

Abstract
Multiple sclerosis (MS) is a common chronic inflammatory demyelinating disease of the central nervous system (CNS) causing progressive disability. Many observations implicate Epstein–Barr virus (EBV) in the pathogenesis of MS, namely universal EBV seropositivity, high anti-EBV antibody levels, alterations in EBV-specific CD8+ T-cell immunity, increased spontaneous EBV-induced transformation of peripheral blood B cells, increased shedding of EBV from saliva and accumulation of EBV-infected B cells and plasma cells in the brain. Several mechanisms have been postulated to explain the role of EBV in the development of MS including cross-reactivity between EBV and CNS antigens, bystander damage to the CNS by EBV-specific CD8+ T cells, activation of innate immunity by EBV-encoded small RNA molecules in the CNS, expression of αB-crystallin in EBV-infected B cells leading to a CD4+ T-cell response against oligodendrocyte-derived αB-crystallin and EBV infection of autoreactive B cells, which produce pathogenic autoantibodies and provide costimulatory survival signals to autoreactive T cells in the CNS. The rapidly accumulating evidence for a pathogenic role of EBV in MS provides ground for optimism that it might be possible to prevent and cure MS by effectively controlling EBV infection through vaccination, antiviral drugs or treatment with EBV-specific cytotoxic CD8+ T cells. Adoptive immunotherapy with in vitro-expanded autologous EBV-specific CD8+ T cells directed against viral latent proteins was recently used to treat a patient with secondary progressive MS. Following the therapy, there was clinical improvement, decreased disease activity on magnetic resonance imaging and reduced intrathecal immunoglobulin production.
Multiple sclerosis (MS) is a common chronic inflammatory demyelinating disease of the central nervous system (CNS), causing progressive disability and affecting 2.5 million people worldwide.1, 2 It is two to three times more common in females than in males. In most cases the disease initially follows a relapsing–remitting course, where there are repeated attacks of neurological dysfunction, each followed by partial or complete recovery, and a period free of new symptoms. Eventually, most patients with relapsing–remitting MS experience progressive neurological deterioration occurring independently of relapses—secondary progressive MS. In 10–20% of patients, MS follows a primary progressive course, with progressive neurologic deterioration from the onset, sometimes with superimposed relapses. A number of immunomodulatory disease-modifying therapies are available for the treatment of relapsing–remitting MS, but currently there is no effective disease-modifying therapy for progressive MS. A large body of evidence indicates that MS is an autoimmune disease,3, 4 but the primary cause of MS and the other human chronic autoimmune diseases is yet unknown. There is now a large body of evidence indicating that infection with the Epstein–Barr virus (EBV) has a major role in the pathogenesis of MS, although its exact role is incompletely understood.5, 6 This review aims to provide (1) an introduction to the biology of EBV infection, (2) an overview of the evidence implicating EBV in the pathogenesis of MS, (3) a discussion of the proposed mechanisms by which EBV infection contributes to the development of MS and (4) strategies aimed at preventing and treating MS by controlling EBV infection.
Epstein–Barr virus
EBV is a double-stranded DNA γ-herpesvirus causing lifelong infection in a high proportion (≈90%) of the world adult population. As a lymphocryptovirus, it possesses the unique ability to infect, activate and clonally expand B lymphocytes, and then persist as a latent infection within these cells. During primary infection, EBV, transmitted via saliva, enters naive B cells in the tonsil by attaching its surface glycoprotein gp350 to complement receptor 2 (CD21) on the surface of mature B cells and follicular dendritic cells.7 Immediately following initial B-cell infection, EBV expresses two homologues of the cellular anti-apoptotic Bcl-2 protein (BALF1 and BHRF1), which are essential for the survival of newly infected cells.8 EBV drives the infected B cell out of the resting state to become an activated B blast and then takes advantage of the normal pathways of B-cell differentiation to enable the B blast to become a latently infected resting memory B cell.9 To achieve this, the virus uses a series of different latency transcription programmes.9 After entering naive B cells, EBV first employs the latency III or ‘growth' programme to express all viral latent proteins, namely the Epstein–Barr nuclear antigens (EBNAs) 1, 2, 3A, 3B, 3C and LP, and the latent membrane proteins (LMP) 1, 2A and 2B. The thereby activated B blast enters a tonsillar germinal centre where it downregulates the expression of the EBNA proteins 2, 3A, 3B, 3C and LP. Ongoing expression of EBNA1, LMP1 and LMP2 (latency II or ‘default' programme) enables the infected B cell to proceed through a germinal centre reaction to become a memory B cell. The EBV-infected memory B cell exits from the germinal centre and circulates in the blood; it expresses no viral proteins except during cell mitosis, when it expresses only EBNA1 (latency I). EBNA1 engages the host cell DNA polymerase, thereby enabling duplication of each EBV genome and transmission of the genome to each daughter cell. The absence of viral protein expression allows the virus to persist as a latent infection in memory B cells despite a healthy immune response. When latently infected memory B cells return to the tonsils, they can terminally differentiate into plasma cells, which initiates the lytic (replicative) transcription programme with the production of infectious virus.10 The released virions infect tonsil epithelial cells where the virus rapidly replicates and sheds continuously into saliva so that it can be transmitted to new hosts.11 Newly formed virus can also infect additional naive B cells in the same host.
Latently infected memory B cells show the characteristic molecular features of classical antigen-selected memory B cells, namely somatic hypermutation and class-switch recombination of their immunoglobulin (Ig) genes.12 In classical B-cell differentiation, naive B cells, activated by antigen through the B-cell receptor (BCR) and by T-cell help through CD40, proliferate and then proceed through a germinal centre reaction. Strikingly, LMP2A and LMP1, which are expressed by EBV during latency II and latency III, mimic the antigen-activated BCR and the activated CD40 receptor, respectively. In vitro, LMP2A has the ability to mimic and replace constitutive BCR signalling, thereby supporting an activated, proliferative state in B cells, which are resistant to apoptosis.13 In transgenic mice, LMP1 can act as a constitutively active CD40 receptor, completely substituting for CD40 signalling and resulting in normal B-cell development, activation and immune responses, including class-switch recombination, germinal centre formation and somatic hypermutation.14 Although LMP2A and LMP1 have the potential to drive infected B cells through a germinal centre reaction independently of antigen and T-cell help,9 in the tonsils they appear to act synergistically with BCR signalling and CD40 signalling, respectively.15 A further difference between the in vitro and in vivo behaviour of EBV-infected B cells is that when EBV infects B cells in vitro it activates them to become lymphoblasts, which proliferate indefinitely to form a B-lymphoblastoid cell line (LCL), whereas in vivo newly infected lymphoblasts in the tonsils of healthy EBV carriers seemingly undergo very limited proliferation before entering the germinal centre where they proliferate extensively and differentiate into memory B cells.16 The continuous proliferation of B lymphoblasts in LCL in vitro may be a consequence of their not having access to a germinal centre environment to downregulate expression of the EBNA proteins 2, 3A, 3B, 3C and LP.16
To evade immune surveillance, EBV encodes several proteins that inhibit discrete stages of the major histocompatibility complex class I and class II antigen presentation pathways.17 Despite this, EBV infection is normally tightly controlled by EBV-specific immune responses, especially by cytotoxic CD8+ T cells, which kill proliferating and lytically infected B cells.18, 19 A recent study in mice with reconstituted human immune system components suggests that innate immune control by natural killer cells also has an important role in restricting lytic EBV replication during primary infection.20 In the developing world, most children are infected within the first 3 years of life and EBV seropositivity reaches 100% within the first 10 years.21 These early primary infections are nearly always asymptomatic. In contrast, in the developed world, up to 50% of children are EBV seronegative at the end of their first decade and then become infected through intimate oral contact in adolescence or young adulthood.21 As many as half of these delayed primary infections are symptomatic, presenting after an incubation period of 4–7 weeks as acute infectious mononucleosis (AIM) (glandular fever), manifested by fever, fatigue, malaise, pharyngitis and lymphadenopathy.22, 23 During the incubation period, the cycle of infection, B-cell activation, germinal centre reaction, lytic replication and reinfection initially proceeds without interference by cytotoxic CD8+ T cells, because it takes time to mount an adaptive immune response. As a result, the number of latently infected memory B cells during AIM can rise to half, or even higher, of the peripheral memory B-cell compartment.24 Eventually, the infection induces a massive expansion of activated EBV-specific CD8+ T cells, which rapidly control the infection by killing a high proportion of the EBV-infected B cells.22 With the rapid decline in the EBV viral load, the number of EBV-specific CD8+ T cells also rapidly declines towards the levels found in persistently infected healthy virus carriers.25, 26 It has been suggested that the difference between asymptomatic primary EBV infection and AIM is the higher number of EBV-infected B cells in the latter, with the symptoms being due to the massive destruction of virus-infected B cells by cytotoxic CD8+ T cells.26 It is unclear why a higher proportion of B cells should be infected when primary infection is delayed beyond childhood to adolescence, or later. Possible explanations include a higher dose of viral inoculum acquired by intimate oral contact, a less-effective natural killer cell response27 and a reduced capacity to mount a rapid effective CD8+ T-cell response in adolescents/adults compared with young children. Notably, the absolute size of the CD8+ T-cell population in healthy individuals decreases threefold between the ages of 2 and 16 years.28 The pivotal role of CD8+ T cells in controlling primary EBV infection is illustrated by the occurrence of potentially fatal B-cell lymphoproliferative disease in immunosuppressed transplant recipients who have low numbers of EBV-specific T cells and high viral loads.29
Evidence for a role of EBV in the pathogenesis of MS
Universal EBV seropositivity in MS
The first evidence for a role of EBV in the pathogenesis of MS came in 1979 when Fraser et al.30 reported that peripheral blood lymphocytes from patients with clinically active MS have an increased tendency to spontaneous in vitro EBV-induced B-lymphocyte transformation. In the following year, Sumaya et al.31 reported that MS patients have a higher frequency of EBV seropositivity and higher serum anti-EBV antibody titres than controls. Subsequent studies have shown that MS patients are almost universally seropositive for EBV, but not for other viruses.32, 33 In a meta-analysis of 13 case–control studies, 99.5% of MS patients were EBV seropositive compared with 94.0% of controls, with EBV seronegativity having an ORMH odds ratio of MS of 0.06 (exact 95% confidence interval: 0.03, 0.13; P<0.000000001).34 A prospective study has demonstrated that among subjects not infected with EBV the risk of developing MS is extremely low, but after EBV infection there is a sharp increase in risk, with an estimated mean interval of 5.6 years between primary EBV infection and onset of MS.35 The above studies suggest that EBV infection is a prerequisite for developing MS but is not sufficient, by itself, to cause MS because the great majority of people infected with EBV do not develop the disease. Indeed, a recent meta-analysis of 25 studies concluded that EBV infection is present in 100% of MS patients when two independent methods are used to determine EBV seropositivity.36 Moreover, a clinical history of AIM increases the risk of MS, with a relative risk of 2.3.37 Notably, fatigue is a frequent and disabling symptom of both AIM23 and MS.38 It may persist for many months after AIM, especially in females,23 and may herald the onset of MS, often by years.39
Since 1981, it has been noted that the epidemiology of MS is consistent with primary EBV infection in adolescence or young adulthood.40 Indeed, delayed primary infection with EBV can explain all the following epidemiological features of MS: the association with higher socio-economic status; the latitudinal variation in prevalence, which increases with distance from the equator; the effects of migration on the risk of acquiring MS; and the occurrence of clusters and epidemics of MS.41
Humoral immunity to EBV in MS
Patients with MS have elevated levels of serum or plasma IgG antibodies against the EBNA complex,42, 43, 44 EBNA1,44, 45, 46, 47, 48, 49, 50 EBNA2,44 EBNA3 (EBNA3A),50 EBNA4 (EBNA3B),50 EBNA6 (EBNA3C),50 LMP1,50 EBV viral capsid antigen,31, 32, 42, 43, 44 EBV capsid protein VP26 (BFRF3),50 EBV early antigen,43, 44, 51 Epstein–Barr virions52 and the EBV lytic protein BRRF2.45 The increased IgG response to Epstein–Barr virions is directed at multiple components of the virions.52 Serum levels of IgA antibodies to EBV early antigen are also increased in MS patients.48
Of the various EBV proteins, EBNA1 stands out as eliciting the most robust antibody response, its dominance being particularly evident in the study of Ruprecht et al.50 who examined IgG reactivity to 1465 peptides representing 8 full-length EBV proteins, including EBNA1, EBNA3, EBNA4, EBNA6, BLRF2, BZLF1, LMP1 and VP26. The response to EBNA1 is directed at multiple different regions of the molecule, especially within its glycine–alanine repeat (amino acid residues 90–328).50 Elevation of anti-EBNA1 IgG in the serum is present not only at the onset of MS49 but even before the onset of MS.35, 44, 53, 54 Moreover, the serum level of anti-EBNA1 IgG correlates with the number of active inflammatory demyelinating lesions in the brain, as indicated by gadolinium enhancement on magnetic resonance imaging (MRI),55, 56 and with the total number of T2 MRI brain lesions;49 it also correlates with future disease progression measured clinically and by MRI.49, 55 Given that EBNA1 is the only EBV protein expressed by homeostatically proliferating EBV-infected memory B cells,57 the dominance of the anti-EBNA1 IgG response in MS may reflect a high frequency of latently infected cells rather than indicating a specific pathogenic role of these antibodies.
In addition to having elevated levels of anti-EBV antibodies in the serum, patients with MS also have elevated levels of these antibodies in the cerebrospinal fluid (CSF), including IgG antibodies to EBNA1,45, 47, 58, 59 viral capsid antigen,43, 59 EBV early antigen,43 Epstein–Barr virions52 and BRRF2.45 Elevation of anti-EBNA1 and anti-viral capsid antigen IgG in the CSF is present at the onset of MS.59 Elevation of anti-EBV antibodies in the CSF could indicate specific intrathecal synthesis of antibody against EBV or simply reflect the transport of increased anti-EBV antibodies from the blood to the CSF. The antibody index makes this distinction, with an index of ⩾1.5 indicating intrathecal synthesis.60 Some studies have found an elevated anti-EBNA1 IgG index in the CSF of the majority of MS patients,58, 59 whereas others have found this in only a small proportion (<10%) of patients.61, 62 In some patients, some of the characteristic CSF oligoclonal IgG bands have reacted to EBV proteins, indicating intrathecal synthesis.45, 58 However, even in MS patients with an elevated anti-EBV antibody index, anti-EBV IgG comprises only a small fraction (median 0.65%) of the total amount of intrathecally synthesized IgG, much less than in patients with EBV-driven cerebral post-transplantation lymphoproliferative disorder (median 27.82%).63 In MS, there is also intrathecal synthesis of antibodies to other viruses, especially measles, rubella and varicella zoster viruses, as indicated by elevated antibody indices.62, 63, 64, 65 Indeed, the contribution of anti-EBV IgG to total intrathecal IgG production in MS is no greater than the contributions of anti-measles virus IgG,63, 66 anti-rubella virus IgG66 or anti-varicella zoster virus IgG.65 The fractions of total intrathecal IgG production attributable to specific responses to measles, varicella zoster and herpes simplex viruses in MS are 20- to 60-fold lower than in encephalitides caused by direct infection of the CNS by these viruses.65, 66 Intrathecal Ig production in MS is clearly not dependent on the humoral response to EBV or any other single virus, but it could be due to antibody production by clonally expanded EBV-infected B cells/plasma cells within the CNS,6, 67 with EBV-infected autoreactive plasma cells being major contributors, as they are in the synovium in rheumatoid arthritis68 and salivary glands in Sjögren's syndrome.69 A possible explanation for the intrathecal production of antibodies against viruses, such as measles, not present in the brain in MS is the non-specific recruitment to the inflamed CNS, and survival there, of circulating plasmablasts and plasma cells producing antibodies against these viruses.70
Cellular immunity to EBV in MS
The first direct evidence for impaired CD8+ T-cell control of EBV in MS came in 1983 when Craig et al.71 showed that MS patients have impaired regression of the outgrowth of EBV-transformed B cells in vitro, a finding they confirmed in 1985,72 although a more recent study using CD23 expression to detect EBV-infected B cells found normal T-cell-mediated regression.73 Further evidence for impaired CD8+ T-cell control of EBV-infected B cells in MS comes from the observations of decreased T-cell control of Ig-secreting B cells after in vitro infection with EBV,74 a decreased frequency of circulating CD8+ T cells producing interferon-γ in response to autologous LCL75 and a decreased CD8+ T-cell proliferative response to LCL.76 One small study of 11 patients found an increased frequency of CD8+ T cells producing interferon-γ in response to LCL,45 but it is important to note that this was the frequency within the CD8+ T-cell population rather than within peripheral blood mononuclear cells (PBMCs): this fails to take account of the generalized CD8+ T-cell deficiency typical of MS77, 78 and an important determinant of the decreased frequency of EBV-specific CD8+ T cells within PBMCs.79 Studies using synthetic EBV peptides or multimers have found normal,49, 80 increased81, 82, 83 or decreased84 CD8+ T-cell responses to EBV in MS patients. There are several possible reasons for the discrepancy between the results obtained using peptides/multimers on the one hand and EBV-infected B cells on the other to measure CD8+ T-cell reactivity to EBV. First, the responses measured by selected peptides or multimers represent only a small proportion of the aggregrate CD8+ T-cell response to EBV-infected B cells, whereas LCL express not only the latent proteins of EBV but also the lytic proteins,85, 86 because a proportion of the cells in LCL are in the lytic phase of infection. Second, the use of peptides and multimers bypasses the normal physiological process of antigen processing; thus, a person might have a high frequency of T cells producing interferon-γ in response to an exogenously added synthetic EBV peptide, which is presented at only a low density on the surface of EBV-infected B cells so that the infected B cells are poorly recognized by peptide-specific T cells.87 Third, multimer-based analysis does not measure T-cell effector function and hence does not discriminate between healthy and exhausted T cells. Another important variable in all these T-cell studies is the stage of the disease process at which blood is collected. One study found the frequency of EBV-specific CD8+ T cells in the blood to decrease with increasing duration of MS, suggesting T-cell exhaustion;82 another found an increased frequency of CD8+ T cells specific for EBV lytic antigens during the active phase of relapsing–remitting MS.83 It has been proposed that a genetic deficiency of CD8+ T cells impairs control of EBV infection in MS patients, leading to a high EBV load and accumulation of EBV-infected autoreactive B cells and plasma cells in the CNS, with an ensuing vicious circle whereby the inherently deficient CD8+ T-cell response is further compromised by EBV-specific T-cell exhaustion as a result of the persistent high EBV load.6, 79 The CD8+ T-cell deficiency in MS predominantly involves the CD62L– effector memory subset,78 which is responsible for immunosurveillance of the CNS and protection against viral infection.88, 89 Further research is needed to determine the cause of the CD8+ T-cell deficiency in MS and whether it is genetically determined.
EBV-specific CD8+ T cells are enriched in the CSF, compared with that in the blood, in early MS but not in patients with other inflammatory neurological diseases.59 This recruitment of CD8+ T cells to the CNS is selective for EBV-specific T cells, because T cells reactive to cytomegalovirus, another herpesvirus, are not enriched in the CSF.59 In view of the fact that antigen-specific CD8+ T cells infiltrate the brain only when their cognate antigen is present,90 these findings suggest that EBV is present in the CNS in MS. Alternatively, EBV-specific CD8+ T cells in the CSF might be recognizing not EBV but a CNS autoantigen with which they cross-react. LCL-specific CD4+ T cells have been isolated from the CSF of MS patients and patients with other neurological diseases.91
There have been few studies of the CD4+ T-cell response to EBV in MS. One study found a normal frequency of LCL-specific CD4+ T cells in the blood,45 whereas another observed an increased frequency of EBNA1-specific CD4+ T cells.92 There is some evidence of CD4+ T cells cross-reacting with EBV and myelin antigens. In the first reports, two CD4+ T-cell clones specific for myelin basic protein and cross-reacting with EBV DNA polymerase were isolated from an MS patient.93, 94 One subsequent study found that 3–4% of EBNA1-specific CD4+ T cells in healthy subjects and MS patients react with peptides derived from myelin proteins,73 whereas another found no evidence of CD4+ T-cell cross-reactivity between LCL and brain antigens.76
EBV load in MS
Studies on the EBV DNA load in the blood have yielded conflicting results. Some studies have found that the EBV DNA load in PBMC or whole blood is normal in MS patients,95, 96, 97, 98, 99, 100 whereas others have found it to be increased49, 101 or non-significantly increased.92 EBV DNA has been detected in the plasma or serum in only a small proportion (0–7.3%) of MS patients, with no significant differences between MS patients and controls,51, 95, 97, 99, 102, 103, 104 with the exception of the study by Wandinger et al.33 who found EBV DNA in the serum of 72.7% of patients with clinically active disease but in none with clinically stable disease. Some studies,33, 105, 106 but not others,95, 96 have suggested an association between clinical disease activity and elevated EBV DNA load in the blood.
The insensitivity of DNA PCR for detecting EBV in PBMC or whole blood is demonstrated by the detection of EBV DNA in PBMC or whole blood in only 26.4–81.3% of MS patients95, 96, 97, 98, 99, 100, 101 despite virtually all (>99%) MS patients being EBV seropositive.34, 36 The insensitivity of this approach is due to the fact that in healthy EBV-seropositive subjects, the frequency of EBV-infected cells within the peripheral blood B-cell population is only ~5 per 106 B cells.107 A further limitation of the use of DNA PCR to detect EBV in PBMC or whole blood is that the method yields the total EBV genome copy number and does not distinguish between genomes in virions and genomes in latently infected cells. This is important because latently infected cells express only very low numbers (two to five copies) of viral genomes per cell, whereas a single cell replicating the virus and producing virions contains thousands of genomes.108 Therefore, a large increase in the EBV DNA load could be due to a large increase in the frequency of latently infected B cells or a small increase in the proportion of infected cells replicating the virus. To overcome these problems, it is necessary to use methods that identify and quantify single infected cells, such as limiting dilution analysis to measure the precursor frequency of spontaneously transforming B cells in the presence of cyclosporine,109 limiting dilution EBV DNA PCR,108 flow cytometric fluorescent in-situ hybridization (ISH) targeting EBV-encoded small RNA (EBER)110 or single-cell reverse transcription-PCR for EBER.111 Limiting dilution analysis of spontaneously transforming B cells has revealed a 3.6-fold higher frequency of EBV-infected B cells in the peripheral blood of patients with rheumatoid arthritis compared with normal controls,109 and limiting dilution EBV DNA PCR has demonstrated a 10-fold higher frequency of EBV-infected B cells in the blood of patients with systemic lupus erythematosus compared with healthy subjects;108 however, these techniques have not been applied to patients with MS. Recently, Maurer et al.,111 using single-cell reverse transcription-PCR for EBER, found increased frequencies of EBV-infected plasma cells and plasmablasts in the peripheral blood of MS patients. Spontaneous EBV-induced transformation of peripheral blood B cells occurs more frequently in MS patients than in healthy subjects;30, 112, 113 this could be due to an increased frequency of EBV-infected B cells in the blood and/or impaired T-cell-mediated regression of EBV-driven B-cell outgrowth in vitro. In two of these studies, increased spontaneous transformation occurred in the presence of cyclosporine to inhibit T-cell activity,112, 113 suggesting an increased frequency of circulating EBV-infected B cells. However, limiting dilution analysis is needed to confirm this. Children with MS have increased shedding of EBV from saliva, suggesting impairment of immunologic control of EBV.114
EBV DNA has been detected in the CSF or cell pellets isolated from the CSF in only small proportions (0–12.5% and 18.2%, respectively) of MS patients, with no significant differences between MS patients and controls.99, 100, 102, 104, 115 Even in patients with a demonstrated high level of EBV infection in B cells and plasma cells within brain tissue, the frequency of detection of EBV DNA in the CSF was low (12.5%).115 The use of single-cell reverse transcription-PCR for EBER1 on individually sorted B cells and plasma cells from the CSF has yielded conflicting results, with one study detecting no EBV-infected cells116 and another study finding an increased frequency of EBV-infected B cells in the CSF of patients with MS compared with patients with non-inflammatory neurological diseases.111
In 2007, Serafini et al.115 reported that a high proportion of the B cells and plasma cells in the brain in MS are infected with EBV. Meningeal B-cell lymphoid follicles containing germinal centres were major sites of EBV persistence. In contrast, EBV-infected B cells were not found in the brain in other inflammatory CNS diseases. They identified EBV-infected cells using EBER-ISH and by immunohistochemistry with antibodies specific for EBV proteins. EBER-ISH is the gold standard for detection of EBV-infected B cells in histological material117 because EBER, which are non-coding RNA transcripts, are present at high copy numbers in all EBV-infected cells. However, subsequent studies by four other groups, two of whom also employed EBER-ISH, concluded that EBV is rare or absent in the MS brain.116, 118, 119, 120 Although it remains controversial whether EBV in present in the brain in MS,121 additional support for the presence of EBV-infected cells in the brain in MS comes from the study of Tzartos et al.122 Using EBER-ISH and immunocytochemistry, they found EBV-infected cells perivascularly in white matter lesions in seven out of seven MS brains, including four with a high frequency of EBER+ cells. Further support for the presence of EBV in the MS brain has been provided by a highly sensitive radioactive ISH technique using radiolabelled EBER probes, which detected EBER+ cells in MS brain samples123 previously reported to be devoid of EBER+ cells by two groups using non-radioactive EBER-ISH protocols.118, 119
An important question is whether the presence of EBV-infected B cells in the brain is specific for MS. In healthy EBV-seropositive subjects, the frequency of EBV-infected B cells in the peripheral blood is ~5 per 106 B cells.107 Thus, there is likely to be a similar low frequency of EBV-infected B cells at any site of tissue inflammation involving B cells, regardless of cause. For EBV-infected B cells in the CNS to be incriminated in the pathogenesis of MS, they should be present at a substantially higher frequency than would occur if the proportion of EBV-infected B cells within the B-cell component of the tissue infiltrate simply reflected that in the blood. This is certainly the case in the studies that have detected EBV-infected cells in the brain in MS.115, 122, 123 Surprisingly, Tzartos et al.122 also found EBER+ cells in the CNS in two cases of stroke, where B-cell infiltration is sparse.124 They did not identify the type of cell staining positive for EBER with the avidin–biotin complex peroxidase technique, and it is possible that the reactivity resulted from the myeloperoxidase activity of neutrophils, which infiltrate the CNS in ischaemic and haemorrhagic stroke.124, 125 Recently, EBV-infected B cells were also detected in a cervical lymph node, as well as in the CNS, of a patient with primary progressive MS.126
Studies on EBV protein expression in the brain in MS have revealed that a high proportion of B cells within the perivascular cuffs of active and chronic active white matter lesions, meningeal infiltrates and ectopic B-cell follicles coexpress LMP1 and LMP2A, whereas few express EBNA2.115, 127 In contrast, expression of the EBV early lytic cycle protein BFRF1 was restricted to B cells, and particularly plasma cells, in acute lesions, and inside and around meningeal B-cell follicles.115 The expression of LMP1 and LMP2A, but not EBNA2, by B cells in meningeal lymphoid follicles indicates that here EBV employs the latency II (‘default') transcription programme,115, 127 which allows infected B cells to proceed through a germinal centre reaction to become latently infected memory B cells.9 It also fits with the observation that in the tonsils of healthy EBV carriers, EBV-infected B cells using this viral transcription programme not only are functionally indistinguishable from classical germinal centre B cells but also are actually located in the germinal centres.15 Further research is needed to determine whether EBV-infected B cells and plasma cells in the MS brain are autoreactive, as recently shown for EBV-infected plasma cells in the synovium in rheumatoid arthritis68 and salivary glands in Sjögren's syndrome.69
EBV strain variation in MS
EBV strain variation could influence susceptibility to MS in two ways.128 First, different strains of EBV may have different intrinsic biological activities, causing variations in infectivity, B-cell activation or lytic potential, which could affect B-cell homeostasis and the development of autoimmunity. Second, variant EBV strains could evoke different antibody and T-cell immune responses, with less protective immunity or more pathogenic immunity through cross-reactivity with CNS antigens. Brennan et al.128 sequenced the genes encoding EBNA1 and BRRF2 in EBV isolates from MS patients and control subjects. They found that several single-nucleotide polymorphisms within the EBNA1 gene, and one within the BRRF2 gene, occurred at marginally different frequencies in EBV strains infecting MS patients compared with controls. Simon et al.129 sequenced the N- and C-terminal regions of the EBNA1 gene and the C-terminal region of the LMP1 gene, and found no significant differences between MS patients and controls. Two other studies also sequenced the LMP1 gene and observed no differences between MS patients and controls.128, 130 In another study, sequencing of the EBNA2 gene revealed no differences between patients with a first clinical diagnosis of CNS demyelination and control subjects.131 Future studies should be aimed at sequencing the entire EBV genome in EBV isolates from MS patients and healthy EBV carriers.
Mechanisms by which EBV might contribute to the development of MS
Several hypotheses have been proposed to explain the role of EBV in the development of MS: (1) the cross-reactivity hypothesis; (2) the bystander damage hypothesis; (3) the αB-crystallin (mistaken self) hypothesis; and (4) the EBV-infected autoreactive B-cell hypothesis.
Cross-reactivity hypothesis
For many years, the most popular hypothesis was the cross-reactivity hypothesis. This postulates that T cells primed by exposure to EBV antigens cross-react with, and attack, CNS antigens.93, 94 In support of this, one study found that 3–4% of EBNA1-specific CD4+ T cells in healthy subjects and MS patients react with peptides derived from myelin proteins,73 although another study found no evidence of CD4+ T-cell cross-reactivity between LCL and brain antigens.76 The EBV cross-reactivity hypothesis does not explain why the autoimmune attack on the CNS is not switched off by the activation-induced apoptosis of autoreactive T cells that normally occurs when autoreactive T cells enter the CNS.132, 133, 134 Furthermore, this hypothesis does not require or explain the presence of EBV-infected B cells in the brain,115, 122, 123 because cross-reactivity is initiated when T cells are exposed to EBV in lymphoid tissue outside the CNS. Although T-cell cross-reactivity between EBV and CNS antigens might contribute to the disease process in MS, it is unlikely to be the main role of EBV in the development of MS.67
Bystander damage hypothesis
The EBV bystander damage hypothesis proposes that the immune attack on the CNS in MS is directed primarily against EBV antigens, particularly lytic antigens, resulting in bystander damage to the CNS.115 With this hypothesis, MS would not be an autoimmune disease, although secondary autoimmune responses could occur as a result of sensitization to CNS antigens released after virus-targeted bystander damage. In support of this hypothesis, CD8+ T cells have been located in close proximity to lytically infected plasma cells in the brain,83, 115 and the frequency of CD8+ T cells specific for EBV lytic antigens is increased in the blood during the active phase of relapsing–remitting MS, as indicated by clinical relapses and/or gadolinium-enhancing MRI brain lesions.83 A key question is whether the inflammation, demyelination and neurological deficits typical of clinical attacks of MS are caused by the entry into the CNS of EBV-specific CD8+ T cells, CNS-reactive T cells or a combination of both. Some patients also show an increased frequency of myelin-reactive T cells in the blood during attacks of MS.135 The bystander damage hypothesis does not explain the evidence for a primary role of autoimmunity in the development of MS.3, 4, 136 It also questions why an EBV-targeted immune response sufficient to cause bystander CNS damage does not delete EBV-infected B cells from the CNS. EBV-directed bystander damage might contribute to the disease process in MS but it is unlikely to constitute the main pathogenic role of EBV in MS.67 Another possible mechanism whereby EBV-infected B cells might contribute to CNS damage has been suggested by Tzartos et al.122 who found that macrophages and microglia express interferon-α in lesions containing EBV-infected cells. They proposed that EBER molecules released by EBV-infected B cells activate innate immunity through stimulation of Toll-like receptor-3 on macrophages and microglia, and that the resulting antiviral state contributes to CNS inflammation.
αB-crystallin or ‘mistaken self' hypothesis
The αB-crystallin or ‘mistaken self' hypothesis proposes that exposure to infectious agents induces the expression of αB-crystallin, a small heat-shock protein, in lymphoid cells and that the immune system mistakes self αB-crystallin for a microbial antigen and generates a CD4+ T-cell response, which then attacks αB-crystallin derived from oligodendrocytes, with resultant demyelination.137 αB-crystallin is reportedly an immunodominant antigen of CNS myelin from MS patients, which is expressed in oligodendrocytes and myelin in early MS lesions, but not in normal white matter.137 An essential aspect of this hypothesis is that infection of the CNS by a microbial agent, which may not be the same as the one inducing αB-crystallin in lymphoid cells, upregulates the expression of αB-crystallin in oligodendrocytes and provides other ‘danger' signals in the CNS, thereby allowing inflammation to develop. Although the hypothesis is not specific for EBV, EBV is a potential candidate because it induces the expression of αB-crystallin in B cells, which present the protein to CD4+ T cells in a human leukocyte antigen-DR-restricted manner.138 By itself, this hypothesis cannot explain the initial development and subsequent persistence of inflammation in the CNS but it might account for how CD4+ T cells target oligodendrocytes and myelin after initiation of CNS inflammation.
EBV-infected autoreactive B-cell hypothesis
The EBV-infected autoreactive B-cell hypothesis of autoimmunity published in 2003 postulates that human chronic autoimmune diseases, including MS, are caused by EBV infection of autoreactive B cells, which accumulate in the target organ where they produce pathogenic autoantibodies and provide costimulatory survival signals to autoreactive T cells that would otherwise die in the target organ by activation-induced apoptosis.67 It also proposes that the accumulation of EBV-infected autoreactive B cells in the target organ is due to a genetically determined defect in the elimination of EBV-infected B cells by the cytotoxic CD8+ T cells that normally keep EBV infection under stringent control. The probability of EBV infecting naive autoreactive B cells is substantial, because at least 20% of human naive B cells are autoreactive.139 The hypothesis makes predictions that have subsequently been verified, namely the presence of EBV-infected B cells in the brain in MS;115, 122, 123 EBV infection of autoreactive memory B cells during AIM;140 EBV infection of autoreactive plasma cells in the synovium in rheumatoid arthritis68 and salivary glands in Sjögren's syndrome;69 decreased CD8+ T-cell immunity to EBV in MS;75 a beneficial effect in MS of rituximab, which kills B cells, including EBV-infected B cells;141 and a beneficial effect of EBV-specific adoptive immunotherapy in MS.142 It also explains why irradiation of the CNS fails to eradicate the production of oligoclonal IgG,143 because the EBV protein BHRF1, a homologue of the anti-apoptotic protein Bcl-2 and produced by both latently and lytically infected cells,144 inhibits radiation-induced B-cell apoptosis.145
Based on the EBV-infected autoreactive B-cell hypothesis,67 the following scenario is postulated to lead to the development of MS6 (Figure 1). EBV infection in people with a genetically determined deficiency of CD8+ T cells results in a high frequency of EBV-infected memory B cells, including autoreactive B cells, which infiltrate the CNS and produce oligoclonal IgG in the CSF. In people possessing class II human leukocyte antigen types (such as DRB1*1501), which predispose to MS, common systemic infections activate CNS-reactive CD4+ T cells that traffic into the CNS where they are reactivated by EBV-infected B cells presenting CNS antigens. These EBV-infected B cells provide costimulatory survival signals to the T cells, thereby inhibiting the activation-induced T-cell apoptosis that normally occurs when autoreactive T cells enter the CNS.132, 133, 134 The autoreactive T cells initiate an immune attack on the CNS, recruiting macrophages and B cells. CNS antigens released by this attack induce spreading of the immune response to other CNS antigens. Repeated T-cell attacks on the CNS supported by local EBV-infected B cells facilitate the development of meningeal B-cell follicles with germinal centres, which generate CNS-reactive B cells and plasma cells producing autoantibodies that cause demyelination and neuronal damage in the underlying cerebral and cerebellar cortex, leading to the progressive phase of MS. At the same time as the EBV-infected autoreactive B cells in the brain are driving the autoimmune attack on the brain by producing pathogenic autoantibodies and providing costimulatory survival signals to autoreactive T cells, the autoimmune process itself could promote the survival, proliferation and differentiation of the EBV-infected autoreactive B cells by releasing CNS antigens and giving CD4+ T-cell help, which would complement the BCR and CD40 receptor signalling already provided by LMP2A and LMP1, respectively, that is, ‘double signalling'.146, 147 This could lead to a vicious circle wherein EBV-infected autoreactive B cells promote autoimmunity, which in turn promotes EBV infection in the CNS. Such extensive double signalling through the BCR and CD40 pathways in the target organ of patients with chronic autoimmune diseases could be a relatively new experience for EBV in its 40 million years of coevolution with primates.
Figure 1.
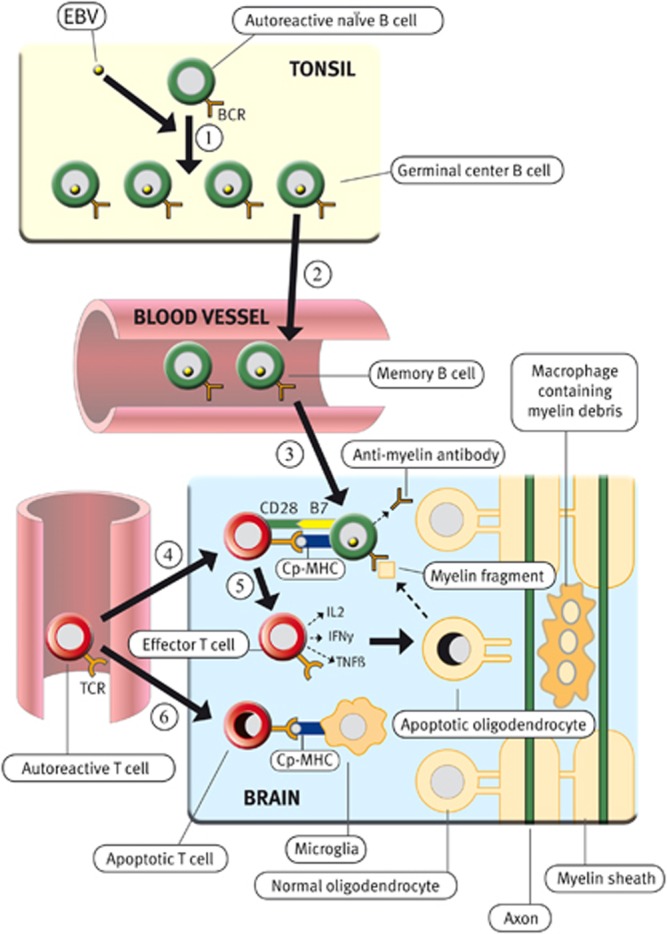
Proposed role of EBV infection in the development of MS. During primary infection, EBV infects autoreactive naïve B cells in the tonsil, driving them to enter germinal centres where they proliferate intensely and differentiate into latently infected autoreactive memory B cells (Step 1), which then exit from the tonsil and circulate in the blood (Step 2). The number of EBV-infected B cells is normally controlled by EBV-specific cytotoxic CD8+ T cells, which kill proliferating and lytically infected B cells, but not if there is a defect in this defence mechanism. Surviving EBV-infected autoreactive memory B cells enter the CNS where they take up residence and produce oligoclonal IgG and pathogenic autoantibodies, which attack myelin and other components of the CNS (Step 3). Autoreactive T cells that have been activated in peripheral lymphoid organs by common systemic infections circulate in the blood and enter the CNS where they are reactivated by EBV-infected autoreactive B cells presenting CNS peptides (Cp) bound to major histocompatibility complex (MHC) molecules (Step 4). These EBV-infected B cells provide costimulatory survival signals (B7) to the CD28 receptor on the autoreactive T cells and thereby inhibit the activation-induced T-cell apoptosis, which normally occurs when autoreactive T cells enter the CNS and interact with non-professional antigen-presenting cells such as astrocytes and microglia, which do not express B7 costimulatory molecules132, 133, 134 (Step 6). After the autoreactive T cells have been reactivated by EBV-infected autoreactive B cells, they produce cytokines such as interleukin-2 (IL2), interferon-γ (IFNγ) and tumour necrosis factor (TNFβ) and orchestrate an autoimmune attack on the CNS with resultant oligodendrocyte and myelin destruction (Step 5). Reproduced from Pender,6 with permission.
Strategies aimed at preventing and treating MS by controlling EBV infection
It has been proposed that effective control of EBV infection will prevent and cure chronic autoimmune diseases, including MS.6, 67, 148, 149 The following strategies of controlling EBV infection have therapeutic potential.
Prevention
Vaccination of healthy EBV-seronegative young adults with recombinant gp350 prevents the development of AIM induced by EBV infection, although it does not prevent asymptomatic infection.150 After vaccination, there was seroconversion to anti-gp350 antibodies persisting for >18 months and accounting for the protective effect, given that anti-gp350 antibody neutralizes EBV infectivity.151 Vaccination of rhesus monkeys with soluble rhesus lymphocryptovirus gp350 not only protects against infection but also reduces viral loads in animals that become infected with virus after challenge.152 As AIM increases the risk of MS,37 vaccination with gp350 might decrease the occurrence of MS by reducing the occurrence of AIM. By reducing the infectivity of EBV, it might prevent MS also in people who would not have developed AIM after EBV infection. Vaccination against EBV latent proteins also has the potential to prevent MS.
Treatment
There are 3 ways to treat MS by controlling EBV infection: (1) B-cell depletion with monoclonal antibodies, (2) antiviral drugs and (3) boosting immunity to EBV. B-cell depletion with rituximab reduces inflammatory brain lesions and clinical relapses in patients with relapsing–remitting MS141 but has the disadvantage of indiscriminately killing, not only EBV-infected B cells but also all uninfected B cells, thereby impairing protective humoral immunity against infectious agents, including EBV.
With regard to antiviral drugs, therapy with aciclovir, which inhibits herpesvirus DNA polymerase, 2.4 g daily for 2 years decreased the relapse rate by 34% in patients with relapsing–remitting MS (P=0.08).153 In a subsequent study, treatment with valaciclovir 3 g daily for 24 weeks reduced the number of new active MRI brain lesions in a subset of MS patients with high levels of MRI-evident disease activity.154 The limited efficacy of aciclovir and valaciclovir in MS might be due to the fact that these drugs act on EBV only when it is using its own DNA polymerase to replicate its DNA. This will apply only to lytically infected cells, but not to latently infected cells, which replicate EBV DNA by using EBNA1 to engage host-cell DNA polymerase. Thus, these antiviral drugs will inhibit EBV in only the minority of the EBV-infected B cells in the brain that are lytically infected but not in the majority that are latently infected.115 An alternative approach is to target the main latent proteins expressed by EBV-infected B cells in the brain in MS, namely LMP1, LMP2A and EBNA1.115, 127 This approach is exemplified by the use of small interfering RNA targeting the LMP1 gene to downregulate LMP1 expression and induce apoptosis in EBV-infected LCL,155 and by the use of small molecule inhibitors of EBNA1.156 Drugs inducing apoptosis of EBV-infected cells by inhibiting EBV-encoded anti-apoptotic proteins such as BHRF1157 might also be beneficial in the treatment of MS.
Improving immunity to EBV in people with MS could be achieved by vaccination with gp350 or EBV latent proteins, administration of humanized or human monoclonal antibody against gp350, or by the infusion of in vitro-expanded autologous EBV-specific CD8+ T cells. Treatment with autologous EBV-specific cytotoxic CD8+ T cells is beneficial in patients with EBV-induced post-transplantation lymphoproliferative disease158 and EBV-associated metastatic nasopharyngeal carcinoma.159 This approach should be feasible in MS because, despite the quantitative deficiency of EBV-specific CD8+ T cells, it is possible to generate EBV-specific CD8+ T-cell lines by in vitro stimulation with autologous LCL.75 Furthermore, EBV-infected B cells from MS patients are not resistant to killing by CD8+ T cells, because EBV-infected LCL from MS patients can be killed normally by HLA-matched EBV-specific CD8+ T-cell clones from healthy subjects, as well as by autologous EBV-specific CD8+ T-cell lines.75
AdE1-LMPpoly is a novel recombinant adenovirus vector encoding multiple CD8+ T-cell epitopes from three EBV latent proteins, namely EBNA1, LMP1 and LMP2A.159 Adoptive immunotherapy with autologous T cells expanded in vitro with AdE1-LMPpoly increases survival in patients with metastatic nasopharyngeal carcinoma, where the EBV-infected carcinoma cells express EBNA1, LMP1 and LMP2A.159 As EBV-infected B cells in the brain in MS express the same three EBV proteins,115, 127 adoptive immunotherapy with AdE1-LMPpoly might be an effective way to increase the number of CD8+ T cells available to eliminate EBV-infected B cells from the CNS in MS. Recently, this approach was used to treat a patient with secondary progressive MS.142 EBV-specific T cells from the patient's blood were expanded by in vitro stimulation with AdE1-LMPpoly and interleukin-2. After expansion, 38.46% of CD8+ T cells, but only 0.22% of CD4+ T cells, reacted to the LMP peptides within AdE1-LMPpoly. The EBV-specific T cells were returned to the patient intravenously at fortnightly intervals. To reduce the risk of aggravating CNS inflammation, an initial dose of 5 × 106 T cells was administered, which is 25% of the median dose used for nasopharyngeal carcinoma,159 with gradual escalation of the dose over the following three infusions to 1 × 107, 1.5 × 107 and 2 × 107 cells. The treatment was successfully completed without significant adverse effects. Following the treatment, the patient experienced a reduction in fatigue and painful lower limb spasms, an improvement in cognition and hand function, and increased productivity at work. These improvements were sustained up to the time of the latest review, 21 weeks after the final T-cell infusion, when neurological examination demonstrated increased voluntary movement of the lower limbs. Following treatment, the frequency of circulating EBV-specific CD8+ T cells increased and there were decreases in intrathecal IgG production and disease activity on brain MRI.142 The beneficial effects of the therapy were attributed to the killing of EBV-infected B cells in the CNS by the adoptively transferred CD8+ T cells.
The adoptive transfer of EBV-specific CD8+ T cells in MS is not without risk. The transferred T cells could aggravate inflammation in the CNS and actually worsen MS, either through cross-reactivity between EBV and CNS antigens or through bystander damage. Clinical trials are needed to determine first the safety and then the efficacy of EBV-specific adoptive immunotherapy in a larger number of patients with progressive MS. In view of the potential risk of aggravating CNS inflammation, this therapy should probably not be tried yet in patients with relapsing–remitting MS, for which a number of disease-modifying therapies are already available.147 Another important question is how long any beneficial effect of EBV-specific adoptive immunotherapy in MS is likely to last. As the therapy does not correct the generalized CD8+ T-cell deficiency that might underlie the impaired CD8+ T-cell immunity to EBV in MS,77, 78 it is likely to be that EBV-specific CD8+ T-cell immunity might eventually wane again after the initial increase from immunotherapy. If such a decrease is accompanied by worsening of MS, consideration should be given to administering a further course of EBV-specific adoptive immunotherapy.
Conclusion
Given the rapidly accumulating evidence for a role of EBV in the pathogenesis of MS, there is ground for optimism that it might be possible to prevent and cure MS by effectively controlling EBV infection. Strategies to control EBV infection include vaccination against EBV, antiviral drugs and adoptive immunotherapy with EBV-specific cytotoxic CD8+ T cells.
Acknowledgments
Our research on the role of EBV in MS has been supported by Multiple Sclerosis Research Australia and the Trish Multiple Sclerosis Research Foundation.
SRB holds a patent on the EBV epitopes included in the AdE1-LMPpoly construct. MPP declares no conflicts of interest.
References
- Pender MP. Neurology 4: multiple sclerosis. Med J Aust. 2000;172:556–562. doi: 10.5694/j.1326-5377.2000.tb124107.x. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Pender MP, Greer JM. Immunology of multiple sclerosis. Curr Allergy Asthma Rep. 2007;7:285–292. doi: 10.1007/s11882-007-0043-x. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Epstein–Barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol. 2010;5:271–277. doi: 10.1007/s11481-010-9201-3. [DOI] [PubMed] [Google Scholar]
- Pender MP. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist. 2011;17:351–367. doi: 10.1177/1073858410381531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerow GR, Mold C, Schwend VK, Tollefson V, Cooper NR. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J Virol. 1987;61:1416–1420. doi: 10.1128/jvi.61.5.1416-1420.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann M, Hammerschmidt W. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005;3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. New Engl J Med. 2004;350:1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol. 2005;79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009;5:e1000496. doi: 10.1371/journal.ppat.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza TA, Stollar BD, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Peripheral B cells latently infected with Epstein-Barr virus display molecular hallmarks of classical antigen-selected memory B cells. Proc Natl Acad Sci USA. 2005;102:18093–18098. doi: 10.1073/pnas.0509311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancao C, Hammerschmidt W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood. 2007;110:3715–3721. doi: 10.1182/blood-2007-05-090142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastelli J, Hömig-Hölzel C, Seagal J, Müller W, Hermann AC, Rajewsky K, et al. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood. 2008;111:1448–1455. doi: 10.1182/blood-2007-10-117655. [DOI] [PubMed] [Google Scholar]
- Roughan JE, Thorley-Lawson DA. The intersection of Epstein-Barr virus with the germinal center. J Virol. 2009;83:3968–3976. doi: 10.1128/JVI.02609-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roughan JE, Torgbor C, Thorley-Lawson DA. Germinal center B cells latently infected with Epstein-Barr virus proliferate extensively but do not increase in number. J Virol. 2010;84:1158–1168. doi: 10.1128/JVI.01780-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressing ME, Horst D, Griffin BD, Tellam J, Zuo J, Khanna R, et al. Epstein-Barr virus evasion of CD8+ and CD4+ T cell immunity via concerted actions of multiple gene products. Semin Cancer Biol. 2008;18:397–408. doi: 10.1016/j.semcancer.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Khanna R, Burrows SR. Role of cytotoxic T lymphocytes in Epstein-Barr virus-associated diseases. Annu Rev Microbiol. 2000;54:19–48. doi: 10.1146/annurev.micro.54.1.19. [DOI] [PubMed] [Google Scholar]
- Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- Chijioke O, Müller A, Feederle R, Barros MHM, Krieg C, Emmel V, et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 2013;5:1489–1498. doi: 10.1016/j.celrep.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson AB, Kieff E.Epstein-Barr virusIn: Knipe DM, Howley PM (ed). Fields Virology Lippincott Williams & Wilkins: Philadelphia; 20012575–2627. [Google Scholar]
- Vetsika EK, Callan M. Infectious mononucleosis and Epstein–Barr virus. Expert Rev Mol Med. 2004;6(23):1–16. doi: 10.1017/S1462399404008440. [DOI] [PubMed] [Google Scholar]
- Macsween KF, Higgins CD, McAulay KA, Williams H, Harrison N, Swerdlow AJ, et al. Infectious mononucleosis in university students in the United Kingdom: evaluation of the clinical features and consequences of the disease. Clin Infect Dis. 2010;50:699–706. doi: 10.1086/650456. [DOI] [PubMed] [Google Scholar]
- Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J Virol. 2004;78:5194–5204. doi: 10.1128/JVI.78.10.5194-5204.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino Y, Morishima T, Kimura H, Nishikawa K, Tsurumi T, Kuzushima K. Antigen-driven expansion and contraction of CD8+ -activated T cells in primary EBV infection. J Immunol. 1999;163:5735–5740. [PubMed] [Google Scholar]
- Hadinoto V, Shapiro M, Greenough TC, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. On the dynamics of acute EBV infection and the pathogenesis of infectious mononucleosis. Blood. 2008;111:1420–1427. doi: 10.1182/blood-2007-06-093278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson AB, Long HM, Palendira U, Münz C, Hislop AD. Cellular immune controls over Epstein–Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol. 2014;35:159–169. doi: 10.1016/j.it.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Comans-Bitter WM, de Groot R, van den Beemd R, Neijens HJ, Hop WCJ, Groeneveld K, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997;130:388–393. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- Smets F, Latinne D, Bazin H, Reding R, Otte JB, Buts JP, et al. Ratio between Epstein-Barr viral load and anti-Epstein-Barr virus specific T-cell response as a predictive marker of posttransplant lymphoproliferative disease. Transplantation. 2002;73:1603–1610. doi: 10.1097/00007890-200205270-00014. [DOI] [PubMed] [Google Scholar]
- Fraser KB, Millar JHD, Haire M, McCrea S. Increased tendency to spontaneous in-vitro lymphocyte transformation in clinically active multiple sclerosis. Lancet. 1979;314:715–717. [PubMed] [Google Scholar]
- Sumaya CV, Myers LW, Ellison GW. Epstein-Barr virus antibodies in multiple sclerosis. Arch Neurol. 1980;37:94–96. doi: 10.1001/archneur.1980.00500510052009. [DOI] [PubMed] [Google Scholar]
- Bray PF, Bloomer LC, Salmon VC, Bagley MH, Larsen PD. Epstein-Barr virus infection and antibody synthesis in patients with multiple sclerosis. Arch Neurol. 1983;40:406–408. doi: 10.1001/archneur.1983.04050070036006. [DOI] [PubMed] [Google Scholar]
- Wandinger K-P, Jabs W, Siekhaus A, Bubel S, Trillenberg P, Wagner H-J, et al. Association between clinical disease activity and Epstein-Barr virus reactivation in MS. Neurology. 2000;55:178–184. doi: 10.1212/wnl.55.2.178. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann Neurol. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- Levin LI, Munger KL, O'Reilly EJ, Falk KI, Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol. 2010;67:824–830. doi: 10.1002/ana.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakpoor J, Disanto G, Gerber JE, Dobson R, Meier UC, Giovannoni G, et al. The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: a meta-analysis. Mult Scler. 2013;19:162–166. doi: 10.1177/1352458512449682. [DOI] [PubMed] [Google Scholar]
- Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol. 2006;59:499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- Krupp LB, Serafin DJ, Christodoulou C. Multiple sclerosis-associated fatigue. Expert Rev Neurother. 2010;10:1437–1447. doi: 10.1586/ern.10.99. [DOI] [PubMed] [Google Scholar]
- Berger JR, Pocoski J, Preblick R, Boklage S. Fatigue heralding multiple sclerosis. Mult Scler. 2013;19:1526–1532. doi: 10.1177/1352458513477924. [DOI] [PubMed] [Google Scholar]
- Warner HB, Carp RI. Multiple sclerosis and Epstein-Barr virus. Lancet. 1981;318:1290. doi: 10.1016/s0140-6736(81)91527-0. [DOI] [PubMed] [Google Scholar]
- Haahr S, Höllsberg P. Multiple sclerosis is linked to Epstein-Barr virus infection. Rev Med Virol. 2006;16:297–310. doi: 10.1002/rmv.503. [DOI] [PubMed] [Google Scholar]
- Larsen PD, Bloomer LC, Bray PF. Epstein-Barr nuclear antigen and viral capsid antigen antibody titers in multiple sclerosis. Neurology. 1985;35:435–438. doi: 10.1212/wnl.35.3.435. [DOI] [PubMed] [Google Scholar]
- Sumaya CV, Myers LW, Ellison GW, Ench Y. Increased prevalence and titer of Epstein-Barr virus antibodies in patients with multiple sclerosis. Ann Neurol. 1985;17:371–377. doi: 10.1002/ana.410170412. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernán MA, Olek MJ, et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA. 2001;286:3083–3088. doi: 10.1001/jama.286.24.3083. [DOI] [PubMed] [Google Scholar]
- Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Büssow K, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115:1352–1360. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundström P, Nyström M, Ruuth K, Lundgren E. Antibodies to specific EBNA-1 domains and HLA DRB1*1501 interact as risk factors for multiple sclerosis. J Neuroimmunol. 2009;215:102–107. doi: 10.1016/j.jneuroim.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Jafari N, van Nierop GP, Verjans GMGM, Osterhaus ADME, Middeldorp JM, Hintzen RQ. No evidence for intrathecal IgG synthesis to Epstein Barr virus nuclear antigen-1 in multiple sclerosis. J Clin Virol. 2010;49:26–31. doi: 10.1016/j.jcv.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Lindsey JW, Hatfield LM, Vu T. Epstein–Barr virus neutralizing and early antigen antibodies in multiple sclerosis. Eur J Neurol. 2010;17:1263–1269. doi: 10.1111/j.1468-1331.2010.03005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lünemann JD, Tintoré M, Messmer B, Strowig T, Rovira A, Perkal H, et al. Elevated Epstein–Barr virus-encoded nuclear antigen-1 immune responses predict conversion to multiple sclerosis. Ann Neurol. 2010;67:159–169. doi: 10.1002/ana.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht K, Wunderlich B, Gieß R, Meyer P, Loebel M, Lenz K, et al. Multiple sclerosis: The elevated antibody response to Epstein–Barr virus primarily targets, but is not confined to, the glycine–alanine repeat of Epstein–Barr nuclear antigen-1. J Neuroimmunol. 2014;272:56–61. doi: 10.1016/j.jneuroim.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Buljevac D, van Doornum GJJ, Flach HZ, Groen J, Osterhaus ADME, Hop W, et al. Epstein-Barr virus and disease activity in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2005;76:1377–1381. doi: 10.1136/jnnp.2004.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JW, Khan U, Ansari W, Powell T, Wang YH, Guirguis MS. The antibody response to Epstein-Barr virions is altered in multiple sclerosis. J Neuroimmunol. 2013;254:146–153. doi: 10.1016/j.jneuroim.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Sundström P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nyström L, et al. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. 2004;62:2277–2282. doi: 10.1212/01.wnl.0000130496.51156.d7. [DOI] [PubMed] [Google Scholar]
- Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. 2005;293:2496–2500. doi: 10.1001/jama.293.20.2496. [DOI] [PubMed] [Google Scholar]
- Farrell RA, Antony D, Wall GR, Clark DA, Fisniku L, Swanton J, et al. Humoral immune response to EBV in multiple sclerosis is associated with disease activity on MRI. Neurology. 2009;73:32–38. doi: 10.1212/WNL.0b013e3181aa29fe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvistad S, Myhr KM, Holmøy T, Bakke S, Beiske AG, Bjerve KS, et al. Antibodies to Epstein-Barr virus and MRI disease activity in multiple sclerosis Mult Scler 2014. doi: 10.1177/1352458514533843 [DOI] [PubMed]
- Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Demonstration of the Burkitt's lymphoma Epstein–Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci USA. 2004;101:239–244. doi: 10.1073/pnas.2237267100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand KH, Houck H, Denslow ND, Heilman KM. Epstein-Barr virus nuclear antigen-1 (EBNA-1) associated oligoclonal bands in patients with multiple sclerosis. J Neurol Sci. 2000;173:32–39. doi: 10.1016/s0022-510x(99)00298-1. [DOI] [PubMed] [Google Scholar]
- Jaquiéry E, Jilek S, Schluep M, Meylan P, Lysandropoulos A, Pantaleo G, et al. Intrathecal immune responses to EBV in early MS. Eur J Immunol. 2010;40:878–887. doi: 10.1002/eji.200939761. [DOI] [PubMed] [Google Scholar]
- Reiber H, Lange P. Quantification of virus-specific antibodies in cerebrospinal fluid and serum: sensitive and specific detection of antibody synthesis in brain. Clin Chem. 1991;37:1153–1160. [PubMed] [Google Scholar]
- Castellazzi M, Tamborino C, Cani A, Negri E, Baldi E, Seraceni S, et al. Epstein–Barr virus-specific antibody response in cerebrospinal fluid and serum of patients with multiple sclerosis. Mult Scler. 2010;16:883–887. doi: 10.1177/1352458510368051. [DOI] [PubMed] [Google Scholar]
- Pohl D, Rostasy K, Jacobi C, Lange P, Nau R, Krone B, et al. Intrathecal antibody production against Epstein-Barr and other neurotropic viruses in pediatric and adult onset multiple sclerosis. J Neurol. 2010;257:212–216. doi: 10.1007/s00415-009-5296-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C, Oltmann A, Stein A, Frenzel K, Schroeter J, Habbel P, et al. Intrathecal EBV antibodies are part of the polyspecific immune response in multiple sclerosis. Neurology. 2011;76:1316–1321. doi: 10.1212/WNL.0b013e318215286d. [DOI] [PubMed] [Google Scholar]
- Reiber H, Ungefehr S, Jacobi C. The intrathecal, polyspecific and oligoclonal immune response in multiple sclerosis. Mult Scler. 1998;4:111–117. doi: 10.1177/135245859800400304. [DOI] [PubMed] [Google Scholar]
- Otto C, Hofmann J, Finke C, Zimmermann M, Ruprecht K. The fraction of varicella zoster virus-specific antibodies among all intrathecally-produced antibodies discriminates between patients with varicella zoster virus reactivation and multiple sclerosis. Fluids Barriers CNS. 2014;11:3. doi: 10.1186/2045-8118-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi C, Lange P, Reiber H. Quantitation of intrathecal antibodies in cerebrospinal fluid of subacute sclerosing panencephalitis, herpes simplex encephalitis and multiple sclerosis: discrimination between microorganism-driven and polyspecific immune response. J Neuroimmunol. 2007;187:139–146. doi: 10.1016/j.jneuroim.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. 2003;24:584–588. doi: 10.1016/j.it.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Croia C, Serafini B, Bombardieri M, Kelly S, Humby F, Severa M, et al. Epstein–Barr virus persistence and infection of autoreactive plasma cells in synovial lymphoid structures in rheumatoid arthritis. Ann Rheum Dis. 2013;72:1559–1568. doi: 10.1136/annrheumdis-2012-202352. [DOI] [PubMed] [Google Scholar]
- Croia C, Astorri E, Murray-Brown W, Willis A, Brokstad KA, Sutcliffe N, et al. Implication of Epstein-Barr virus infection in disease-specific autoreactive B cell activation in ectopic lymphoid structures of Sjögren's syndrome. Arthritis Rheumatol. 2014;66:2545–2557. doi: 10.1002/art.38726. [DOI] [PubMed] [Google Scholar]
- Bonnan M. Does disease-irrelevant intrathecal synthesis in multiple sclerosis make sense in the light of tertiary lymphoid organs. Front Neurol. 2014;5:27. doi: 10.3389/fneur.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig JC, Haire M, Millar JHD, Fraser KB, Merrett JD. Immunological control of Epstein–Barr virus-transformed lymphocytes in multiple sclerosis. Clin Immunol Immunopathol. 1983;29:86–93. doi: 10.1016/0090-1229(83)90009-0. [DOI] [PubMed] [Google Scholar]
- Craig JC, Hawkins SA, Swallow MW, Lyttle JA, Patterson VH, Merrett JD, et al. Subsets of T lymphocytes in relation to T lymphocyte function in multiple sclerosis. Clin Exp Immunol. 1985;61:548–555. [PMC free article] [PubMed] [Google Scholar]
- Lünemann JD, Jelčić I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J Exp Med. 2008;205:1763–1773. doi: 10.1084/jem.20072397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig JC, Haire M, Merrett JD. T-cell-mediated suppression of Epstein-Barr virus-induced B lymphocyte activation in multiple sclerosis. Clin Immunol Immunopathol. 1988;48:253–260. doi: 10.1016/0090-1229(88)90019-0. [DOI] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Lenarczyk A, Pfluger CMM, Burrows SR. Decreased T cell reactivity to Epstein-Barr virus infected lymphoblastoid cell lines in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2009;80:498–505. doi: 10.1136/jnnp.2008.161018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JW, Hatfield LM. Epstein-Barr virus and multiple sclerosis: Cellular immune response and cross-reactivity. J Neuroimmunol. 2010;229:238–242. doi: 10.1016/j.jneuroim.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Pfluger CMM, Burrows SR. Decreased CD8+ T cell response to Epstein-Barr virus infected B cells in multiple sclerosis is not due to decreased HLA class I expression on B cells or monocytes. BMC Neurology. 2011;11:95. doi: 10.1186/1471-2377-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Pfluger CMM, Burrows SR.Deficiency of CD8+ effector memory T cells is an early and persistent feature of multiple sclerosis Mult Scler 2014. doi: 10.1177/1352458514536252 [DOI] [PMC free article] [PubMed]
- Pender MP, Csurhes PA, Pfluger CMM, Burrows SR. CD8 T cell deficiency impairs control of Epstein–Barr virus and worsens with age in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2012;83:353–354. doi: 10.1136/jnnp-2011-300213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronen F, Ruprecht K, Weissbrich B, Klinker E, Kroner A, Hofstetter HH, et al. Frequency analysis of HLA-B7-restricted Epstein-Barr virus-specific cytotoxic T lymphocytes in patients with multiple sclerosis and healthy controls. J Neuroimmunol. 2006;180:185–192. doi: 10.1016/j.jneuroim.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Höllsberg P, Hansen HJ, Haahr S. Altered CD8+ T cell responses to selected Epstein-Barr virus immunodominant epitopes in patients with multiple sclerosis. Clin Exp Immunol. 2003;132:137–143. doi: 10.1046/j.1365-2249.2003.02114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain. 2008;131:1712–1721. doi: 10.1093/brain/awn108. [DOI] [PubMed] [Google Scholar]
- Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, et al. Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog. 2013;9:e1003220. doi: 10.1371/journal.ppat.1003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilek S, Schluep M, Harari A, Canales M, Lysandropoulos A, Zekeridou A, et al. HLA-B7-restricted EBV-specific CD8+ T cells are dysregulated in multiple sclerosis. J Immunol. 2012;188:4671–4680. doi: 10.4049/jimmunol.1103100. [DOI] [PubMed] [Google Scholar]
- Pudney VA, Leese AM, Rickinson AB, Hislop AD. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J Exp Med. 2005;201:349–360. doi: 10.1084/jem.20041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan FE, Elhassen D, Purcell AW, Burrows JM, Borg NA, Miles JJ, et al. The immunogenicity of a viral cytotoxic T cell epitope is controlled by its MHC-bound conformation. J Exp Med. 2005;202:1249–1260. doi: 10.1084/jem.20050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Smith KD, Kurilla MG, Lutz CT. Cytotoxic CD8+ T cells recognize EBV antigen but poorly kill autologous EBV-infected B lymphoblasts. Immunodominance is elicited by a peptide epitope that is presented at low levels in vitro. J Immunol. 1997;159:1844–1852. [PubMed] [Google Scholar]
- Bergmann CC, Altman JD, Hinton D, Stohlman SA. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J Immunol. 1999;163:3379–3387. [PubMed] [Google Scholar]
- Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonnière L, et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on α4 integrin. Brain. 2011;134:3560–3577. doi: 10.1093/brain/awr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007;204:2023–2030. doi: 10.1084/jem.20070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmøy T, Vartdal F. Cerebrospinal fluid T cells from multiple sclerosis patients recognize autologous Epstein-Barr virus-transformed B cells. J Neurovirol. 2004;10:52–56. doi: 10.1080/13550280490261671. [DOI] [PubMed] [Google Scholar]
- Lünemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Münz C, et al. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain. 2006;129:1493–1506. doi: 10.1093/brain/awl067. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang HLE, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. 2002;3:940–943. doi: 10.1038/ni835. [DOI] [PubMed] [Google Scholar]
- Álvarez-Lafuente R, De Las Heras V, Bartolomé M, García-Montojo M, Arroyo R. Human herpesvirus 6 and multiple sclerosis: a one-year follow-up study. Brain Pathol. 2006;16:20–27. doi: 10.1111/j.1750-3639.2006.tb00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Ordoñez G, Pineda B. Varicella-zoster virus at relapses of multiple sclerosis. J Neurol. 2007;254:493–500. doi: 10.1007/s00415-006-0402-x. [DOI] [PubMed] [Google Scholar]
- Lindsey JW, Hatfield LM, Crawford MP, Patel S. Quantitative PCR for Epstein–Barr virus DNA and RNA in multiple sclerosis. Mult Scler. 2009;15:153–158. doi: 10.1177/1352458508097920. [DOI] [PubMed] [Google Scholar]
- Lucas RM, Ponsonby A-L, Dear K, Valery P, Pender MP, Burrows JM, et al. Current and past Epstein-Barr virus infection in risk of initial CNS demyelination. Neurology. 2011;77:371–379. doi: 10.1212/WNL.0b013e318227062a. [DOI] [PubMed] [Google Scholar]
- Cocuzza CE, Piazza F, Musumeci R, Oggioni D, Andreoni S, Gardinetti M, et al. Quantitative detection of Epstein-Barr virus DNA in cerebrospinal fluid and blood samples of patients with relapsing-remitting multiple sclerosis. PLoS ONE. 2014;9:e94497. doi: 10.1371/journal.pone.0094497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Ordoñez G, Pineda B, Flores J. The participation of varicella zoster virus in relapses of multiple sclerosis. Clin Neurol Neurosurg. 2014;119:44–48. doi: 10.1016/j.clineuro.2013.12.020. [DOI] [PubMed] [Google Scholar]
- Ben Fredj N, Rotola A, Nefzi F, Chebel S, Rizzo R, Caselli E, et al. Identification of human herpesviruses 1 to 8 in Tunisian multiple sclerosis patients and healthy blood donors. J Neurovirol. 2012;18:12–19. doi: 10.1007/s13365-011-0056-z. [DOI] [PubMed] [Google Scholar]
- Martin C, Enbom M, Söderström M, Fredrikson S, Dahl H, Lycke J, et al. Absence of seven human herpesviruses, including HHV-6, by polymerase chain reaction in CSF and blood from patients with multiple sclerosis and optic neuritis. Acta Neurol Scand. 1997;95:280–283. doi: 10.1111/j.1600-0404.1997.tb00210.x. [DOI] [PubMed] [Google Scholar]
- Villoslada P, Juste C, Tintore M, Llorenç V, Codina G, Pozo-Rosich P, et al. The immune response against herpesvirus is more prominent in the early stages of MS. Neurology. 2003;60:1944–1948. doi: 10.1212/01.wnl.0000069461.53733.f7. [DOI] [PubMed] [Google Scholar]
- Franciotta D, Bestetti A, Sala S, Perucca P, Jarius S, Price RW, et al. Broad screening for human herpesviridae DNA in multiple sclerosis cerebrospinal fluid and serum. Acta Neurol Belg. 2009;109:277–282. [PubMed] [Google Scholar]
- Ferrante P, Omodeo-Zorini E, Zuffolato MR, Mancuso R, Caldarelli-Stefano R, Puricelli S, et al. Human T-cell lymphotropic virus tax and Epstein-Barr virus DNA in peripheral blood of multiple sclerosis patients during acute attack. Acta Neurol Scand Suppl. 1997;169:79–85. doi: 10.1111/j.1600-0404.1997.tb08155.x. [DOI] [PubMed] [Google Scholar]
- Höllsberg P, Kusk M, Bech E, Hansen HJ, Jakobsen J, Haahr S. Presence of Epstein–Barr virus and human herpesvirus 6B DNA in multiple sclerosis patients: associations with disease activity. Acta Neurol Scand. 2005;112:395–402. doi: 10.1111/j.1600-0404.2005.00516.x. [DOI] [PubMed] [Google Scholar]
- Babcock GJ, Decker LL, Freeman RB, Thorley-Lawson DA. Epstein-Barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J Exp Med. 1999;190:567–576. doi: 10.1084/jem.190.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross AJ, Hochberg D, Rand WM, Thorley-Lawson DA. EBV and systemic lupus erythematosus: a new perspective. J Immunol. 2005;174:6599–6607. doi: 10.4049/jimmunol.174.11.6599. [DOI] [PubMed] [Google Scholar]
- Tosato G, Steinberg AD, Yarchoan R, Heilman CA, Pike SE, De Seau V, et al. Abnormally elevated frequency of Epstein-Barr virus-infected B cells in the blood of patients with rheumatoid arthritis. J Clin Invest. 1984;73:1789–1795. doi: 10.1172/JCI111388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Miyake K, Yamauchi Y, Nishiyama K, Iwata S, Iwatsuki K, et al. Identification of Epstein-Barr virus (EBV)-infected lymphocyte subtypes by flow cytometric in situ hybridization in EBV-associated lymphoproliferative diseases. J Infect Dis. 2009;200:1078–1087. doi: 10.1086/605610. [DOI] [PubMed] [Google Scholar]
- Maurer M, Kuhle J, Fischer K, Kappos L, Hartung H-P, Goebels N. Increased frequency of Epstein-Barr virus infected B cells in cerebrospinal fluid and peripheral blood of patients with multiple sclerosis. Mult Scler. 2013;19 ((11 Suppl:394–395. [Google Scholar]
- Munch M, Møller-Larsen A, Christensen T, Morling N, Hansen HJ, Haahr S. B-lymphoblastoid cell lines from multiple sclerosis patients and a healthy control producing a putative new human retrovirus and Epstein–Barr virus. Mult Scler. 1995;1:78–81. doi: 10.1177/135245859500100204. [DOI] [PubMed] [Google Scholar]
- Tørring C, Andreasen C, Gehr N, Bjerg L, Petersen T, Höllsberg P. Higher incidence of Epstein–Barr virus-induced lymphocyte transformation in multiple sclerosis. Acta Neurol Scand. 2014;130:90–96. doi: 10.1111/ane.12249. [DOI] [PubMed] [Google Scholar]
- Yea C, Tellier R, Chong P, Westmacott G, Marrie RA, Bar-Or A, et al. Epstein-Barr virus in oral shedding of children with multiple sclerosis. Neurology. 2013;81:1392–1399. doi: 10.1212/WNL.0b013e3182a841e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B, et al. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology. 2010;74:1127–1135. doi: 10.1212/WNL.0b013e3181d865a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulley ML, Tang W. Laboratory assays for Epstein-Barr virus-related disease. J Mol Diagn. 2008;10:279–292. doi: 10.2353/jmoldx.2008.080023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein–Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. 2009;132:3318–3328. doi: 10.1093/brain/awp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peferoen LAN, Lamers F, Lodder LNR, Gerritsen WH, Huitinga I, Melief J, et al. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. 2010;133:e137. doi: 10.1093/brain/awp296. [DOI] [PubMed] [Google Scholar]
- Torkildsen Ø, Stansberg C, Angelskår SM, Kooi E-J, Geurts JJG, van der Valk P, et al. Upregulation of immunoglobulin-related genes in cortical sections from multiple sclerosis patients. Brain Pathol. 2010;20:720–729. doi: 10.1111/j.1750-3639.2009.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM. NeuroproMiSe EBV Working GroupEpstein-Barr virus in the multiple sclerosis brain: a controversial issue—report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011;134:2772–2786. doi: 10.1093/brain/awr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzartos JS, Khan G, Vossenkamper A, Cruz-Sadaba M, Lonardi S, Sefia E, et al. Association of innate immune activation with latent Epstein-Barr virus in active MS lesions. Neurology. 2012;78:15–23. doi: 10.1212/WNL.0b013e31823ed057. [DOI] [PubMed] [Google Scholar]
- Serafini B, Muzio L, Rosicarelli B, Aloisi F. Radioactive in situ hybridization for Epstein-Barr virus-encoded small RNA supports presence of Epstein-Barr virus in the multiple sclerosis brain. Brain. 2013;136:e233. doi: 10.1093/brain/aws315. [DOI] [PubMed] [Google Scholar]
- Mǎrgǎritescu O, Mogoantǎ L, Pirici I, Pirici D, Cernea D, Mǎrgǎritescu C. Histopathological changes in acute ischemic stroke. Rom J Morphol Embryol. 2009;50:327–339. [PubMed] [Google Scholar]
- Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Aloisi F, Stigliano E. Epstein-Barr virus in the central nervous system and cervical lymph node of a patient with primary progressive multiple sclerosis. J Neuropathol Exp Neurol. 2014;73:729–731. doi: 10.1097/NEN.0000000000000082. [DOI] [PubMed] [Google Scholar]
- Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol. 2010;69:677–693. doi: 10.1097/NEN.0b013e3181e332ec. [DOI] [PubMed] [Google Scholar]
- Brennan RM, Burrows JM, Bell MJ, Bromham L, Csurhes PA, Lenarczyk A, et al. Strains of Epstein–Barr virus infecting multiple sclerosis patients. Mult Scler. 2010;16:643–651. doi: 10.1177/1352458510364537. [DOI] [PubMed] [Google Scholar]
- Simon KC, Yang X, Munger KL, Ascherio A. EBNA1 and LMP1 variants in multiple sclerosis cases and controls. Acta Neurol Scand. 2011;124:53–58. doi: 10.1111/j.1600-0404.2010.01410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JW, Patel S, Zou J. Epstein–Barr virus genotypes in multiple sclerosis. Acta Neurol Scand. 2008;117:141–144. doi: 10.1111/j.1600-0404.2007.00917.x. [DOI] [PubMed] [Google Scholar]
- Lay M-L, Lucas RM, Toi C, Ratnamohan M, Ponsonby A-L, Dwyer DE, et al. Epstein-Barr virus genotypes and strains in central nervous system demyelinating disease and Epstein-Barr virus-related illnesses in Australia. Intervirology. 2012;55:372–379. doi: 10.1159/000334693. [DOI] [PubMed] [Google Scholar]
- Tabi Z, McCombe PA, Pender MP. Apoptotic elimination of Vβ8.2+ cells from the central nervous system during recovery from experimental autoimmune encephalomyelitis induced by the passive transfer of Vβ8.2+ encephalitogenic T cells. Eur J Immunol. 1994;24:2609–2617. doi: 10.1002/eji.1830241107. [DOI] [PubMed] [Google Scholar]
- White CA, McCombe PA, Pender MP. The roles of Fas, Fas ligand and Bcl-2 in T cell apoptosis in the central nervous system in experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998;82:47–55. doi: 10.1016/S0165-5728(97)00187-2. [DOI] [PubMed] [Google Scholar]
- Pender MP. Genetically determined failure of activation-induced apoptosis of autoreactive T cells as a cause of multiple sclerosis. Lancet. 1998;351:978–981. doi: 10.1016/S0140-6736(05)60642-3. [DOI] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Greer JM, Mowat PD, Henderson RD, Cameron KD, et al. Surges of increased T cell reactivity to an encephalitogenic region of myelin proteolipid protein occur more often in patients with multiple sclerosis than in healthy subjects. J Immunol. 2000;165:5322–5331. doi: 10.4049/jimmunol.165.9.5322. [DOI] [PubMed] [Google Scholar]
- Greer JM, Csurhes PA, Muller DM, Pender MP. Correlation of blood T cell and antibody reactivity to myelin proteins with HLA type and lesion localization in multiple sclerosis. J Immunol. 2008;180:6402–6410. doi: 10.4049/jimmunol.180.9.6402. [DOI] [PubMed] [Google Scholar]
- Van Noort JM, Bajramovic JJ, Plomp AC, van Stipdonk MJB. Mistaken self, a novel model that links microbial infections with myelin-directed autoimmunity in multiple sclerosis. J Neuroimmunol. 2000;105:46–57. doi: 10.1016/s0165-5728(00)00181-8. [DOI] [PubMed] [Google Scholar]
- Van Sechel AC, Bajramovic JJ, van Stipdonk MJB, Persoon-Deen C, Geutskens SB, van Noort JM. EBV-induced expression and HLA-DR-restricted presentation by human B cells of αB-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol. 1999;162:129–135. [PubMed] [Google Scholar]
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- Tracy SI, Kakalacheva K, Lünemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. Persistence of Epstein-Barr virus in self-reactive memory B cells. J Virol. 2012;86:12330–12340. doi: 10.1128/JVI.01699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. New Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Smith C, Beagley L, Hooper KD, Raj M, et al. Epstein–Barr virus-specific adoptive immunotherapy for progressive multiple sclerosis. Mult Scler. 2014;20:1541–1544. doi: 10.1177/1352458514521888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourtellotte WW, Potvin AR, Baumhefner RW, Potvin JH, Ma BI, Syndulko K, et al. Multiple sclerosis de novo CNS IgG synthesis. Effect of CNS irradiation. Arch Neurol. 1980;37:620–624. doi: 10.1001/archneur.1980.00500590044005. [DOI] [PubMed] [Google Scholar]
- Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, et al. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in Burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 2009;5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy NJ, Hazlewood SA, Huen DS, Rickinson AB, Williams GT. The Epstein–Barr virus gene BHRF1, a homologue of the cellular oncogene Bcl-2, inhibits apoptosis induced by gamma radiation and chemotherapeutic drugs. Adv Exp Med Biol. 1996;406:83–97. doi: 10.1007/978-1-4899-0274-0_9. [DOI] [PubMed] [Google Scholar]
- Pender MP. Does Epstein–Barr virus infection in the brain drive the development of multiple sclerosis. Brain. 2009;132:3196–3198. doi: 10.1093/brain/awp312. [DOI] [PubMed] [Google Scholar]
- Pender MP, Khanna R. Epstein–Barr virus-specific adoptive immunotherapy: a new horizon for multiple sclerosis treatment. Immunotherapy. 2014;6:659–661. doi: 10.2217/imt.14.43. [DOI] [PubMed] [Google Scholar]
- Pender MP. Preventing and curing multiple sclerosis by controlling Epstein–Barr virus infection. Autoimmun Rev. 2009;8:563–568. doi: 10.1016/j.autrev.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Pender MP. CD8+ T-cell deficiency, Epstein-Barr virus infection, vitamin D deficiency and steps to autoimmunity: a unifying hypothesis. Autoimmune Dis. 2012;2012:189096. doi: 10.1155/2012/189096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Léonard P, Moreels A, et al. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis. 2007;196:1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- Haque T, Johannessen I, Dombagoda D, Sengupta C, Burns DM, Bird P, et al. A mouse monoclonal antibody against Epstein-Barr virus envelope glycoprotein 350 prevents infection both in vitro and in vivo. J Infect Dis. 2006;194:584–587. doi: 10.1086/505912. [DOI] [PubMed] [Google Scholar]
- Sashihara J, Hoshino Y, Bowman JJ, Krogmann T, Burbelo PD, Coffield VM, et al. Soluble rhesus lymphocryptovirus gp350 protects against infection and reduces viral loads in animals that become infected with virus after challenge. PLoS Pathog. 2011;7:e1002308. doi: 10.1371/journal.ppat.1002308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycke J, Svennerholm B, Hjelmquist E, Frisen L, Badr G, Andersson M, et al. Acyclovir treatment of relapsing-remitting multiple sclerosis. A randomized, placebo-controlled, double-blind study. J Neurol. 1996;243:214–224. doi: 10.1007/BF00868517. [DOI] [PubMed] [Google Scholar]
- Bech E, Lycke J, Gadeberg P, Hansen HJ, Malmeström C, Andersen O, et al. A randomized, double-blind, placebo-controlled MRI study of anti-herpes virus therapy in MS. Neurology. 2002;58:31–36. doi: 10.1212/wnl.58.1.31. [DOI] [PubMed] [Google Scholar]
- Mei YP, Zhu XF, Zhou JM, Huang H, Deng R, Zeng YX. siRNA targeting LMP1-induced apoptosis in EBV-positive lymphoma cells is associated with inhibition of telomerase activity and expression. Cancer Lett. 2006;232:189–198. doi: 10.1016/j.canlet.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Li N, Thompson S, Schultz DC, Zhu W, Jiang H, Luo C, et al. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS ONE. 2010;5:e10126. doi: 10.1371/journal.pone.0010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procko E, Berguig GY, Shen BW, Song Y, Frayo S, Convertine AJ, et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014;157:1644–1656. doi: 10.1016/j.cell.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B, Goss JA, Hammer MM, Zhang L, Lopez T, Gee AP, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs) Blood. 2006;108:2942–2949. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Tsang J, Beagley L, Chua D, Lee V, Li V, et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. 2012;72:1116–1125. doi: 10.1158/0008-5472.CAN-11-3399. [DOI] [PubMed] [Google Scholar]