

Abstract
Herpes simplex virus-1 (HSV-1) is a human pathogen that utilizes several strategies to circumvent the host immune response. An immune evasion mechanism employed by HSV-1 is retention of interleukin-1β (IL-1β) in the intracellular space, which blocks the pro-inflammatory activity of IL-1β. Here, we report that HSV-1 infected keratinocytes actively release the also pro-inflammatory IL-1α, preserving the ability of infected cells to signal danger to the surrounding tissue. The extracellular release of IL-1α is independent of inflammatory caspases. In vivo recruitment of leukocytes to early HSV-1 micro-infection sites within the epidermis is dependent upon IL-1 signalling. Following cutaneous HSV-1 infection, mice unable to signal via extracellular IL-1α exhibit an increased mortality rate associated with viral dissemination. We conclude that IL-1α acts as an alarmin essential for leukocyte recruitment and protective immunity against HSV-1. This function may have evolved to counteract an immune evasion mechanism deployed by HSV-1.
Pathogenic microorganisms have evolved to avoid detection by the host in which they proliferate. Their immune evasion strategies are essential for establishing infections; however, parallel to the pathogens acquiring mechanisms to avoid recognition by the host's immune system, the host co-evolves to continue to sense and eliminate microorganisms.
Herpes simplex virus-1 (HSV-1) is a human pathogen that establishes primary and secondary infections of skin and mucosal epithelial cells, and latency in sensory nerve ganglia 1. HSV-1 achieves productive local infection of mammalian cells by inhibiting both inflammatory and apoptotic pathways. The virus can spread to the brain or internal organs such as the liver and lungs; however, such cases are rare as the host normally develops strong and effective immune protection against dissemination of the virus 1,2. Complications from HSV-1 infections are typically only observed in immune compromised individuals and neonates where the immune system is still developing 3-5. However, when either encephalitis or disseminated disease occur, the morbidity and mortality rates are high.
Interleukin-1 (IL-1) represents two pleiotropic cytokines, IL-1α and IL-1β, essential for both innate and adaptive immune responses Reviewed in 6. IL-1α is expressed in high levels in keratinocytes, whereas myeloid cells such as macrophages are a major source of IL-1β. Interestingly, while both IL-1α and IL-1β signal through the extracellular domain of the transmembrane IL-1 receptor type 1 (IL-1R1), both cytokines are initially synthesized as intracellular pro-proteins. The pro-IL-1α and pro-IL-1β can be cleaved by the cysteine proteases calpain and caspase-1, respectively 6. Processing of pro-IL-1β to the mature IL-1β is required for functional activity and extracellular release of the cytokine 6. In contrast, pro-IL-1α is functionally active and may be released from dying cells 6. As such it is believed to act as an alarmin; a protein stimulating recruitment of inflammatory cells to sites of tissue damage after the protein is released from the dying cells 7. It has been shown that necrotic cells release IL-1α in vitro and that these cells can recruit neutrophils when administered in vivo 8,9. However, an alarmin function of IL-1α during a natural physiological response involving in vivo induced cell death has not been reported previously.
Caspase-1, which cleaves pro-IL-1β, is activated following assembly of inflammasomes, multimeric protein complexes activated by microorganisms and tissue damage 10. Caspase-4 and caspase-5 (mouse homologue: caspase-11) are functionally related to caspase-1, as they also initiate inflammation 10. Interestingly, while many activators of the inflammasomes, including pathogens, lead to cleavage and secretion of IL-1β, HSV-1 infected cells fail to release IL-1β 11,12, despite inflammasome assembly and caspase-1 activation 12-14. In fibroblasts, at least, this appears to be due to degradation of IFI16 initiated by the immediate early HSV-1 protein ICP0 and retention of caspase-1 within actin fibres 12. Hence, HSV-1 appears to elude IL-1β dependent immune responses by preventing secretion of IL-1β. The mechanisms whereby IL-1α is released from cells are less well understood and whether HSV-1 targets these as an immune evasion strategy is unknown.
Despite extensive studies of HSV-1's immune evasion strategies in fibroblasts and myeloid cells, less is known about how the virus is sensed in epithelial cells in the skin and mucosal membranes where it causes tissue damage and blisters through cytopathic effects. We demonstrate here that skin keratinocytes release IL-1α following HSV-1 infection in an inflammatory caspase independent manner. This release is important for recruitment of leukocytes to the epidermis during the very early stages of skin infection. Furthermore, IL-1R1 deficient mice are more susceptible to dissemination of the virus and the resulting lethal outcomes. Hence, IL-1α acts as an alarmin signalling danger in response to HSV-1 infection. This mechanism may have evolved to counter the IL-1β targeting HSV-1 immune evasion strategy.
Results
HSV-1 infected keratinocytes release IL-1α
HSV-1 evades the inflammatory functions of IL-1β by preventing secretion of the mature cytokine 11,12. If a similar immunosuppressive strategy is in place to neutralize the related function of IL-1α is unknown. Since keratinocytes express approximately 10-fold more IL-1α than IL-1β 15, 16, 17 and are one of the major cell types in which HSV-1 replicates, we were interested in the role of these cells in signalling danger to the host. Using direct and indirect approaches, it has previously been demonstrated that keratinocytes release IL-1α following infection with the HSV-1 isolates KOS and F 18,19. TLR3 plays an important role in protecting the central nervous system from HSV-1 infections, presumably thorough detection of double stranded RNA that may be produced during the viral life-cycle 20,21. We previously reported that poly(I:C), a synthetic double stranded RNA analogue, induced chemokine expression in keratinocytes and that this induction is partially dependent upon IL-1α release from the cells 22. As predicted, based on these observations, we found elevated levels of IL-1α in medium from cells treated with poly(I:C) (Fig. 1a). Similar levels of IL-1α were present in medium from HSV-1 NS isolate infected human (Fig. 1a) and mouse (Supplementary Fig. 1) primary keratinocytes 6 and 24-hours post-infection (human Fig. 1a; 24-hours mouse Supplementary Fig. 1). While increased IL-1β levels were detected after poly(I:C) treatment, analogous changes in IL-1β levels were not detected after HSV-1 infection (Fig. 1b). The IL-1β released by keratinocytes in response to poly(I:C) has previously been shown to be the inactive pro-IL-1β form 23. Hence, our data demonstrates that IL-1α, but not IL-1β, is successfully released from keratinocytes infected with the HSV-1 NS isolate.
Figure 1. HSV-1 infected keratinocytes release IL-1α.

Primary human keratinocytes were treated with medium only, 25 μg ml-1 poly(I:C), or HSV-1 as indicated. After one hour cells were rinsed with PBS and fresh medium without agents was added. Cultures were allowed to incubate further for 1 (white bars), 2 (grey), 6 (striped) or 24 (black) hours before conditioned medium (a-b) and cells (c) were collected. (a-b) Extracellular levels of IL-1α (a) and IL-1β (b) protein (in the culture medium) were determined by ELISA. (c) IL-1α mRNA levels were determined using the ΔΔCt method with GAPDH as the internal reference gene and medium only treated cells as the external reference point. Data points (n = 2) are means ± s.d. from one representative experiment of at least 3 independent experiments with similar outcomes. *, P < 0.05 (compared to medium only, t-test); **, P < 0.01.
Released IL-1α is derived from a pre-formed pool of protein
In agreement with a previous report examining IL-1α mRNA expression in mouse keratinocytes infected with the HSV-1 KOS isolate 19, we found that levels of the IL-1α mRNA were significantly down-regulated 24 hours post-infection with the HSV-1 NS isolate (Fig. 1c). The absence of an increase in IL-1α mRNA expression correlating with the levels of IL-1α protein detected in the medium (Fig. 1a) suggests that the released IL-1α is primarily derived from a pre-formed pool of protein. Analyses of cellular IL-1α further supported this interpretation, as levels of IL-1α decreased in a time and viral titer (MOI) dependent manner (Fig. 2a). Significantly higher levels of intracellular than extracellular IL-1α (compare Fig. 2a to Fig. 1a) were detected. This likely reflects autocrine functionality of IL-1α leading to internalization and degradation of the cytokine following receptor engagement 24-29 . In summary, our data (Figs. 1-2) show that pre-formed cellular IL-1α is released into the extracellular milieu in response to HSV-1 infection.
Figure 2. IL-1α release precedes cell death and correlates with HSV-1 replication.

Mouse (a-d and f-g) and human (e) keratinocytes were treated with poly(I:C) or infected with HSV-1 as described in Fig. 1 and indicated in each panel. (a) Cellular levels of IL-1α and GAPDH were determined by ELISA. Cellular IL-1α levels are shown standardized against levels of GAPDH. (b) Viability of cells was determined by measuring intracellular levels of ATP. (c) Loss of membrane potential was measured via levels of lactate dehydrogenase (LDH) in the culture medium. (d) Activity of the apoptosis executioner caspases-3/7 was determined enzymatically. (b-d) Cell viability/death readouts were standardized against medium only treated cells at the equivalent time-point. (e) HSV-1 cytopathic effects were detected using brightfield and fluorescence microscopy and nuclear staining with Hoechst and propidium iodide. A giant multinucleated cell containing 4 nuclei is outlined. Additional giant multinucleated cells containing > 2 nuclei are indicated with white block arrows. Horizontal red line arrow indicates a cell with full Hoechst stain brightness surrounded by cells with reduced brightness. White line arrows indicate cells at different stages of nuclear condensation associated with apoptosis. Representative images from 2 independent experiments are shown. Scale bars = 200 μm. (f) Numbers of virus particles in the cell culture medium were determined by quantitative PCR. (g) Equal volumes of cellular protein extracts were examined by Western blotting. The HSV-1 protein gD was detected using serum from HSV-1 infected mice. Levels of host cell proteins were indicated by detection of GAPDH (rabbit anti-GAPDH, FL-335). Two independent samples per time-point and treatment are shown. Full images of Western blots are shown in Supplementary Fig. 2. (a-d and f) Data points (n = 2) are means ± s.d. from one representative experiment of at least 3 independent experiments with similar outcomes. *, P < 0.05 (compared to medium only, t-test); **, P < 0.01; ***, P < 0.001.
Extracellular release of IL-1α precedes cell death execution
HSV-1 is well known to be cytopathic to epithelial cells, including keratinocytes. We found that viability of HSV-1 infected keratinocytes was significantly reduced after 24 hours (Fig. 2b). This loss of viability correlated with loss of membrane potential (Fig. 2c) and activation of the apoptosis executioner caspases-3/7 (Fig. 2d). Formation of multinucleated giant cells could be detected after 12 hours at MOI < 1 (Fig. 2e, MOI 0.2, block arrows). At the 24-hour time-point most HSV-1 infected cells exhibited clear signs of cytopathic effects. However, it is noteworthy that most cells still maintained membrane potential as indicated by the exclusion of propidium iodide (Fig. 2e). Decreased brightness of the Hoechst stain, an indication of apoptosis initiation (Fig. 2e, red arrow indicates a cell with full brightness), could also be detected in many HSV-1 infected cells (Fig. 2e).
Interestingly, the apparent mixed cell death events were detected only at time-points past that at which the first IL-1α was released from the cells (Fig. 1a and Fig. 2a). This shows that the release of IL-1α is an active process preceding execution of cell death programs.
Release of IL-1α correlates with viral replication
The observation that IL-1α is released from cells earlier (Fig. 1a and Fig. 2a) than the cells die (Fig. 2b-e) prompted us to examine timing of viral replication in our model system. Viral particles could be detected in culture medium from infected cells as early as 4 hours post-infection and levels increased over time (Fig. 2f). In agreement with this, trace levels of the HSV-1 glycoprotein D (gD) could be detected in cells at the 4-hour time-point, when cells were infected with the highest viral titer used (Fig. 2g and Supplementary Fig. 2). Higher levels of HSV-1 gD were observed at later time-points where gD could also be detected in cells infected with lower HSV-1 titers (Fig. 2g and Supplementary Fig. 2). At the 24-hour time-point the levels of HSV-1 gD correlated with the MOI, except for the highest titer used (MOI=2.0). The observed lower levels of HSV-1 gD at the latter data point correlated with decreased levels of host cell glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and is consistent with the reduced viability of cells (Fig. 2b) and initiation of cell death (Figs. 2c-d). The timing of viral protein (Fig. 2g and Supplementary Fig. 2) and particle (Fig. 2f) production correlated with the release of IL-1α (Fig. 1a and Fig. 2a). Hence, our observations suggest that the release of IL-1α is initiated by events associated with replication of HSV-1 in the host cell.
HSV-1 activates inflammatory caspases in keratinocytes
It has been reported that the inflammasome is activated in response to HSV-1 infection in fibroblasts in vitro and in the eye in vivo 13,14. However, keratinocytes have not been examined previously. Analyses of the human inflammatory caspases revealed that one or more of these were significantly activated 6 hours post-infection (Fig. 3). Similar observations were made using mouse keratinocytes (Supplementary Fig. 3). Therefore, our data document that HSV-1 does activate inflammasomes in keratinocytes.
Figure 3. Inflammatory caspases are activated in HSV-1 infected keratinocytes.

Human keratinocytes were treated with vehicle or HSV-1. After 6 hours cells were incubated for 2 hours with a fluorescent probe for the active site of the inflammatory caspases 1, 4 and 5. Unbound probe was removed by washing the cells with fresh medium and activated inflammatory caspases visualized using a fluorescence microscope (a, representative images shown, scale bars = 200 μm) or quantified using a plate reader (b, means ± s.d.). ***, P < 0.001 (compared to medium only, t-test). One representative experiment of 3 independent experiments is shown.
IL-1α can be released independently of inflammatory caspases
HSV-1 is believed to activate innate immune mechanisms via double stranded RNA generated during the viral life-cycle and poly(I:C) has been widely used to simulate these responses (e.g. 30,31). Inflammatory caspases have been linked to poly(I:C) induced release of pro-IL-1β 23 and IL-36γ 15. Since both poly(I:C) and HSV-1 trigger release of IL-1α (Fig. 1), we hypothesized that these responses were also dependent upon inflammatory caspases. As predicted, we found that poly(I:C) induced release of IL-1α could be blocked by a caspase inhibitor specific to the inflammatory caspases (Fig. 4a). Surprisingly, IL-1α levels in medium from HSV-1 infected wild type cells and cells deficient of both caspase-1 and caspase-11, the inflammatory caspases in mice, were not significantly different (Fig. 4b). This demonstrates that HSV-1 induced IL-1α is independent of the inflammatory caspases and further suggests that HSV-1 and poly(I:C) activate innate immune mechanisms in keratinocytes through, at least in part, different pathways.
Figure 4. Poly(I:C), but not HSV-1, stimulated IL-1α release is inflammatory caspase dependent.

(a) Human primary keratinocytes were pre-treated with vehicle (white bars) or an inflammatory caspase specific inhibitor (black bars) for 30 min. before addition of medium or 25 μg ml-1 poly(I:C). Culture medium was collected after 12 hours and IL-1α levels determined by ELISA. (b) Mouse primary keratinocytes isolated from wild type (white bars) and inflammatory caspase deficient (black bars) mice were infected with HSV-1 (0.2 MOI) as described in Fig. 1. Control cells were treated with medium only. IL-1α released into the culture medium 24 hours post-infection was quantified using a mouse IL-1α specific ELISA. (a and b) Data points (n = 3) represent means ± s.d. Each experiment was repeated at least twice with similar outcomes. *, P < 0.05 (compared to medium only, t-test); **, P < 0.01.
IL-1 does not affect primary and secondary infections
To explore the physiological role of HSV-1 induced IL-1α release from skin keratinocytes, we employed the flank model of HSV-1 infection. Briefly, in this model mice are scratch inoculated with HSV-1 on the skin (primary infection site). From here, the virus spreads first through retrograde nerve migration to the dorsal root ganglion, and subsequently through anterograde migration back into the skin, where it causes secondary skin disease along the affected nerves. At the virus dose utilized here, death may occur from day 10-14 post-infection due to viral dissemination to vital organs such as the brain, liver and lungs. Death is sometimes preceded by weight loss (Supplementary Fig. 4). Infected wild type and IL-1R1 KO mice revealed no strain specific differences in viral titers at the primary infection sites 2 days post-infection (Supplementary Fig. 5). This demonstrates that wild type and the IL-1R1 KO mice are equally susceptible to the primary HSV-1 infection. Secondary lesions along the sensory nerves started to appear on day 5 in both wild type and IL-1R1 KO mice (see below). Total areas of lesions were comparable in wild type and IL-1R1 KO mice (Supplementary Fig. 6). Hence, HSV-1 appears to spread through the nerves of wild type and IL-1R1 KO mice at a similar rate.
Inflammation is indistinguishable in processed skin
IL-1 has pleiotropic effects upon the immune system 6. Therefore, we examined markers of immunological activation in full thickness skin, comprising both dermis and epidermis, from primary and secondary lesions (Fig. 5). IL-1α can activate T cells, including γδ T cells, and hence modulate IL-17A expression 32. Surprisingly, there were no statistically significant differences between the wild type and the IL-1R1 KO mice in terms of TCR δ chain (an indirect measure of γδ T cell numbers) and IL-17A mRNA expression (Fig. 5a).
Figure 5. Skin inflammation appears independent of IL-1R1 in processed full thickness skin.

Wild type (black circles) and IL-1R1 KO (white boxes) mice (n=4-7 per group) were infected with 1.5 x 106 PFU HSV-1 on flank skin. Five days post-infection the primary and secondary skin lesions were collected separately and expression of cell type marker (TCR δ chain (a), Ly6C and Ly6G (b)) and cytokine (IL-17A (a) and IL-6 (c)) mRNAs examined by real-time RT-PCR. The mRNA levels were standardized against GAPDH and expressed as fold change as described in Fig. 1. No statistically significant (t-test) differences between wild type and IL-1R1 KO mice were identified.
Neutrophils and monocytes/macrophages are among the cell types initially recruited to sites of sterile inflammation reviewed in 33. IL-1α is known to have neutrophil and monocyte targeting pro-inflammatory activity 22,32,34,35; hence, we examined recruitment of these cell types into HSV-1 infected skin of wild type and IL-1R1 KO mice using the granulocyte markers Ly6C and Ly6G. Again, we were surprised to find no statistically significant differences between wild type and IL-1R1 KO mice (Fig. 5b). We further examined expression of the pro-inflammatory cytokine IL-6. While levels of the IL-6 mRNA were found to vary dramatically from mouse to mouse, we observed no statistically significant differences in IL-6 expression between wild type and IL-1R1 KO mice (Fig. 5c). While these expression analyses are only indirect indications of leukocyte recruitment into the skin, they suggest that overall skin inflammation was indistinguishable in wild type and IL-1R1 KO mice.
IL-1 signalling promotes spatial recruitment of leukocytes
The outcome of the expression analyses (Fig. 5) prompted us to examine the 3-dimensional localisation of inflammation within lesions (Fig. 6). As mentioned earlier we did not find that external appearances of lesions progressed differently in wild type and IL-1R1 KO mice (Fig. 6, photographs). Microscopically, however, differences between wild type and IL-1R1 KO mice were identified. Primary lesions exhibited extensive leukocyte recruitment into the skin and the epidermis was absent (Fig. 6a). Interestingly, while in wild type mice a dense infiltrate could be detected where the epidermis previously was, this inflammation appeared more diffuse in IL-1R1 KO mice (Fig. 6a).
Figure 6. IL-1R1 controls spatial leukocyte recruitment to the epidermis.


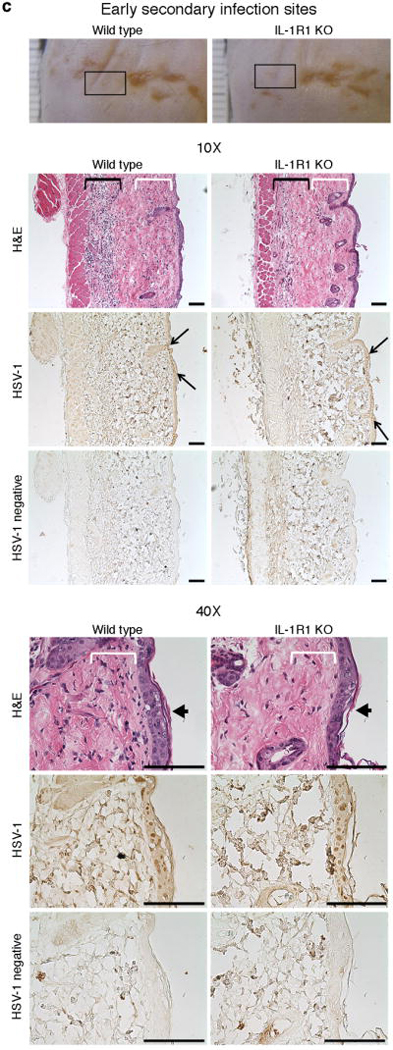

Wild type and IL-1R1 KO mice were scratch inoculated with 1.5 x 106 PFU HSV-1. Five days post-infection primary infection sites (a, white arrows), advanced secondary lesions (b, boxes), and early secondary infection sites (c-d, boxes) were collected, fixed and examined by hematoxylin-eosin (H&E) staining. (c) Consecutive tissue sections were additionally stained with HSV-1 antiserum (HSV-1) or secondary antibody only (HSV-1 negative). (c) White and black brackets indicate positions of sub-epidermal and -cutaneous leukocyte infiltration, respectively. Black line arrows demarcate borders of early secondary infection sites. Black block arrows indicate centres of early secondary infection sites. Representative images from one of two experiments are shown (n = 3-5 per group per experiment). Black scale bars = 100μm. White scale bar = 10μm.
To avoid confounding by the potential effects of viral scratch inoculation and secondary leukocyte recruitment, we further examined inflammation in and around sites of secondary lesions (Fig. 6b, boxes) and early secondary infection sites (Fig. 6c-d, boxes). The latter had yet to develop into lesions, i.e. the epidermis was intact. Interestingly, sites of early epidermal HSV-1 infection, where the virus had not yet destroyed the cells (Fig. 6c, black line arrows), contained high numbers of leukocytes just below the epidermis in wild type mice (Fig. 6c, white brackets; magnified in Fig. 6d). Based on morphology, these cells appeared to be primarily neutrophils and macrophages (Fig. 6d). These cell populations were greatly reduced in IL-1R1 KO mice (Fig. 6c-d, white brackets). It is noteworthy that the timing of leukocyte recruitment in vivo to infected sites, before destruction of the epidermis (Fig. 6c-d), correlates with the release of IL-1α from infected keratinocytes before detectable cell death (Figs. 1-2). Hence, in vivo release of IL-1α from HSV-1 infected cells appears to play an essential role in spatial recruitment of leukocytes to sites immediately adjacent to the HSV-1 infection; in particular, the very early stages of infection before and/or during cell death and tissue damage.
IL-1R1 provides protection against lethal outcomes
HSV-1 infections can lead to serious complications with a potential fatal outcome 3-5. Therefore, we examined the prospective mortality rates of wild type and IL-1R1 KO mice following flank HSV-1 infection. Approximately 25% of the wild type mice died from the infection during a 16-day period (Fig. 7a). The median survival time of the IL-1R1 KO mice was only 13 days (Fig. 7a) and approximately only 40% of the mice were alive at day 16. This represents a statistically significant reduction in survival of the IL-1R1 KO mice compared to wild type mice and demonstrates that IL-1R1 signalling plays an important role in protecting the host against HSV-1 infections.
Figure 7. IL-1R1, but not inflammatory caspases, provides protection from lethal outcome.

Wild type (black lines, a and b), IL-1R1 KO (dotted line, a) and inflammatory caspase KO (dotted line, b) mice were infected with 1.5 x 106 PFU HSV-1 on the flank and survival monitored for 16 days. Data shown are pooled from 4 (a) and 3 (b) independent experiments. n indicates the total number of mice per strain used. **, P < 0.01; #, P > 0.05 (Mantel-Cox and Gehan-Breslow-Wilcoxon tests).
Both IL-1α and IL-1β can signal via IL-1R1. While IL-1α is active in its pro-form, IL-1β requires processing (e.g., by caspase-1) for functional activity and can be released from pyroptotic cells in a caspase-11 dependent manner 6,36 and refs. therein. Mice deficient of the inflammatory caspases, caspase-1 and caspase-11, exhibited the same survival rate as wild type mice (Fig. 7b). Consequently, a role for inflammatory caspase dependent IL-1β in IL-1R1 mediated protection (Fig. 7a) can be excluded. Hence, our data suggest that the protection conferred by IL-1R1 is due to IL-1α engagement.
Wild type and IL-1R1 KO mice show similar disease symptoms
It has previously been reported that TLR3 deficient mice have an increased HSV-2 infectivity of astrocytes 21. The same study indicated that the TLR3 KO mice developed constipation, bladder retention and paralysis following vaginal infection with HSV-2 21. In our flank model, HSV-1 infected mice developed a severe bowel dysfunction syndrome without any bias towards either strain (Supplementary Fig. 7). We did not find bladder retention to be associated with HSV-1 infection (Supplementary Fig. 7), nor did we observe paralysis. Weight differences of wild type and IL-1R1 KO survivors at the end of the experiment (day 16, Fig. 8a) were not statistically significant. The wild type and IL-1R1 KO moribund mice, although their average weight was lower than that of survivors, also had similar weights (Fig. 8a). Furthermore, serum IL-6 levels were comparable in moribund wild type and IL-1R1 KO mice (Supplementary Fig. 8). Hence, IL-1R1 KO mice appear to develop disease similar to that of wild type mice.
Figure 8. Wild type and IL-1R1 KO mice exhibit similar biodistribution of HSV-1.

Wild type (circles, n = 19) and IL-1R1 KO (squares, n = 14) mice were infected with 1.5 x 106 PFU HSV-1 and weight and behaviour monitored for up to 16 days. Moribund mice were identified based on a dramatic and abnormal decrease in activity and euthanized before collection of tissues. (a) Weight at the end of the experiment (day of euthanasia for moribund mice and day 16 for survivors) is represented as percentage of weight at the beginning of the experiment. (b) HSV-1 viral loads were determined by qPCR in moribund mice. Grey boxes indicate the limit of detection. Each colour represents a single mouse. Data shown are pooled from two independent experiments. No statistically significant (t-test) differences between wild type and IL-1R1 KO mice were identified.
IL-1R1 deficiency increases viral dissemination
To better understand the nature of the increased mortality of the IL-1R1 KO mice, viral loads in organs from moribund mice were determined. Somewhat expected given the neurotropic behaviour of the HSV-1 NS strain, HSV-1 could be detected in the brains of moribund mice (Fig. 8b). However, we did not observe a statistically significant difference in brain viral burden between wild type and IL-1R1 KO mice (Fig. 8b).
In humans, HSV can cause disseminated disease involving internal organs such as the lungs and liver 3-5. It also has been reported that different strains of HSV can proliferate in the spleens of infected mice 37. Here, we detected HSV-1 in the lungs, liver and spleen of moribund wild type and IL-1R1 KO mice (Fig. 8b). Interestingly, no trends toward statistically significant differences in viral titers between wild type and IL-1R1 KO mice were detected (Fig. 8b). Since wild type and IL-1R1 KO mice exhibit similar biodistribution of HSV-1 following flank infection (Fig. 8b), our data demonstrate that IL-1R1 does not regulate susceptibility to infection in target organs per se. Instead, the protective function of IL-1R1 (Fig. 7a) appears to be mediated by limiting viral dissemination from the skin or nerves to other organs, i.e. more IL-1R1 KO than wild type mice succumb to fatal disease (Figs. 7a and 8).
Discussion
IL-1α is believed to be an alarmin as the protein is released from dying cells and has pro-inflammatory properties 7,9; however, direct documentation of this function in vivo has been elusive. Our data demonstrate that the cytopathic HSV-1 triggers extracellular release of IL-1α, but not IL-1β, by keratinocytes from a pre-formed pool of the cytokine (Figs. 1-2). During in vivo HSV-1 infection IL-1R1 signalling is required for recruitment of leukocytes to sites immediately adjacent to infected cells in the epidermis (Fig. 6), a tell-tale sign of alarmin activity 7. Hence, our study demonstrates, for the first time, the function of IL-1α as an alarmin during viral skin infections.
Only microorganisms that avoid detection by the host's immune response succeed as pathogens. HSV-1 has developed several immune evasion strategies; one of which involves blocking the function of caspase-1 and extracellular release of the pro-inflammatory mature IL-1β 11,12. The ability of HSV-1 infected keratinocytes to release the alarmin IL-1α preserves the capacity of the infected cells to communicate the present danger and may represent an evolutionary adaptation of the host to counter viral immune evasion strategies. Such co-evolution of the host may explain why most HSV-1 infections are self-limiting and typically only immune deficient individuals are at risk of developing serious complications such as disseminated disease and encephalitis 3-5.
Cells can die through many different pathways and mechanisms. As of 2012, there were at least 12 biochemically distinct types of cell death 38. HSV-1 is cytopathic to keratinocytes; however, the process of cell death remains unclear. It has been reported that HSV-1 initiates apoptosis in the early stages of cell infection 31,39-43. However, as the viral cycle progresses the virus expresses proteins involved in blocking the execution of apoptosis 31,39-43. The latter is believed to be an immune evasion mechanism 31,39, and the initiated cell death may in fact be part of the host's response aimed at eliminating the infection 31,39-43. Similarly, autophagy and necroptosis may be initiated and inhibited by HSV-1 31,44. We previously reported that poly(I:C), a double stranded RNA analogue often utilized to mimic viral infections 30,31, activates pyroptosis in keratinocytes 15. Pyroptosis is an inflammatory form of programmed cell death dependent upon the inflammatory caspases. We here found that poly(I:C) triggered IL-1α release was inflammatory caspase dependent (Fig. 4a). Surprisingly, extracellular IL-1α accumulation induced by HSV-1 was independent of the inflammatory caspases (Fig. 4b). This could suggest that the initiating events are different, e.g. poly(I:C) engages TLR3 23 whereas HSV-1 activates IFI16 45. Alternatively, the activating event leading to extracellular release of IL-1α is the same and the differential requirement for inflammatory caspases is due to the ability of HSV-1 to modulate the inflammasome pathway 11,12 leading to divergence of the pathways. Cellular release of IL-1α correlated with viral replication (Fig. 2f-g) and IL-1α appeared in cell culture medium prior to detectable signs of cell death (Figs. 1-2). However, while the release of IL-1α appears to precede cell lysis, it may still be linked to cell death as an early, programmed, step of self-destruction. Further studies will be required to elucidate how keratinocytes die and release IL-1α following infection with HSV-1.
Recruitment of immune cells to sites of infection is a pre-requisite for initiation of adaptive immunity. We found that IL-1α•IL-1R1 signalling promotes leukocyte placement next to HSV-1 positive cells in the skin (Fig. 6). In addition, IL-1R1 is crucial for preventing lethal systemic disease (Figs. 7a and 8), indicating a role in regulating protective immunity. IL-1R1 is the receptor for both IL-1α and IL-1β. IL-1β must be proteolytically processed, for example, by caspase-1 6, and undergo unconventional protein secretion in a frequently caspase-11 dependent manner 36. Ablation of caspase-1 and caspase-11 did not affect survival of mice in our experimental system (Fig. 7b). This excludes caspase-1/caspase-11 dependent IL-1β release from contributing to the observed IL-1R1 protective function (Fig. 7a). However, alternative pathways, e.g., caspase-8 46 and cathepsin C 47, for IL-1β activation have been discovered in recent years. Yet, HSV-1 infected fibroblasts and macrophages fail to release processed IL-1β due to a poorly understood mechanism deployed by the virus 11,12. In addition, we observed that keratinocytes do not secrete IL-1β in response to HSV-1 (Fig. 1b). Hence, IL-1β release from HSV-1 infected cells appears unlikely as a contributing factor in our infection model.
It remains a possibility that caspase-1/caspase-11 independent release of IL-1β from non-HSV-1 infected cells, for example, in response to tissue damage caused by the virus, could contribute to the here reported IL-1R1-dependent survival (Fig. 7a). A definitive conclusion regarding the possible role of IL-1β in this system will require use of IL-1α and IL-1β KO mice. Such studies may also reveal the specific mechanism(s) of IL-1R1-dependent protective immunity against HSV-1. Evolutionary development of multiple pathways for IL-1β activation may represent host counter measures for viral immune evasion strategies.
Inhibition of IL-1 signalling via transgenic over-expression of the IL-1 receptor antagonist, IL-1Ra, has been shown to protect against ocular damage caused by HSV-1 strain RE infection 48. This protective effect was mediated via a reduction in neutrophil recruitment and angiogenesis 48. In HSV-1 strain NS infected skin we observed a dramatic influx of neutrophils in advanced lesions (Fig. 6), and it is very likely that the recruited neutrophils contributed to the tissue damage leading to skin lesions (Fig. 6). However, we did not detect differences in lesion development and progression between wild type and IL-1R1 KO mice (Fig. 6 and Supplementary Fig. 6). As discussed above, the immune system has several levels of apparent redundancies built into it to counter immune evasion mechanisms developed by pathogens. Since the eye is an immune privileged site there may be less redundancy in the pro-inflammatory mechanisms here compared to non-privileged sites such as the skin. Consequently, a tissue protective effect of inhibiting IL-1•IL-1R1 signalling may be more readily detected in an immune privileged site, e.g. the eye, with fewer pro-inflammatory pathways.
Here, our reported data demonstrate that keratinocytes are capable of signalling danger to the surrounding tissue via IL-1α (Fig. 1). This alarm signal is essential for recruitment of leukocytes to sites of infection (Fig. 6), and development of protective immunity (Fig. 7). Future studies exploring how IL-1α is released from infected cells and how this cytokine supports immune functions may lead to novel immune enhancement strategies for immunocompromised patients with, or at risk of, disseminated disease and/or encephalitis.
Methods
Cell cultures and protein extracts
Primary human neonatal keratinocytes (Life Technologies) were maintained in EpiLife Medium supplemented with EpiLife Defined Growth Supplement and gentamicin (25 ug ml-1, Life Technologies). Alternatively the cells were maintained in Defined Keratinocyte Serum Free Medium (Life Technologies).
Mouse primary keratinocytes were isolated from newborn pups (24-72 hours). The epidermis and dermis were separated after incubation with 0.25% trypsin at 37°C for 45 minutes or over night at 4°C. Epidermal sheets were minced and nutated at 4°C for 45 min in culture medium. Cells were plated on rat-tail type I collagen-coated (4.3 μg cm-1, BD Bioscience) plates and maintained in Keratinocyte Serum-Free Medium (Life Technologies) supplemented with mouse epidermal growth factor (10 ng ml-1, Sigma-Aldrich), calcium chloride (45 μM, final concentration), bovine pituitary extract (140 μg ml-1, Life Technologies) and gentamicin.
African green monkey kidney epithelial cells (Vero, generously provided by Dr. Harvey M. Friedman, University of Pennsylvania, Philadelphia, PA) were maintained in DMEM (Life Technologies) supplemented with 50 mM HEPES, 2 mM glutamine, 5% fetal bovine serum and 10 μg ml-1 gentamicin.
Cellular protein extracts were prepared by freeze-thawing cells three times in M-PER Mammalian Protein Extraction Reagent (Thermo Scientific) supplemented with 1% Mammalian Cells Protease Inhibitor Cocktail (Sigma-Aldrich) and 10 mM EDTA.
Production of virus and infections
The clinical HSV-1 isolate NS (kindly provided by Dr. Harvey M. Friedman) was produced in Vero cells. Viral titers were determined by plaque assays. Briefly, 10-fold serial dilutions were prepared on ice in DMEM serum free medium. Vero cells were grown in a 12-well plate and washed with phosphate saline buffer (PBS) with calcium and magnesium immediately before infection. Subsequently, cells were incubated with 200 μl virus from the serial dilutions at 37°C for 1 hour. After incubation, the cells were washed and overlayed with 1 ml per well of a 1:1 mixture of 2-fold medium (DMEM low glucose powder (Sigma) with supplements) and 1.2% low melting temperature agarose. Plaques were counted approximately 48 hours post-infection.
For experimental infections, cells were incubated with viral multiplicity of infection (MOI) as indicated for 1 hour in serum/protein free medium. Cells were rinsed with PBS before addition of virus free complete culture medium. Cells and conditioned medium were collected at the indicated time points.
Detection and imaging of cell death and viability
Lactate dehydrogenase (LDH) in cell culture medium was measured using the Cytotoxicity Detection Kit (Roche) according to the manufacturer's instructions. Caspase-3/7 activity and cell viability (intracellular ATP) was monitored using the Caspase-Glo 3/7 and CellTiter-Glo, respectively, luciferase assay systems (Promega) following the manufacturer's instructions.
For imaging, cells were cultured on glass bottom dishes (MatTek) coated with rat tail collagen type I (BD Biosciences). Nuclei were sequentially stained with Hoechst 33342 and propidium iodide (ImmunoChemistry Technologies).
Inflammatory caspase activity and inhibition
Activation of the human inflammatory caspases, caspase-1, -4 and -5 (mouse homologues caspase-1 and -11) was monitored using FAM-YVAD-FMK according to the manufacturers (ImmunoChemistry Technologies) instructions. Inflammatory caspases were inhibited with Caspase-1 Inhibitor VI (EMD Biosciences). The inhibitor was solubilized in DMSO (vehicle control) and added to cultures 30 min prior to stimulation with medium only or poly(I:C) (Sigma-Aldrich). Final concentration of the inhibitor was 20-50 μM.
Expression analyses
Levels of human and mouse IL-1α, IL-1β and IL-6 protein were determined using appropriate species specific ELISAs according to the manufacturers' (R&D Systems and Peprotech) instructions. GAPDH protein levels were determined using a direct ELISA approach as previously described 15. Briefly, cell lysates were coated onto 96-well plates and GAPDH detected by sequential incubation with rabbit anti-GAPDH (FL-335, 6 pg ml-1) (Santa Cruz Biotechnology), goat anti-rabbit IgG-HRP (250 ng ml-1, Santa Cruz Biotechnology), and ABTS chromogen (Thermo Scientific).
RNA was isolated from cell cultures using RNeasy (Qiagen) according to the manufacturer's instructions. Skin was homogenized in QIAzol using a Bio-Gen PRO200 homogenizer equipped with interchangeable Multi-Gen 7XL probes (PRO Scientific) and RNA isolated using RNeasy Plus Universal kit (Qiagen) according to the manufacturer's instructions. Relative gene expression was determined by real-time PCR using the comparative ΔΔCt method and GAPDH as the internal reference/housekeeping gene. RNA (1 μg per sample) was reverse transcribed at 37°C for 1.5 hours using 1 μg oligo(dN)6, 5.5 U RNAguard Ribonuclease Inhibitor (GE Healthcare) and 10 U AMV reverse transcriptase (Promega) according to the manufacturer's instructions. Inactivation of AMV was performed at 95°C for 10 mins. The following primers were used for analyses of gene expression in human cells: hIL-1α-forward, 5′-CAGTGAAATTTGACATGGGTG-3′; hIL-1α-reverse, 5′-CAGGCATCTCCTTCAGCAG-3′; hGAPDH-forward, 5′-GGTCGGAGTCAACGGATTTG -3′; hGAPDH-reverse, 5′-TGGGTGGAATCATATTGGAAC-3′. For analyses of gene expression in mouse skin the following primers were used: mIL-1α-forward, 5′-TGAGTCGGCAAAGAAATCAAG-3′; mIL-1α-reverse, 5′-AGTGAGCCATAGCTTGCATC-3′; mTCR delta-forward, 5′-GGAGACTACGGTTCCTGAAAG-3′; mTCR delta-reverse, 5′-AAATGTGGTGGTGAAGGGT-3′; mIL-17A-forward, 5′-TTTTCAGCAAGGAATGTGGA-3′; mIL-17A-reverse, 5′-TTCATTGTGGAGGGCAGA-3′; mIL-6-forward, 5′-GATGGATGCTACCAAACTGG-3′; mIL-6-reverse, 5′-TCTGAAGGACTCTGGCTTTG-3′; mLy6c1-forward, 5′-GGATGGACACTTCTCACACTAC-3′; mLy6c1-reverse, 5′-GCAGTCCCTGAGCTCTTTC-3′; mLy6G-forward, 5′-GCACATGAAAGAGGCAGTATTC-3′; mLy6G-reverse, 5′-GAGACATTGAACAGCACACATAG-3′; mGAPDH-forward, 5′-CTTGTGCAGTGCCAGCC-3′; mGAPDH-reverse, 5′-GCCCAATACGGCCAAATCC-3′.
Equal volumes of cellular protein extracts were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% milk-protein and HSV-1 proteins detected using serum (1/100 dilution) from mice infected with HSV-1. GAPDH (detected with FL-335, 67 ng ml-1) was used as the loading control.
In vivo HSV-1 infections
C57BL/6, IL-1 receptor type 1 (IL-1R1) KO and caspase-1/11 (inflammatory caspases) KO mice were obtained from Jackson Laboratory and bred in house under specific pathogen free conditions. Both KO strains were on the C57BL/6 genetic background. The Temple University Institutional Animal Care and Use Committee approved all animal procedures. Male and female mice of ages 7-12 weeks were used. Mice were matched for age and sex in each individual experiment. Mice were denuded the day before infections. Mice were scratch inoculated with 1.5 × 106 PFU HSV-1 on the right flank. Development of secondary lesions along the sensory nerves was photo-documented and lesion sizes determined using ImageJ. Blood was collected by cardiac puncture immediately after euthanasia.
Titration of HSV-1 in tissue
Primary infection sites were collected using 8 mm biopsy punches. Each lesion was divided in two and one half used for DNA isolation. Organs were frozen and crushed into a powder with autoclaved mortar and pestle over dry ice. DNA was extracted from skin and organs using the DNeasy Blood & Tissue kit from Qiagen according to the manufacturer's instructions. Viral titers at the primary infection sites and organs were determined using equal amounts of total DNA and quantitative real-time PCR with the primer/probe set 5′-CGACCAACTACCCCGAT-3′ (forward primer), 5′-CACTATGACGACAAACAAAATCAC-3′ (reverse primer) and VIC-CAGTTATCCTTAAGGTCTC-MGBNFQ (probe). PCR amplification was performed using TaqMan Gene Expression Master Mix (Applied Biosystems) on an Eppendorf Realplex2 Mastercycler for primary infection site analysis, and an Applied Biosystems StepOnePlus Real-Time PCR System for organ tissue analysis.
Histology and immunohistochemistry
Mouse skin was fixed in 4% formaldehyde, buffered in phosphate-buffered saline, and processed for standard hematoxylin-eosin (H&E) staining at the Histotechnology Facility (Wistar Institute). For immunohistochemistry, paraffin sections were deparaffinized in xylene and passed through decreasing concentrations of alcohol. Sections were microwaved in 10 mM Sodium Citrate (pH 6.0) then allowed to cool to room temperature. Slides were treated with 3% hydrogen peroxide followed by blocking with 5% goat serum, 20% egg whites, and biotin solution (Vector). HSV-1 was detected using serum collected 8 days post-HSV-1 infection of wild-type mice. Serum was added as primary antibody and incubated at room temperature for 1 hour. ECL anti-mouse IgG, horseradish peroxidase linked whole antibody (GE Healthcare) was used as secondary antibody (1/100 in PBS) and allowed to incubate for 30 minutes at room temperature. Bound antibodies were visualized with DAB Plus substrate and chromogen (Ultravision Plus Detection System, Thermo Scientific). Slides were mounted with Shandon-Mount (Thermo Scientific).
Image acquisition
Images were acquired using an Olympus BX60 microscope equipped with Olympus 10×/0.25 and 40×/0.65 lenses and fluorescence capability. Digital acquisition was achieved using SPOT Insight camera (2 mega sample) and software (version 4.5) from Diagnostic Instruments. Imaging processes were Mac operated at room temperature.
Statistical analyses
Statistical significance was determined using t, Mantel-Cox and Gehan-Breslow-Wilcoxon tests. Data are shown as arithmetic means ± standard deviations (s.d.) unless specified otherwise. All experiments were performed at least 3 times with similar outcomes.
Supplementary Material
Acknowledgments
This study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health award R56AI103096 to L. E. Jensen. We would like to thank Dr. Ryan D. King for instructions on performing in vivo HSV-1 inoculations, and Russell Delgiacco for histological services. We also thank Drs. Florina Olaru and Mireia Uribe-Herranz for additional technical support.
Footnotes
Author Contributions: K.A.M, S.L.M and L.E.J performed experiments and analyzed data. J.C.S developed the HSV-1 TaqMan assay. L.E.J. designed the study. K.A.M. provided all artistic illustrations. K.A.M and L.E.J. prepared the manuscript and all authors reviewed and commented on the manuscript.
Competing Financial Interests statement: The authors declare no competing financial interests.
References
- 1.Egan KP, Wu S, Wigdahl B, Jennings SR. Immunological control of herpes simplex virus infections. J Neurovirol. 2013;19:328–345. doi: 10.1007/s13365-013-0189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chew T, Taylor K, Mossman K. Innate and adaptive immune responses to herpes simplex virus. Viruses. 2009;1:979–1002. doi: 10.3390/v1030979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knezevic A, Martic J, Stanojevic M, Jankovic S, Nedeljkovic J, Nikolic L, Pasic S, Jankovic B, Jovanovic T. Disseminated neonatal herpes caused by herpes simplex virus types 1 and 2. Emerg Infect Dis. 2007;13:302–304. doi: 10.3201/eid1302.060907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teh BW, Worth LJ. Disseminated herpes simplex virus infection following epidermal growth factor tyrosine kinase inhibitor therapy for non-small-cell lung carcinoma. Intern Med J. 2012;42:1269–1270. doi: 10.1111/j.1445-5994.2012.02956.x. [DOI] [PubMed] [Google Scholar]
- 5.Riediger C, Sauer P, Matevossian E, Müller MW, Büchler P, Friess H. Herpes simplex virus sepsis and acute liver failure. Clin Transplant. 2009;23:37–41. doi: 10.1111/j.1399-0012.2009.01108.x. [DOI] [PubMed] [Google Scholar]
- 6.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 7.Yang D, Wei F, Tewary P, Howard OMZ, Oppenheim JJ. Alarmin-induced cell migration. Eur J Immunol. 2013;43:1412–1418. doi: 10.1002/eji.201243138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 9.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 10.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 11.Miettinen JJ, Matikainen S, Nyman TA. Global secretome characterization of herpes simplex virus 1-infected human primary macrophages. J Virology. 2012;86:12770–12778. doi: 10.1128/JVI.01545-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virology. 2013;87:5005–5018. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soberman RJ, MacKay CR, Vaine CA, Ryan GB, Cerny AM, Thompson MR, Nikolic B, Primo V, Christmas P, Sheiffele P, Aronov L, Knipe DM, Kurt-Jones EA. CD200R1 supports HSV-1 viral replication and licenses pro-inflammatory signaling functions of TLR2. PLoS ONE. 2012;7:e47740. doi: 10.1371/journal.pone.0047740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M, Covar J, Zhang NY, Chen W, Marshall B, Mo J, Atherton SS. Virus spread and immune response following anterior chamber inoculation of HSV-1 Lacking the Beclin-binding domain (BBD) J Neuroimmunol. 2013;260:82–91. doi: 10.1016/j.jneuroim.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lian LH, Milora KA, Manupipatpong KK, Jensen LE. The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of interleukin-36γ. J Invest Dermatol. 2012;132:1346–1353. doi: 10.1038/jid.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauder DN, Carter CS, Katz SI, Oppenheim JJ. Epidermal cell production of thymocyte activating factor (ETAF) J Invest Dermatol. 1982;79:34–39. doi: 10.1111/1523-1747.ep12510569. [DOI] [PubMed] [Google Scholar]
- 17.Luger TA, Stadler BM, Katz SI, Oppenheim JJ. Epidermal cell (keratinocyte)-derived thymocyte-activating factor (ETAF) J Immunol. 1981;127:1493–1498. [PubMed] [Google Scholar]
- 18.Mikloska Z, Danis VA, Adams S, Lloyd AR, Adrian DL, Cunningham AL. In vivo production of cytokines and β (C-C) chemokines in human recurrent herpes simplex lesions — do herpes simplex virus-infected keratinocytes contribute to their production? J Infect Dis. 1998;177:827–838. doi: 10.1086/515236. [DOI] [PubMed] [Google Scholar]
- 19.Enk C, Sprecher E, Becker Y. Interleukin-1 alpha gene-transcription in murine keratinocytes is inhibited by HSV-1 infection. Arch Virol. 1991;121:141–151. doi: 10.1007/BF01316750. [DOI] [PubMed] [Google Scholar]
- 20.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Héron B, Vallée L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 21.Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnæs-Hansen F, Ulhøi BP, Holm TH, Mogensen TH, Owens T, Nyengaard JR, Thomsen AR, Paludan SR. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest. 2012;122:1368–1376. doi: 10.1172/JCI60893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uribe-Herranz M, Lian LH, Hooper KM, Milora KA, Jensen LE. IL-1R1 signaling facilitates Munro's microabscess formation in psoriasiform imiquimod-induced skin inflammation. J Invest Dermatol. 2013;133:1541–1549. doi: 10.1038/jid.2012.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimstad Ø, Husebye H, Espevik T. TLR3 mediates release of IL-1b and cell death in keratinocytes in a caspase-4 dependent manner. J Dermatol Sci. 2013;72:45–53. doi: 10.1016/j.jdermsci.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Windheim M, Hansen B. Interleukin-1-induced activation of the small GTPase Rac1 depends on receptor internalization and regulates gene expression. Cell Signal. 2014;26:49–55. doi: 10.1016/j.cellsig.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Brissoni B, Agostini L, Kropf M, Martinon F, Swoboda V, Lippens S, Everett H, Aebi N, Janssens S, Meylan E, Felberbaum-Corti M, Hirling H, Gruenberg J, Tschopp J, Burns K. Intracellular trafficking of interleukin-1 receptor I requires Tollip. Curr Biol. 2006;16:2265–2270. doi: 10.1016/j.cub.2006.09.062. [DOI] [PubMed] [Google Scholar]
- 26.Horuk R. Differences in internalization and intracellular processing of interleukin-1 associated with the two forms of interleukin-1 receptor found in B-cells and T-cells. Biochem J. 1991;273:79–83. doi: 10.1042/bj2730079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizel SB, Kilian PL, Lewis JC, Paganelli KA, Chizzonite RA. The interleukin 1 receptor. Dynamics of interleukin 1 binding and internalization in T cells and fibroblasts. J Immunol. 1987;138:2906–2912. [PubMed] [Google Scholar]
- 28.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtis BM, Widmer MB, deRoos P, Qwarnstrom EE. IL-1 and its receptor are translocated to the nucleus. J Immunol. 1990;144:1295–1303. [PubMed] [Google Scholar]
- 30.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Núñez G. Critical role for Cryopyrin/Nalp3 in activation of Caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 31.Dufour F, Bertrand L, Pearson A, Grandvaux N, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus 1 and 2 protect cells against poly(I :C)-induced apoptosis. J Virology. 2011;85:8689–8701. doi: 10.1128/JVI.00362-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol. 2014;14:289–301. doi: 10.1038/nri3646. [DOI] [PubMed] [Google Scholar]
- 33.Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol. 2014;14:232–246. doi: 10.1038/nri3641. [DOI] [PubMed] [Google Scholar]
- 34.Olaru F, Jensen LE. Staphylococcus aureus stimulates neutrophil targeting chemokine expression in keratinocytes through an autocrine IL-1α signaling loop. J Invest Dermatol. 2010;130:1866–1876. doi: 10.1038/jid.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanmiguel JC, Olaru F, Li J, Mohr E, Jensen LE. Interleukin-1 regulates keratinocyte expression of T cell targeting chemokines through interleukin-1 receptor associated kinase-1 (IRAK1) dependent and independent pathways. Cell Signal. 2009;21:685–694. doi: 10.1016/j.cellsig.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, Dietrich A, Lipinski MM, Henry T, Clapham DE, Yuan J. Caspase-11 controls interleukin-1β release through degradation of TRPC1. Cell Reports. 2014;6:1122–1128. doi: 10.1016/j.celrep.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopp SJ, Karaba AH, Cohen LK, Banisadr G, Miller RJ, Muller WJ. Pathogenesis of neonatal herpes simplex 2 disease in a mouse model is dependent on entry receptor expression and route of inoculation. J Virology. 2013;87:474–481. doi: 10.1128/JVI.01849-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2008;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen ML, Blaho JA. Advances in Virus Research. Vol. 69. Academic Press; 2006. Apoptosis during herpes simplex virus infection in; pp. 67–97. [DOI] [PubMed] [Google Scholar]
- 40.Aubert M, Pomeranz L, Blaho J. Herpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cells. Apoptosis. 2007;12:19–35. doi: 10.1007/s10495-006-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraft RM, Nguyen ML, Yang XH, Thor AD, Blaho JA. Caspase 3 activation during herpes simplex virus 1 infection. Virus Res. 2006;120:163–175. doi: 10.1016/j.virusres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Patenode C, Roizman B. US3 protein kinase of HSV-1 cycles between the cytoplasm and nucleus and interacts with programmed cell death protein 4 (PDCD4) to block apoptosis. PNAS. 2011;108:14632–14636. doi: 10.1073/pnas.1111942108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krzyzowska M, Shestakov A, Eriksson K, Chiodi F. Role of Fas/FasL in regulation of inflammation in vaginal tissue during HSV-2 infection. Cell Death Dis. 2011;2:e132. doi: 10.1038/cddis.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavignac Y, Esclatine A. Herpesviruses and Autophagy: Catch Me If You Can! Viruses. 2010;2:314–333. doi: 10.3390/v2010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 2012;5:173–183. doi: 10.1038/mi.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1{beta} maturation by caspase-8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kono H, Orlowski GM, Patel Z, Rock KL. The IL-1—dependent sterile inflammatory response has a substantial caspase-1—independent component that requires cathepsin C. J Immunol. 2012;189:3734–3740. doi: 10.4049/jimmunol.1200136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biswas PS, Banerjee K, Kim B, Rouse BT. Mice transgenic for IL-1 receptor antagonist protein are resistant to herpetic stromal keratitis: possible role for IL-1 in herpetic stromal keratitis pathogenesis. J Immunol. 2004;172:3736–3744. doi: 10.4049/jimmunol.172.6.3736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.