateral 4th mammary fat pad of NOD/SCID mice. Mice were supplemented with E2 alone or estradiol plus progestin (E2+MPA). Tumors produced by 29aZIP cells were significantly larger than those produced by SCR-ZIP cells, in both E2 alone or E2+MPA treated mice (Figure 4A,B). Tumors in mice treated with MPA were significantly smaller than those of E2 treated mice, although Ki67 staining did not show any significant difference in proliferation in any group (Figure 4C). Interestingly, during the first two weeks, tumors from 29aZIP cells in MPA-treated mice grew significantly larger than tumors in E2-treated mice, suggesting that 29aZIP had an initial growth advantage in the presence of MPA at the onset of tumor growth.
Figure 4. Stable inhibition of miR-29a increases tumor growth.

Ovariectomized female NOD/SCID mice were injected with 1x 106 T47D-SCRZIP or T47D-29aZIP cells in the left and right 4th mammary fat pad respectively. Pellets containing either estrogen alone (E2) or in combination with MPA (E2+MPA) were implanted subcutaneously at time of tumor cell injection (n=8 per group) and tumor size measured using a caliper weekly after injection. A. Tumor growth in E2 treated mice. B. Tumor growth in E2+MPA treated mice. For A and B, data represent mean± SEM. *P<0.05 paired t-test compared to SCR control. Images show representative tumors for each group at the end of the experiment. C. Representative immunohistochemistry showing Ki67 staining SCRZIP and 29aZIP tumors. Magnification 20X. Tumors grown in MPA show decreased cellularity compared to E2-treated tumors, but no differences in the percentage of Ki67 positive cells was observed among SCR-ZIP and 29aZIP tumors.
KLF4 is upregulated by P4 and is a direct target of miR-29
KLF4, which encodes Krüppel-like factor 4, is a predicted target of miR-29, and is required for induction of pluripotent stem-cells (41), self-renewal of ES cells, and maintenance of breast CSCs (32). In addition, KLF4 is upregulated by P4 via PR in T47D cells (42). To determine if KLF4 is involved in the promotion of CK5+ cells in response to P4, we performed time course analysis of KLF4 expression in T47D cells following P4 stimulation. KLF4 mRNA and protein levels increased rapidly after P4 stimulation (Figure 5A,B), at the same time decreases in miR-29a were observed (Figure 1C). The increase in KLF4 preceded the promotion of CK5+ cells in response to P4 (Figure 5B), and CD44hi cells isolated from P4 treated T47D cells showed significantly higher KLF4 and CK5 mRNA levels (Figure 5C).
Figure 5. KLF4, a transcription factor required for maintenance of breast cancer stem-cells, is upregulated by progesterone prior to the increase in CK5+ cells.

A. Time course of progestin-induced increases in KLF4 and CK5 protein level. Western blot of KLF4 and CK5 protein expression at 6, 18 and 24h following treatment of T47D cells with 100 nM P4. α-tubulin was used as loading control. KLF4 expression normalized to α-tubulin and relative to OH-treated control for each time point is shown in the graph on the right (n=2). B. Levels of KLF4 and CK5 mRNA were measured following treatment with 100 nM P4 in T47D cells relative to β-actin mRNA levels and normalized to 0h. Bars =mean± SEM from biological triplicates. Asterisks indicate statistically significant data compared to 0h. *P<0.05 (ANOVA followed by Bonferoni post-hoc t-tests). C. Cells were treated with 100 nM P4 for 24h, immunolabeled, then sorted for expression of CD44hi and CD44lo populations. CK5 and KLF4 mRNA levels were normalized to β-actin mRNA and expressed relative to levels in CD44lo cells. ***P<0.0001 paired t-test.
To determine whether KLF4 is a direct target of miR-29, we cloned the KLF4 3′UTR containing the predicted miR-29 binding site downstream of luciferase in a reporter construct, and measured luciferase activity in presence of miR-29 mimic. The predicted binding site for miR-29a and b in the KLF4 3′UTR is depicted in Figure 6A (left). Since transfection of miR-29 can activate p53 and induce apoptosis at low concentrations (43), we normalized our luciferase reporter experiments to the expression of a non-targetable Renilla control. KLF4 is targeted by miR-200c (44), we therefore used exogenous miR-200c as a control to repress KLF4 3′UTR. As shown in Figure 6A (middle) miR-29a, miR-29b, and miR-200c mimics decreased luciferase activity to a similar extent, and blocking miR-29 by adding miR-29a inhibitor blocked this effect. These results demonstrate direct targeting of the KLF4 3′UTR by both the miR-200c and miR-29 family. To further demonstrate that downregulation of miRNAs by P4 regulates the KLF4 3′UTR, we transfected the luciferase reporter containing the KLF4 3′UTR into T47D cells and treated for 48 h with either vehicle or P4. P4 treatment increased luciferase activity from the KLF4-3′UTR, and this effect was partially blocked by adding miR-29a mimic (Figure 6A, right). This indicates that the P4-mediated decrease in a miRNA, specifically miR-29a, relieved targeting of the KLF4-3′UTR resulting in an increase in luciferase activity. Neither P4 nor the miR-29a mimics affected luciferase expression in the vector control (Supplementary Figure 4).
Figure 6. Progesterone-mediated downregulation of miR-29 relieves repression of KLF4.
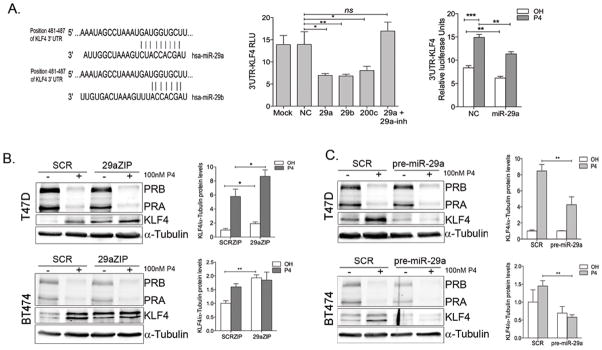
A. Left: Predicted binding sites for miR-29a and b in the KLF4 3′UTR. The KLF4 3′UTR containing the putative miR-29 and miR-200c binding sites cloned downstream of luciferase (pMIRGLO-KLF4) was transfected into T47D cells with either 50 nM negative control (NC), miR-29a (29a), miR29b (29b), miR-200c or miR-29a in combination with miR-29a inhibitor (29a +29a inh) and luciferase activity measured 24h after transfection. Data represents relative luciferase activity normalized to renilla-luciferase present in the pmiRGLO vector. Bars are mean ± SEM. *P<0.05, **P<0.01: ***P<0.001 Right: T47D cells transfected with negative control (NC) or miR-29 mimics (miR-29) in combination with pMIRGLO-KLF4 were treated with either ethanol (OH) or 100 nM P4 for 48h. P4 alone can relieve repression of the 3′UTR-KLF4 and miR-29a partially blocks this effect. B. T47D and BT474 cells stably expressing SCR-ZIPs or 29a-ZIPs were treated for 24h with vehicle or 100nM P4. Western blot of endogenous KLF4 protein in 29aZIP cells compared to SCR-ZIPs (quantified on the right) in T47D and BT474 cells with KLF4 expression normalized to α-tubulin and relative to expression of SCR-ZIP cells treated with vehicle in biological triplicate samples. *P<0.05. C. Stable overexpression of miR29a (pre-miR-29a) decreases P4 induced KLF4 in T47D and BT474 cells.
These data support that KLF4 expression depends on the balance between transcriptional activation (by PR) and post-transcriptional repression (by miR-29). To decipher the contribution of miR-29 to the repression of KLF4 in luminal breast cancer cells, we quantified KLF4 protein levels in 29aZIP T47D and BT474 cells in the absence and presence of P4 (Figure 6B). Compared to SCR-ZIP controls, 29aZIP cells had increased baseline levels of KLF4 in both T47D and BT474 cell lines, suggesting that repression of KLF4 translation by miR-29 plays a role in maintaining low KLF4 levels in the absence of P4 (Figure 6B). Furthermore, P4 stimulation resulted in significantly higher KLF4 levels in 29aZIPs cells.
To further unveil the role of miR-29a in the regulation of KLF4 expression, we overexpressed miR-29a in T47D and BT474 cells using a lentiviral vector carrying its precursor sequence (pre-miR-29a). Western blot analyses showed that miR-29a significantly blocked the upregulation of KLF4 in response to P4 in T47D and BT474 cells (Figure 6C), indicating that blocking the P4-mediated downregulation of miR-29a by addition of excess miR-29a precursor limits the amount of KLF4 protein induced by P4.
Both KLF4 upregulation and miR-29 downregulation are required for maximal P4-mediated expansion of the CK5+ population
To measure the contribution of KLF4 and miR-29 on expression of the CK5+ promoter, we used T47D cells stably expressing a luciferase reporter driven by the human KRT5-promoter (KRT5-luciferase). In this system, there is no direct repression of luciferase through the 3′UTR, thus the luciferase activity is an indirect measure of the CK5 promoter activation in CK5+ cells. P4 stimulation led to a significant activation of KRT5-luciferase, which was blocked by the P4 antagonist RU486 (Supplementary Figure 5). Transient overexpression of miR-29a and b (using miRNA mimics) or KLF4 inhibition (using siKLF4) resulted in significantly lower KRT5-luciferase activation in response to P4 compared to vehicle control (Figure 7A). Furthermore, western blot analysis showed that partial knockdown of KLF4 expression using siRNA decreased CK5 levels after 24 h P4 treatment (Figure 7B), and significantly decreased the percentage of CD44+ cells basally and in response to P4 (Figure 7C) in T47D cells. Taken together, these results demonstrate that KLF4 upregulation and miR-29 downregulation contribute to the maximal expansion of the stem-like population by progestins.
Figure 7. miR-29 repression and KLF4 upregulation contribute to maximal promotion of CK5+ and CD44+ cells.

A. Transient miR-29 and siKLF4 transfection decreases P4 induced activation of CK5 promoter. T47D cells stably expressing a luciferase reporter driven by the KRT5-promoter were plated at 10000 cells/well in 96-well plates. After 24h cells were transfected with 50 nM miR-29a (29a) or miR-29b mimics or negative control (NC), 5 nM on-target pool siKLF4 or siNC in combination with a renilla-luciferase plasmid (pRL-SV40) using Dual-transfection reagent. Cells were treated for additional 24h with ethanol (OH) or 100 nM P4 and luciferase activity measured 24h after transfection and normalized to renilla-luciferase control. *P<0.05 B. T47D cells were transfected with 5 or 10 nM siKLF4-smart pool or si-NC control in combination with pGFP plasmid to control for transfection efficiency. After 24h, cells were treated with either ethanol (OH) or 100 nM P4 for 24h and western blotting was performed to detect KLF4 and CK5. C. T47D were transfected with 10nM siNC or si-KLF4. After 24h, cells were treated with vehicle (OH) or 100nM P4 for 24h and CD44+ expression was measured by flow cytometry. Left: Representative flow-charts. Right. Percentage of CD44+ cells from biological triplicates *P<0.05.
DISCUSSION
In cancers, acquisition of stem-like phenotype can occur through multiple means including EMT, selection during prolonged therapy, or local and systemic signals. This study identified a novel mechanism by which progestins stimulate the expansion of breast cancer stem-like cells by both direct and miRNA-mediated control of a potent reprogramming factor.
MiR-29 family members are downregulated in many human cancers as compared to their normal tissue of origin (36,45,46), including invasive breast cancer (47). Expression of miR-29 is associated with more differentiated luminal-subtype human and mouse mammary tumors and is repressed in less differentiated basal-like tumors (48,49). Accordingly, we demonstrate that a decrease in miR-29 levels plays a role in the progestin-mediated acquisition of more de-differentiated CD44+/CK5+ cells. Thus, alterations in miRNA expression constitute one downstream molecular mechanism by which P4 cues cancer cell de-differentiation.
The three miR-29 mature forms are predicted to target the same mRNAs due to the homology in their seed sequence. We found that miR-29a is the most abundant of the three mature forms in luminal breast cancer cells, and the most decreased in the CD44+ cell fraction, suggesting that miR-29a might play the major functional role in the P4-mediated expansion of the CD44+ fraction. In support of this hypothesis, inhibition of miR-29a significantly potentiated the increase in CD44+ and CK5+ cells in response to P4, whereas inhibition of miR-29b or miR-29c did not. Furthermore, miR-29a is more stable than miR-29b in cycling cells (38,50), which could also explain the relatively high abundance of miR-29a.
Decreased levels of mature miR-29 correlated with lower levels of miR-29 precursors, suggesting that miR-29 downregulation results from transcriptional suppression. c-Myc downregulates miR-29a to enhance reprogramming of mouse embryonic fibroblasts (51). Indeed, we demonstrate that progestin-downregulation of miR-29a is transcriptional and also mediated by c-Myc. C-Myc was transiently upregulated by progestins during the first 1h and returned to basal levels by 3h in T47D cells. This parallels the early downregulation of miR-29a after P4 treatment and its return to basal levels by 24h. Likewise, c-Myc upregulation lasted longer (6h) and resulted in more marked miR-29 repression in BT474 cells. Early and transient expression of c-Myc effectively initiates cellular reprogramming to induce pluripotent stem cells (iPSCs) but is dispensable at later stages in mature iPSCs (52). Our data indicate that similar to c-Myc, early downregulation of miR-29a in response to progestins is involved in reprogramming of luminal breast cancer cells.
Sustained miR-29 inhibition increased tumor growth indicating that miR-29 acts as tumor suppressor in T47D cells in vivo. Tumor suppressor roles attributed to miR-29 include reduction of cell proliferation (37), induction of apoptosis (39) and decreased cancer cell invasion and metastasis by targeting extracellular matrix proteins (36). MiR-29 repression did not correlate with increased proliferation in vivo as determined by Ki67 staining, suggesting that the increased growth of 29aZIP tumors results, at least in part, from their increased tumor initiating ability. Indeed, sustained miR-29 repression resulted in increased mammosphere formation in vitro and tumor initiating ability in vivo, suggesting that the small but statistically insignificant increase in the CD44+ population observed in 29aZIP cells in the absence of P4 (Figure 3A, OH-treated cells) might result in significant increases in tumor initiating ability. The tumor initiating-ability of 29aZIP cells was even greater when pre-treated with P4, further supporting a role for miR-29a repression in enhancing the P4-mediated expansion of the stem-like population in luminal breast cancer cells.
We describe for the first time the direct targeting of KLF4 by miR-29 and a role for KLF4 in the P4-mediated expansion of CK5+ and CD44+ cells in ER+PR+ breast cancer. KLF4 is highly expressed in CSC enriched populations in breast cancers and KLF4 knockdown results in decreased tumorigenesis (32). P4 directly upregulates KLF4 in T47D cells (42) indicating that KLF4 expression involves both transcriptional and post-transcriptional regulation. Importantly, P4 treatment is sufficient to relieve repression of the KLF4 3′UTR, corroborating that suppression of miRNAs targeting KLF4 significantly facilitates the progestin-mediated increase in KLF4 expression. We recently reported a similar mechanism for other genes transcriptionally regulated by liganded-PR binding to the promoter, and post-transcriptionally controlled via downregulation of miRNAs targeting their 3′UTR (30).
It is interesting that a potent reprogramming transcription factor for normal cells might also cause reprogramming in cancers. KLF4 directly activates telomerase activity in human ESC and CSCs (31). Additionally, in breast cancer cells, KLF4 maintains the stem cell phenotype and increases cell motility via activation of the Notch pathway (32). Further studies are necessary to determine if these or other pathways activated by KLF4 play a role in the promotion of CK5+ cells.
In this study, stable miR-29 inhibition alone was not sufficient to induce CK5+ cells in the absence of progestins suggesting that progestin-mediated transcriptional activation of KLF4 (and perhaps additional factors) is necessary for the induction of the CK5+ population. Transient inhibition of the pluripotent factor c-Myc did not significantly affect the promotion of CD44+ cells by P4 (not shown) indicating that c-Myc is not solely responsible for this reprogramming. Recently KLF5, another KLF family member upregulated by P4 was found to partially mediate the increase of CK5+ cells in T47D cells in vitro (53). Our finding that KLF4 knockdown or miR-29 overexpression diminishes the P4-mediated CK5 promoter activation and increase in CD44+ cells indicates that upregulation of KLF4 and repression of miR-29 by P4 both contribute to the maximal expansion of the stem-like population in luminal breast cancer cells.
In conclusion, progestins upregulate KLF4 at the transcriptional and post-transcriptional level through downregulation of its repressor, miR-29 (Figure 8). miR-29 repression augments the P4-mediated expansion of the CK5+ CD44+ population. Other progestin-regulated miRNAs may contribute to progestin-dependent reprogramming of a subpopulation of cells in ER+PR+ breast cancers and we are actively investigating this hypothesis. Understanding the molecular mechanisms by which cancer cells can revert to stem-like state and how hormones affect this process will ultimately help us prevent this undesirable plasticity.
Figure 8. Current model.
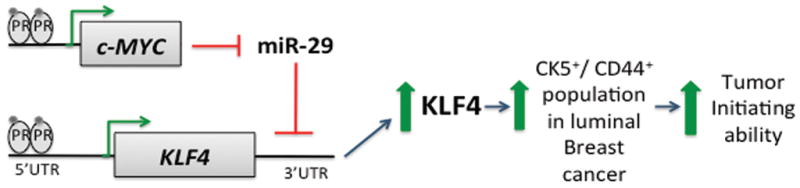
Progestins acting through PR induce transcriptional activation of c-MYC and KLF4. C-MYC is rapidly upregulated to repress miR-29a/b1, thus relieving post-transcriptional repression of KLF4 mRNA. Downregulation of miR-29, and upregulation of KLF4 contribute to maximal expansion of CK5 and CD44+ cells, which in turns increase tumor initiating capability.
METHODS
Cell lines
T47D cells were cultured in DMEM 10% FBS, L-glutamine and penicillin/streptomycin. BT474 cells were maintained in RPMI, 5% FBS, L-glutamine and penicillin/streptomycin.
Stable cell lines
Stable inhibition of miR-29a, b or c was obtained with lentiviral vectors expressing complementary sequences to each mature miRNA (pMIRZIPs, System Biosciences, CA). For overexpression, lentiviral vectors carried precursor sequences for miR-29a, miR-29b1 and miR-29c, respectively. A scrambled, non-silencing, SCR-ZIP vector was used as the negative control. Stable expressing cells were selected based on GFP-based cell sorting (for pMIRNA vectors) or puromycin selection (miRZIP vectors).
Reagents
Primary antibodies included KLF4 (Santa Cruz, CA); α-tubulin (Sigma-Aldrich, St. Louis, MO), CK5 (Vector laboratories, Burlingame, CA), progesterone receptor (Dako, Carpinteria, CA), Ki67 (Dakocytomation, Denmark); c-Myc (Roche, Indianapolis, IN), Dicer (Sigma-Aldrich). For western blot, secondary antibodies were Alexa-fluor 680 Anti-Rabbit or Anti-Mouse IgG (Invitrogen, NY) detected using an Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE). For immunohistochemistry, secondary antibodies were anti-mouse Alexa 488 or anti-rabbit Alexa 555 (Invitrogen). miRNA mimics, miRNA inhibitors, on target-pool siRNAs were obtained from Dharmacon (Lafayette, CO, USA); dual transfection reagent (Invitrogen) and Dual Luciferase Reporter assay system (Promega, Madison, WI).
Flow Cytometry
Cells were labeled with antibodies CD44-APC, CD24-PE (BD Biosciences, Franklin Lakes, NJ) at a concentration of 10 × 106 cells/mL under optimized conditions and were subjected to FACS analysis and sorting on a Moflo XDP 100 (Beckman-coulter). Data analysis was performed using Kaluza® Analysis Software.
RNA Extraction and qRT-PCR
Total RNA from cultured cell lines was isolated using Trizol. To isolate RNA from sorted cells (<10,000), cells were collected in lysis buffer and RNA isolated with the RNAqueous-Micro kit (Ambion, Austin, TX) following manufacturer instructions. cDNA from mature miR-29a, miR-29b and miR-29c was synthesized using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Carlsband, CA) and qRT-PCR was performed using TaqMan MicroRNA Assays (Applied Biosystems). RNU6B was used for normalization. cDNA for quantification of pri-miR-29a, CK5 and KLF4 mRNA levels was synthesized using the MuLV reverse transcriptase (Applied Biosystems). β-actin or GAPDG was used for normalization. The relative mRNA or miRNA levels were calculated using the comparative Ct method (ΔΔCt).
Tumor growth and limiting dilution tumor formation in vivo
Tumor xenografts were developed by injecting indicated amount of cells in 50% Matrigel Basement Membrane Matrix (BD Biosciences, San Jose, CA) into the fourth mouse mammary fad pad of ovariectomized female Non-obese diabetic (NOD)/SCID mice (for tumor growth) or nu/nu mice (for limiting dilution experiments)(Jackson Labs, Maine, USA). Pellets containing either estrogen alone (2 mg) or in combination with MPA (10 mg) were implanted subcutaneously at time of tumor cell injection (54). Tumors were measured weekly using a digital caliper and tumor volume was estimated using the formula lw2/2. At termination of the experiment, mice were euthanized and tumors were excised and weighed. These experiments were performed under an approved University of Colorado Institutional Animal Care and Use Committee protocol.
3D-matrigel colony formation assay
Cells were plated at 1000 cells/well in 8-well chambers (Lab-Tek Chamberslide) in Matrigel pre-coated wells and allowed to grow for 2 to 3 weeks. For quantification of colonies, three fields per well in duplicate wells were collected using a Nikon digital camera (Zeiss) at 4x objective. All images were exported as tiff files, into ImageJ 1.44o, developed by Wayne Rasband for the NIH (http://imagej.nih.gov/ij). Images were first thresholded using the default method in the HSB (hue/saturation/balance) color space. The colonies were analyzed using the analyze particles script. To distinguish colonies from single cells, only particles with size 200-infinity were included in analysis. The “analyze particles” script generates a list of number and cross-sectional area of the colonies that was used to calculate colonies diameters.
Digital imaging
For immunofluorescence analysis, images were collected using a Nikon digital camera and images were exported as tiff files to Adobe Photoshop. Within each figure, minor adjustments were made to brightness and contrast to more accurately portray the appearance of the actual samples. For the experimental and control panels within each figure or subfigure, all manipulations and adjustments were performed identically and in parallel. Within Photoshop, color bitmaps were changed from RGB to CMYK.
Statistical Analysis
Statistics were done using Graphpad Prism 5.0 software. Two-tailed Student’s t tests or ANOVA followed by Bonferroni post hoc tests were used. P<0.05 were considered significant.
Supplementary Material
A. Expression of CK5 in response to P4 in BT474 cells. Cells were pretreated with 10nM E2 for 48h and then treated with 100nM P4 for the indicated times. CK5 mRNA levels were measured by qRT-PCR. Data represents CK5 mRNA levels normalized to β-actin and relative to time 0. Bars are mean ± SEM. *P<0.05. B. Representative immunostaining of CK5+ cells (green) in ZR-75-1 and MCF-7 cells after 24h treatment with vehicle (OH) or 100nM MPA (MPA). Cells were pretreated with 10nM E2 as in B. DAPI stains nuclei (Blue). C. Increase in CD44% cells in response to P4 in T47D cells. T47D cells treated with vehicle (OH) or 100nM P4 for 24h, were labeled and CD44 expression measured by flow cytometry. Bars are mean ± SEM of seven biological replicates and three independent experiments **P<0.001. Paired t-test.
T47D and BT474 cells were treated for 3, 6, and 18h with either 100 nM P4 or vehicle (OH). Data represent miRNA levels normalized to RNU6 and relative to levels at time 0 (expression levels for each miRNA at time 0 were normalized to 1). Bars = mean ± SEM. BT474 cells were pre-treated with 10nM 17-β-estradiol for 48h to induce PR expression prior to progestin stimulation.
A. Top: si-MYC prevents P4-induced upregulation of c-myc in BT474 cells. BT474 cells were pre-treated for 48h with 10nM E2 and then transfected with either 20nM siNC (−) or siMYC (+). After 24h, cells were treated with 100nM P4 for the indicated times. 150ug of total cell lysates were used to measure c-Myc and CK5/6 expression by western blot. α-tubulin is used as a loading control. Bottom: c-Myc inhibition abolishes downregulation of mature miR-29 (left) and pri-miR-29a (right) 18h after P4 treatment. BT474 cells were pretreated with 10nM E2 for 48h, and then transfected with either 20nM siNC or 20nM siMYC. After 24h cells were treated with vehicle (OH) or 100nM P4 for 18h. Data represent miR-29a or pri-miR-29a levels normalized to RNU6B (for miR-29a) and GAPDH (for pri-mir-29a) and relative to siNC-OH-treated cells. *P<0.05. B. P4 inhibitor RU486 prevents P4-mediated repression of miR-29a. BT474 cells were treated for 18h with either vehicle (OH), 100nM P4 (P4) or 100nM P4 in combination with 100nM RU486 (P4+RU486) for the indicated times. qRT-PCR shows miR-29a levels normalized to RNU6B and relative to OH-treated cells. *P<0.05. C. P4 does not affect Dicer protein levels in T47D cells. T47D cells were treated with 100nM P4 for 6, 18 and 24h, and Dicer protein expression was detected by western blot. α-tubulin is used as a loading control.
pmiRGLO-empty vector was transfected into T47D cells with either 50 nM negative control (NC) or miR-29a (m29a) and luciferase activity measured 48h after transfection. Neither P4 nor miR-29a significantly affects luciferase expression of p-MIR-GLO vector. Data represents relative luciferase activity normalized to renilla-luciferase present in the pmiR-GLO vector. Bars are mean ± SEM.
T47D cells stably expressing a luciferase reporter driven by the KRT5-promoter (KRT5-luciferase) or the control luciferase reporter driven by a CMV-promoter (CMV-luciferase) were plated at 10000 cells/well in 96-well plates. After 24h, cells were transfected with a renilla-luciferase control (used for normalization of transfection efficiency in reporter experiments) and treated with either ethanol (OH), 100nM P4 alone (P4) or in combination with P4 antagonist 100 nM RU486 (P4+ RU486). Luciferase activity was measure 24h later. The KRT5-promoter only drives the expression of luciferase in response to P4, and the P4 antagonist RU486 abolishes this effect. The luciferase vector control driven by CMV is not responsive to P4 or RU486. Data represents firefly luciferase normalized to renilla-luciferase (relative luciferase units). Bars=mean± SEM. *P<0.05
Acknowledgments
We thank the University of Colorado Cancer Center Cytometry and Cell Sorting Shared Resource Facility supported by P30CA046934. The human cytokeratin 5 promoter (KRT5) was a gift from Elaine Fuchs (The Rockefeller University). NIH F31CA165668-01 supported ENH. DOD BCRP Postdoctoral Fellowship W81XWH-11-1-0101 (DMC) and DOD Idea Award BCRP W81XWH-11-1-0210 (CAS, JKR) supported this work.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DMC performed most studies. JFS, PH created BT474 stable cell lines and performed RT-PCRs in BT474 cells. ENH quantified immunofluorescence images. SDA created the KRT5-luciferase reporter. NSS performed IHC. DMC, JFS, CAS, BMJ and JKR contributed intellectual input towards the design, implementation and interpretation of results. DMC wrote the manuscript. BMJ, CAS, JKR provided editorial assistance. All authors read and approved the final manuscript.
References
- 1.Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proceedings of the National Academy of Sciences. 2003;100(17):9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ismail PM, Amato P, Soyal SM, DeMayo FJ, Conneely OM, O’Malley BW, et al. Progesterone involvement in breast development and tumorigenesis—as revealed by progesterone receptor “knockout” and “knockin” mouse models. Steroids. 2003 Nov;68(10–13):779–87. doi: 10.1016/s0039-128x(03)00133-8. [DOI] [PubMed] [Google Scholar]
- 3.Lange CA. Challenges to defining a role for progesterone in breast cancer. Steroids. 2008 Oct;73(9–10):914–21. doi: 10.1016/j.steroids.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lydon JP, Ge G, Kittrell FS, Medina D, O’Malley BW. Murine Mammary Gland Carcinogenesis Is Critically Dependent on Progesterone Receptor Function. Cancer Research. 1999;59(17):4276–4284. [PubMed] [Google Scholar]
- 5.Magnusson C, Baron JA, Correia N, Bergström R, Adami HO, Persson I. Breast-cancer risk following long-term oestrogen- and oestrogen-progestin-replacement therapy. Int J Cancer. 1999 May 5;81(3):339–44. doi: 10.1002/(sici)1097-0215(19990505)81:3<339::aid-ijc5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000 Feb 16;92(4):328–32. doi: 10.1093/jnci/92.4.328. [DOI] [PubMed] [Google Scholar]
- 7.Schairer C, Lubin J, Troisi R, Sturgeon S, Brinton L, Hoover R. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA. 2000 Jan 26;283(4):485–91. doi: 10.1001/jama.283.4.485. [DOI] [PubMed] [Google Scholar]
- 8.Asselin-Labat M-L, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010 Jun 10;465(7299):798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 9.Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010 Jun 10;465(7299):803–7. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- 10.Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI, et al. DNA Replication Licensing and Progenitor Numbers Are Increased by Progesterone in Normal Human Breast. Endocrinology. 2009 Jul 1;150(7):3318–3326. doi: 10.1210/en.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proceedings of the National Academy of Sciences. 2008 Apr 15;105(15):5774–5779. doi: 10.1073/pnas.0706216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010 Nov 4;468(7320):98–102. doi: 10.1038/nature09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Böcker W, Moll R, Poremba C, Holland R, Van Diest PJ, Dervan P, et al. Common adult stem cells in the human breast give rise to glandular and myoepithelial cell lineages: a new cell biological concept. Lab Invest. 2002 Jun;82(6):737–46. doi: 10.1097/01.lab.0000017371.72714.c5. [DOI] [PubMed] [Google Scholar]
- 14.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009 Aug;15(8):907–13. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 15.Haughian JM, Pinto MP, Harrell JC, Bliesner BS, Joensuu KM, Dye WW, et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proceedings of the National Academy of Sciences of the United States of America [Internet] 2011 Oct 3; doi: 10.1073/pnas.1106509108. [cited 2011 Oct 21]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21969591. [DOI] [PMC free article] [PubMed]
- 16.Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Research and Treatment. 2010 Jul 28;128:45–55. doi: 10.1007/s10549-010-1078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shcherbata HR, Hatfield S, Ward EJ. The MicroRNA Pathway Plays a Regulatory Role in Stem Cell Division. Cell Cycle. 2006 Jan 16;5:172–5. doi: 10.4161/cc.5.2.2343. [DOI] [PubMed] [Google Scholar]
- 18.Zhang T. microRNA-150 inhibits human CD133-positive liver cancer stem cells through negative regulation of the transcription factor c-Myb. International Journal of Oncology [Internet] 2011 Oct 24; doi: 10.3892/ijo.2011.1242. [cited 2011 Nov 13]; Available from: http://www.spandidos-publications.com/10.3892/ijo.2011.1242. [DOI] [PubMed]
- 19.Yu F, Deng H, Yao H, Liu Q, Su F, Song E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. 2010 Jul 22;29(29):4194–204. doi: 10.1038/onc.2010.167. [DOI] [PubMed] [Google Scholar]
- 20.Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13(2):R45. doi: 10.1186/bcr2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radisky DC. miR-200c at the nexus of epithelial-mesenchymal transition, resistance to apoptosis, and the breast cancer stem cell phenotype. Breast Cancer Res. 2011;13(3):110. doi: 10.1186/bcr2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011 Feb;17(2):211–5. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tessel MA, Krett NL, Rosen ST. Steroid receptor and microRNA regulation in cancer. Curr Opin Oncol. 2010 Nov;22(6):592–7. doi: 10.1097/CCO.0b013e32833ea80c. [DOI] [PubMed] [Google Scholar]
- 24.Cochrane DR, Cittelly DM, Richer JK. Steroid receptors and microRNAs: relationships revealed. Steroids. 2011 Jan;76(1–2):1–10. doi: 10.1016/j.steroids.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Cittelly DM, Das PM, Salvo VA, Fonseca JP, Burow ME, Jones FE. Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis. 2010 Dec;31(12):2049–57. doi: 10.1093/carcin/bgq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cittelly DM, Das PM, Spoelstra NS, Edgerton SM, Richer JK, Thor AD, et al. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol Cancer. 2010;9:317. doi: 10.1186/1476-4598-9-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011 Mar 3;30(9):1082–97. doi: 10.1038/onc.2010.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin S-L, Chang DC, Chang-Lin S, Lin C-H, Wu DTS, Chen DT, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA. 2008 Oct 1;14(10):2115–2124. doi: 10.1261/rna.1162708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin S-L, Chang DC, Lin C-H, Ying S-Y, Leu D, Wu DTS. Regulation of Somatic Cell Reprogramming Through Inducible Mir-302 Expression. Nucl Acids Res. 2011 Feb 1;39(3):1054–65. doi: 10.1093/nar/gkq850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cochrane DR, Jacobsen BM, Connaghan K, Howe EN, Bain DL, Richer JK. Progestin regulated miRNAs that mediate progesterone receptor action in breast cancer. Molecular and Cellular Endocrinology. 2012;355(1):15–24. doi: 10.1016/j.mce.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong C, Hou P, Tseng S, Chien C, Wu K, Chen H, et al. Krüppel-Like Transcription Factor 4 Contributes to Maintenance of Telomerase Activity in Stem Cells. STEM CELLS. 2010 Sep 1;28(9):1510–7. doi: 10.1002/stem.477. [DOI] [PubMed] [Google Scholar]
- 32.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011 May 5;30(18):2161–72. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Andrianakos R, Yang Y, Liu C, Lu W. Kruppel-like Factor 4 (Klf4) Prevents Embryonic Stem (ES) Cell Differentiation by Regulating Nanog Gene Expression. J Biol Chem. 2010 Mar 19;285(12):9180–9. doi: 10.1074/jbc.M109.077958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci USA. 2010 Oct 19;107(42):18115–20. doi: 10.1073/pnas.1006732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen T, Kuo C, Nicholl MB, Sim M-S, Turner RR, Morton DL, et al. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics. 2011 Mar;6(3):388–94. doi: 10.4161/epi.6.3.14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sengupta S, den Boon JA, Chen I-H, Newton MA, Stanhope SA, Cheng Y-J, et al. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proceedings of the National Academy of Sciences. 2008 Apr 15;105(15):5874–5878. doi: 10.1073/pnas.0801130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proceedings of the National Academy of Sciences. 2007 Oct 2;104(40):15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang H-W, Wentzel EA, Mendell JT. A Hexanucleotide Element Directs MicroRNA Nuclear Import. Science. 2007 Jan 5;315(5808):97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 39.Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, Fernandez-Zapico ME. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem. 2010 Aug 1;110(5):1155–64. doi: 10.1002/jcb.22630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore MR, Zhou J-L, Blankenship KA, Strobl JS, Edwards DP, Gentry RN. A sequence in the 5 flanking region confers progestin responsiveness on the human c-myc gene. The Journal of Steroid Biochemistry and Molecular Biology. 1997 Jul;62(4):243–52. doi: 10.1016/s0960-0760(97)00036-8. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006 Aug 25;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 42.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential Gene Regulation by the Two Progesterone Receptor Isoforms in Human Breast Cancer Cells. Journal of Biological Chemistry. 2002 Feb 15;277(7):5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 43.Park S-Y, Lee JH, Ha M, Nam J-W, Kim VN. miR-29 miRNAs activate p53 by targeting p85[alpha] and CDC42. Nat Struct Mol Biol. 2009 Jan;16(1):23–9. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 44.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009 Dec;11(12):1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 45.Pekarsky Y, Santanam U, Cimmino A, Palamarchuk A, Efanov A, Maximov V, et al. Tcl1 Expression in Chronic Lymphocytic Leukemia Is Regulated by miR-29 and miR-181. Cancer Research. 2006 Dec 15;66(24):11590–11593. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 46.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006 Mar;9(3):189–98. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 47.Iorio MV, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA Gene Expression Deregulation in Human Breast Cancer. Cancer Research. 2005;65(16):7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 48.Zhu M, Yi M, Kim CH, Deng C, Li Y, Medina D, et al. Integrated miRNA and mRNA expression profiling of mouse mammary tumor models identifies miRNA signatures associated with mammary tumor lineage. Genome Biology. 2011 Aug 16;12(8):R77. doi: 10.1186/gb-2011-12-8-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin S-F, Dunning MJ, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8(10):R214. doi: 10.1186/gb-2007-8-10-r214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z, Zou J, Wang G-K, Zhang J-T, Huang S, Qin Y-W, et al. Uracils at nucleotide position 9–11 are required for the rapid turnover of miR-29 family. Nucleic Acids Res. 2011 May;39(10):4387–95. doi: 10.1093/nar/gkr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang C-S, Li Z, Rana TM. microRNAs modulate iPS cell generation. RNA. 2011 Aug;17(8):1451–60. doi: 10.1261/rna.2664111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009 Jan 23;136(2):364–77. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu R, Zhou Z, Zhao D, Chen C. The Induction of KLF5 Transcription Factor by Progesterone Contributes to Progesterone-Induced Breast Cancer Cell Proliferation and Dedifferentiation. Molecular Endocrinology. 2011 Jul 1;25(7):1137–1144. doi: 10.1210/me.2010-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sartorius CA, Harvell DME, Shen T, Horwitz KB. Progestins Initiate a Luminal to Myoepithelial Switch in Estrogen-Dependent Human Breast Tumors without Altering Growth. Cancer Research. 2005 Nov 1;65(21):9779–9788. doi: 10.1158/0008-5472.CAN-05-0505. [DOI] [PubMed] [Google Scholar]
- 55.Bu W, Chen J, Morrison GD, Huang S, Creighton CJ, Huang J, et al. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normal-like breast cancer. Oncogene. 2011 May 2;30(43):4399–409. doi: 10.1038/onc.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Expression of CK5 in response to P4 in BT474 cells. Cells were pretreated with 10nM E2 for 48h and then treated with 100nM P4 for the indicated times. CK5 mRNA levels were measured by qRT-PCR. Data represents CK5 mRNA levels normalized to β-actin and relative to time 0. Bars are mean ± SEM. *P<0.05. B. Representative immunostaining of CK5+ cells (green) in ZR-75-1 and MCF-7 cells after 24h treatment with vehicle (OH) or 100nM MPA (MPA). Cells were pretreated with 10nM E2 as in B. DAPI stains nuclei (Blue). C. Increase in CD44% cells in response to P4 in T47D cells. T47D cells treated with vehicle (OH) or 100nM P4 for 24h, were labeled and CD44 expression measured by flow cytometry. Bars are mean ± SEM of seven biological replicates and three independent experiments **P<0.001. Paired t-test.
T47D and BT474 cells were treated for 3, 6, and 18h with either 100 nM P4 or vehicle (OH). Data represent miRNA levels normalized to RNU6 and relative to levels at time 0 (expression levels for each miRNA at time 0 were normalized to 1). Bars = mean ± SEM. BT474 cells were pre-treated with 10nM 17-β-estradiol for 48h to induce PR expression prior to progestin stimulation.
A. Top: si-MYC prevents P4-induced upregulation of c-myc in BT474 cells. BT474 cells were pre-treated for 48h with 10nM E2 and then transfected with either 20nM siNC (−) or siMYC (+). After 24h, cells were treated with 100nM P4 for the indicated times. 150ug of total cell lysates were used to measure c-Myc and CK5/6 expression by western blot. α-tubulin is used as a loading control. Bottom: c-Myc inhibition abolishes downregulation of mature miR-29 (left) and pri-miR-29a (right) 18h after P4 treatment. BT474 cells were pretreated with 10nM E2 for 48h, and then transfected with either 20nM siNC or 20nM siMYC. After 24h cells were treated with vehicle (OH) or 100nM P4 for 18h. Data represent miR-29a or pri-miR-29a levels normalized to RNU6B (for miR-29a) and GAPDH (for pri-mir-29a) and relative to siNC-OH-treated cells. *P<0.05. B. P4 inhibitor RU486 prevents P4-mediated repression of miR-29a. BT474 cells were treated for 18h with either vehicle (OH), 100nM P4 (P4) or 100nM P4 in combination with 100nM RU486 (P4+RU486) for the indicated times. qRT-PCR shows miR-29a levels normalized to RNU6B and relative to OH-treated cells. *P<0.05. C. P4 does not affect Dicer protein levels in T47D cells. T47D cells were treated with 100nM P4 for 6, 18 and 24h, and Dicer protein expression was detected by western blot. α-tubulin is used as a loading control.
pmiRGLO-empty vector was transfected into T47D cells with either 50 nM negative control (NC) or miR-29a (m29a) and luciferase activity measured 48h after transfection. Neither P4 nor miR-29a significantly affects luciferase expression of p-MIR-GLO vector. Data represents relative luciferase activity normalized to renilla-luciferase present in the pmiR-GLO vector. Bars are mean ± SEM.
T47D cells stably expressing a luciferase reporter driven by the KRT5-promoter (KRT5-luciferase) or the control luciferase reporter driven by a CMV-promoter (CMV-luciferase) were plated at 10000 cells/well in 96-well plates. After 24h, cells were transfected with a renilla-luciferase control (used for normalization of transfection efficiency in reporter experiments) and treated with either ethanol (OH), 100nM P4 alone (P4) or in combination with P4 antagonist 100 nM RU486 (P4+ RU486). Luciferase activity was measure 24h later. The KRT5-promoter only drives the expression of luciferase in response to P4, and the P4 antagonist RU486 abolishes this effect. The luciferase vector control driven by CMV is not responsive to P4 or RU486. Data represents firefly luciferase normalized to renilla-luciferase (relative luciferase units). Bars=mean± SEM. *P<0.05