

Abstract
A novel neuroprotective small molecule was discovered using a target-agnostic in vivo screen in living mice. This aminopropyl carbazole, named P7C3, is orally bioavailable, crosses the blood–brain barrier, and is non-toxic at doses several fold higher than the efficacious dose. The potency and drug-like properties of P7C3 were optimized through a medicinal chemistry campaign, providing analogues for detailed examination. Improved versions, such as (−)-P7C3-S243 and P7C3-A20, displayed neuro-protective properties in rodent models of Parkinson’s disease, amyotrophic lateral sclerosis, traumatic brain injury and age-related cognitive decline. Derivatives appended with immobilizing moieties may reveal the protein targets of the P7C3 class of neuroprotective compounds. Our results indicate that unbiased, in vivo screens might provide starting points for the development of treatments for neurodegenerative diseases as well as tools to study the biology underlying these disorders.
Introduction
Neurodegenerative diseases and disorders are physically, emotionally and financially devastating for patients and their families, and are also associated with great costs to caregivers and society. Furthermore, the prevalence of neurodegenerative diseases is increasing as a consequence of an aging population. Unfortunately, we lack pharmacologic agents that arrest disease progression for patients suffering from neurodegenerative disorders including Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Huntington’s disease, and traumatic brain injury (TBI) as well as normal age-related cognitive decline. Current therapies seek to minimize symptoms or provide palliative care, but none arrest the neuronal cell death that underlies these conditions. Indeed, current front-line therapy for ALS, Riluzole, extends lifespan by only 2–3 months and fails to block the rapid neuromuscular decline that characterizes the disease.1 Similarly, despite a flurry of drug candidates entering clinical trials for PD, the most recently successful new approach to disease management involves surgical implantation of an electrode within the thalamus, a procedure with substantial risk and variable results.2 Patients with related afflictions face similarly bleak treatment options.
In their efforts to identify new therapies, the pharmaceutical industry has historically invested substantial time and resources into in vitro, target-directed drug discovery programs. For instance, researchers have focused on β- and γ-secretase to treat AD, but those attempts have not yet proven successful in patients.3 Similarly, the PD community has advanced antagonists of the adenosine A2A receptor, but efforts to date have failed to show efficacy in pivotal trials.4 Perhaps the most spectacular demonstration of the potential limitations of the target-directed discovery approach involves the current state of Huntington’s disease, in which there are still no effective forms of treatment for patients two decades after genetic linkage analysis defined the underlying genetic locus fully responsible for HD.
The absence of effective treatments for a variety of neurodegenerative diseases and the cessation of research in neuroscience by several large pharmaceutical companies provides an opportunity for innovative approaches to drug discovery within academia. Typical target-driven research programs start with specific hypotheses regarding the role of certain enzymes, receptors or channels, and then biochemical assays that interrogate the function are implemented to screen for small molecule modulators. Active compounds that emerge from these screens are then profiled in cell culture, animal models of disease and, ultimately, in human patients. The targeted proteins and pathways reflect the current understanding of specific diseases, but they also reflect the biases of investigators about presumed mechanisms of action, and thus inadvertently close off discovery of previously unanticipated and possibly more effective mechanisms of treating disease. In this context, phenotypic screening strategies offer an attractive alternative approach to drug discovery.
Phenotypic screening
Phenotypic screening for drug leads involves evaluating small molecules for a biological effect at a cell or organismal level.5 These assays return compounds with a desired biological outcome without bias concerning mechanism. Phenotypic screens can prove advantageous when no consensus exists regarding suitable biological targets or when investigators seek early indications of efficacy. While uncertainty surrounding the mechanism of action of potential hits presents a challenge, several compensating considerations can favour phenotypic screening strategies. First, hits that are identified cause a desired biological outcome rather than simply binding or inhibiting a specific target. This characteristic presents a relative advantage compared to target-driven approaches involving unvalidated targets. Second, for compounds to score as hits, they must have suitable physical properties to engage their targets within the cellular or organismal milieu, display suitable toxicity profiles, and remain chemically stable in the context of the experiment. These features facilitate transition into preclinical animal models. Finally, determining how biologically active small molecules function may reveal previously unanticipated protein targets and biochemical pathways relevant to disease.
Neurodegenerative diseases appeared to present an ideal setting for phenotypic screening. They typically involve multiple cell types with uncertain relationships. They also frequently display multifaceted patterns of genetic predisposition and a complex interplay of genetic and environmental causes. As a consequence, few treatments for neurodegenerative diseases have been discovered by targeting pre-determined receptors or enzymes. In fact, even cell cultures have failed to recapitulate the complex biology of, for example, Parkinson’s disease or traumatic brain injury (TBI). Accordingly, we were drawn to a screening strategy that relied on in vivo pharmacology within live rodents to discover new lead compounds for treating neurodegenerative disorders.
Discovery of a neuroprotective chemical
Given the absence of viable biochemical or cell-based assays that reflect the complexity of neurodegenerative diseases, we elected to pursue an unbiased in vivo screen to identify neuroprotective small molecules. Our assay built on the observation that all adult mammals, including humans, form new neurons within the hippocampus.6 This process, which occurs within the subgranular zone of the dentate gyrus, appears important for learning, memory and neuronal plasticity.7 It involves an initial cell division event from a neural stem cell to generate a neural precursor cell (Fig. 1A). In mice, this precursor cell matures over about four weeks into a functional neuron that is incorporated into the dentate gyrus granular layer. For reasons that are not fully understood, however, the vast majority of neural precursor cells normally die before reaching full maturity. Hippocampal neurogenesis can be monitored experimentally by marking newly born cells with bromodeoxyuridine (BrdU), a thymidine analogue that is incorporated into newly synthesized DNA and that can be readily detected by standard immunohistochemistry. BrdU therefore provides a means to monitor the effects of environmental factors, genetic manipulation or small molecules on both neural precursor cell proliferation and neuron survival.8
Fig. 1.
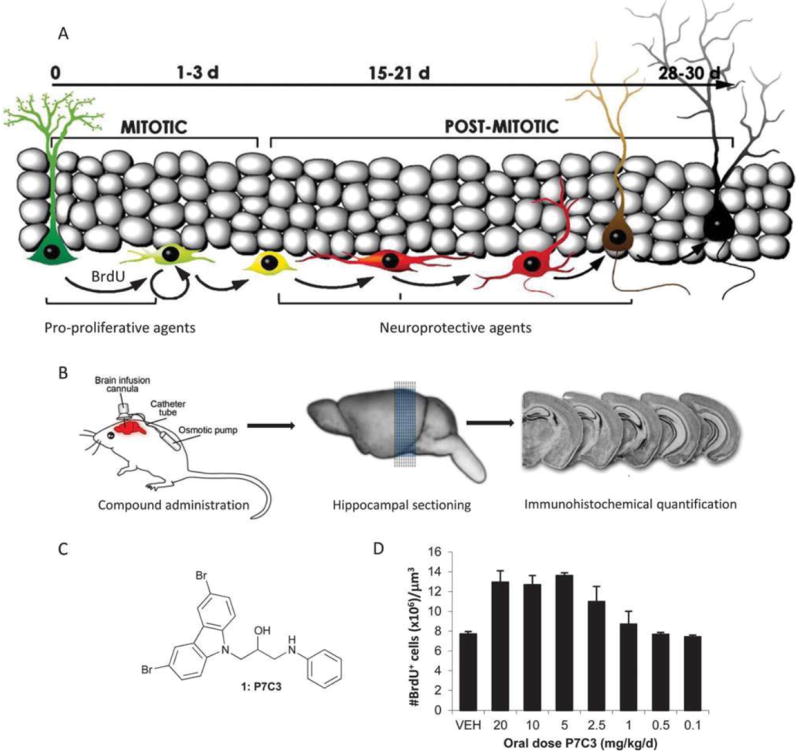
Unbiased screen for neuroprotective small molecules. (A) Schematic of hippocampal neurogenesis illustrating the incorporation of BrdU into new born cells, the month-long period of maturation and roles of pro-proliferative and neuroprotective agents. Modified from ref. J. M. Encinas, A. Vaahtokari, G. Enikolopov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8233. Copyright (2006) National Academy of Sciences, U. S. A. (B) Outline of the in vivo screen. Drugs were infused directly into the brain of live mice over 7 days. Subsequent sectioning and immunohistochemical staining revealed newly formed neurons (black cells). (C) Chemical structure of active component of pool 7. (D) Increase in number of BrdU + cells following oral dosing with P7C3.
We performed an unbiased, in vivo screen in which we monitored the number of newly formed neuronal precursor cells within the dentate gyrus following administration of selected chemicals.9 The goal was to reveal compounds that increased either proliferation or survival of hippocampal neural precursor cells, as both effects represented potential opportunities to augment the net magnitude of hippocampal neurogenesis. We chose a one week duration for the assay based on our pulse-chase BrdU studies, which demonstrated that 40% of hippocampal neural precursor cells die within the first five days of their birth. Furthermore, because hippocampal neurogenesis is augmented by both social interaction and voluntary exercise, test mice were housed individually and without access to running wheels, starting one week before pump implantation. This ensured that the net magnitude of hippocampal neurogenesis was at as low a basal level as possible, thus widening our window for discovery of efficacious agents.
To initiate our screen, we selected 1000 test chemicals from the UT Southwestern small molecule library, which was assembled from a variety of commercial sources. These molecules were selected by applying filters to remove high and low molecular weight compounds, and to favour chiral compounds and those with H-bond donating and accepting capacity. We additionally avoided chemically reactive moieties to minimize the likelihood of covalent protein modification or metabolic instability. These 1000 chemicals were randomly grouped into 100 pools of 10 each, which were tested as mixtures. To avoid complications arising from variable penetration of the blood–brain barrier, compounds were injected directly into the cerebroventricular system of living mice as a solution in artificial cerebrospinal fluid (Fig. 1B). This method of delivery was accomplished with a subcutaneously implanted osmotic minipump, which delivered a 10 μM solution at a rate of 0.5 μL h-1 through a customized cannula that terminated in the left lateral ventricle. In the unlikely scenario of 100% absorbance of compounds into brain tissue and 0% clearance throughout the infusion period, each compound would be present at low micromolar concentration at the end of one week. Since the actual amount of compound in the brain was likely to be only a fraction of this, we reasoned that our method likely administered compounds at sub-micromolar concentrations. Concurrent with compound treatment, mice were given daily IP doses of BrdU (50 mg kg-1 d-1) to label newly formed neurons.
After a 1 week treatment period, mice were sacrificed and transcardially perfused. Using antibodies to BrdU, the number of BrdU + newborn neural precursor cells was quantified in every fifth 40 μm thick coronal section throughout the extent of the hippocampus. Importantly, the number of BrdU + cells was quantified in the hemisphere contralateral to the site of cannula placement, such that active chemicals had to achieve their desired effect by virtue of traveling through the ventricular system to the hippocampus on the opposite side of the brain. The number of BrdU + cells was then normalized to the volume of dentate gyrus for standardization across test animals. Using this assay, we identified several pools of chemicals that approximately doubled the number of newborn hippocampal neurons that remained at the end of the 1 week testing period. This activity was equal in magnitude to the effect of direct infusion of fibroblast growth factor, which the brain makes endogenously to support hippocampal neurogenesis. Deconvolution of these mixtures revealed that the third compound (C3) within the seventh pool (P7) appeared particularly attractive (Fig. 1C).
This hit, which we named P7C3, was orally bioavailable and readily crossed the blood–brain barrier. Using an oral dosing regime, P7C3 increased neuron number when dosed at 2.5 mg kg-1 d-1 and reached maximal efficacy at 5 mg kg-1 d-1 (Fig. 1D). Similarly, P7C3 increased the number of new neurons within the hippocampus of aged rats, and this enhancement was correlated with improvements in memory and learning as reflected by performance in the Morris water maze.9 Finally, the compound appeared non-toxic to embryonic, weaning and adult mice. For these reasons, we selected P7C3 for more detailed study as a possible drug lead for treating neurodegenerative diseases.
P7C3 is neuroprotective
The in vivo neurogenesis assay was designed to identify compounds that either increased neural stem cell proliferation or increased survival of neural precursor cells over the 1 week assay. To distinguish between these possibilities, we performed a pulse-chase experiment in which new neurons were labelled with a single injection of BrdU after P7C3 had been administered for sufficient time to achieve steady state levels in the brain. Dosing with P7C3 was then continued for a 30 day period. Immunohistochemistry revealed that there was no increase in the number of BrdU + cells one hour after labelling, indicating that P7C3 had no effect on cell proliferation. By contrast, significant differences between P7C3 and vehicle treated groups emerged on day 5. By this time point around 40% of cells born by day 1 normally die, but drug-treated animals showed 25% more BrdU + cells than the control arm. By day 30 the difference had increased to 500%.9 Additionally, mouse and rat brain slices revealed that treatment with P7C3 decreased the amount of apoptosis within the hippocampus as determined by immunohistochemical staining with antibodies to cleaved caspase 3, a definitive marker of cellular commitment to apoptosis. Taken together, the data indicate that P7C3 increases the net magnitude of neurogenesis by blocking apoptosis – i.e. it is neuroprotective – rather than by increasing cell proliferation. Moreover, through a mechanism not fully understood, P7C3 appears to block aberrant neuronal apoptosis selectively. For instance, we have observed no effect on the normal apoptotic program associated with neural pruning, nor are mice that received P7C3 in utero born with webbed digits or expanded brain tissue as a consequence of halted apoptosis during normal development.9 Finally, we have not observed any carcinogenicity associated with P7C3, again suggesting that it does not universally halt cell death.
Towards a neuroprotective drug
The discovery and evaluation of P7C3 launched three research programs within our respective laboratories. First, we sought improved chemical matter in terms of potency, toxicity, drug-like characteristics and intellectual property. Second, we wanted to determine if P7C3 could protect mature neurons from death in addition to protecting hippocampal neural precursor cells. Whereas impaired hippocampal neurogenesis is associated with several neuropsychiatric diseases,10 protection of mature neurons could offer a strategy to treat a broader range of neurodegenerative diseases for which there are currently no effective forms of treatment. Finally, we were eager to determine the molecular basis by which P7C3 exerts its neuro-protective effects.
Chemical optimization of P7C3
We initiated a medicinal chemistry program around P7C3 with three objectives. First, we wanted to improve the activity of P7C3, both in terms of potency (lowest dose showing activity) and efficacy (maximal effect). Second, we wanted to improve the drug-like properties of P7C3 by removing the aniline ring, increasing polarity and identifying a single-enantiomer compound. Third, we wanted to identify regions of the compound that would tolerate introduction of functionality to aid mode-of-action studies. Our initial efforts were aimed at determining which components of P7C3 were required for activity. This investigation was facilitated by the rapid assembly of the P7C3 scaffold from a heterocycle, an epoxide and a nucleophile such as a phenol, aniline or thiophenol (eqn (1)).11 Alternatively, epoxide 2 could be opened with sodium azide. Following reduction, (hetero)aromatic rings could be introduced under Cu-catalysed amination conditions.12 Fig. 2 shows a representative set of analogues, grouped as compounds that diminished activity in the in vivo hippocampal neuroprotection assay, maintained activity, or improved activity relative to P7C3. We found that removing the N-phenyl ring (4) reduced activity, as did removing the central hydroxyl group or adding an additional methylene between the hydroxyl and the carbazole (5). One bromine could be replaced with a methyl group (9), but replacing both bromines resulted in a compound that was nearly inactive under these assay conditions (6). Likewise, replacing the bromines with chlorines, iodines, CF3 groups or cyclopropyl groups abrogated activity at the concentrations tested (not shown). Methylation of the aniline NH was detrimental, but encouragingly, the NH could be replaced with an oxygen (8) or a sulfone SO2 (14) without loss of activity. In an important discovery, we found that activity resided predominantly in a single enantiomer, as (S)-8 was as active as P7C3 whereas its enantiomer, (R)-9, was essentially devoid of activity. We also discovered that that carbazole scaffold was not specifically required for activity as the corresponding carboline 11 was as neuroprotective as P7C3.
Fig. 2.
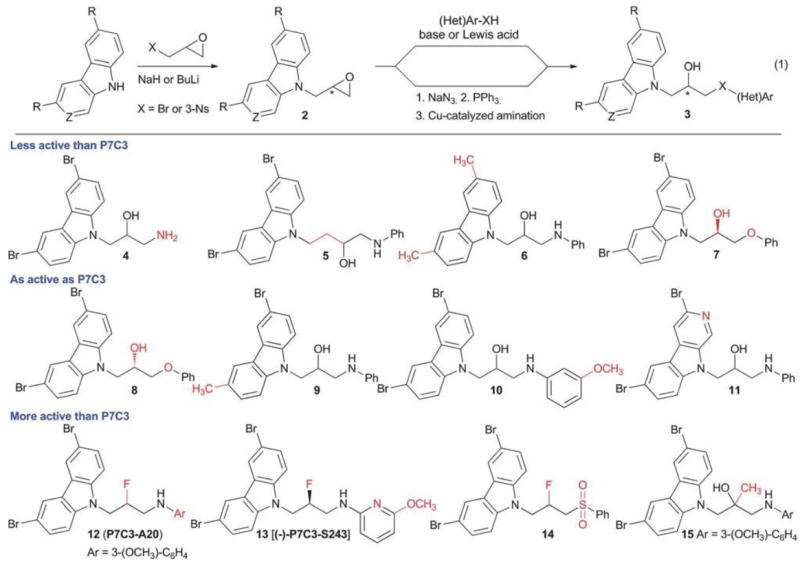
Chemical optimization of P7C3. Derivatives of P7C3 can be accessed rapidly using epoxides as lynchpins. Fluorination of the secondary alcohol provides additional analogues. Compounds are grouped as those that were less active than P7C3, equivalent to P7C3 or more active than P7C3 in the in vivo hippocampal neurogenesis assay via ICV administration. Changes from P7C3 are highlighted in red.
Several changes to the P7C3 scaffold were found to improve activity. Converting the central hydroxyl to a fluorine and introducing an OCH3 on the aniline ring provided 12, also known as P7C3-A20. As described below, this compound has been profiled extensively through experiments facilitated by a chromatography-free synthesis that provided hundred gram batches of the compound.13 The enhanced activity of P7C3-A20 reflects a synergistic contribution of both the fluorine and the methoxy group; either fluorination or addition of – OCH3 (10) alone had little effect on activity. Recently, we discovered (−)-P7C3-S243, an analogue with increased polarity relative to P7C3-A20, and one in which the aniline moiety has been replaced with aminopyridine functionality.11 (−)-P7C3-S243 can be synthesized as a single enantiomer starting from optically active glycidol 3-nosylate14 and utilizing the combination of C4F9S02F and [Bu4N][Ph3SiF2] to effect a stereospecific fluorination.15 As shown below, this compound has also been evaluated in animal models of neurodegeneration. Finally, converting the secondary alcohol of P7C3 to a tertiary alcohol, as in 15, increased activity in our in vivo hippocampal neuroprotection assay. Several additional analogues that may aid mode-of-action studies are described in a following section.
P7C3-A20 and (−)-P7C3-S243 were found to protect newly born hippocampal neurons in a dose-dependent manner following IP injection (Fig. 3). By contrast, the enantiomer (+)-P7C3-S243 showed much lower neuroprotective efficacy. Moreover, both P7C3-A20 and P7C3-S243 were found to penetrate the blood–brain barrier, with the latter partitioning nearly equally between brain and plasma when dosed orally. Both chemicals have long half-lives in the presence of cultured hepatocytes and in vivo (T1/2 > 6 h).10,11 Furthermore, neither compound appears toxic: P7C3-A20 has been dosed at up to 40 mg kg-1 d-1 for 30 days without causing changes in behavior, weight or appearance, and (−)-P7C3-S243 has been administered at up to 5 mg kg-1 d-1 (highest dose tested to date) for 21 days without adverse effects. Neither P7C3-A20 nor (−)-P7C3-S243 inhibits the hERG channel or shows toxicity towards cultured human cell lines (IC50 > μ10 M). Given the favourable potency and toxicity profile of these two neuroprotective agents, they were selected for more detailed interrogation.
Fig. 3.
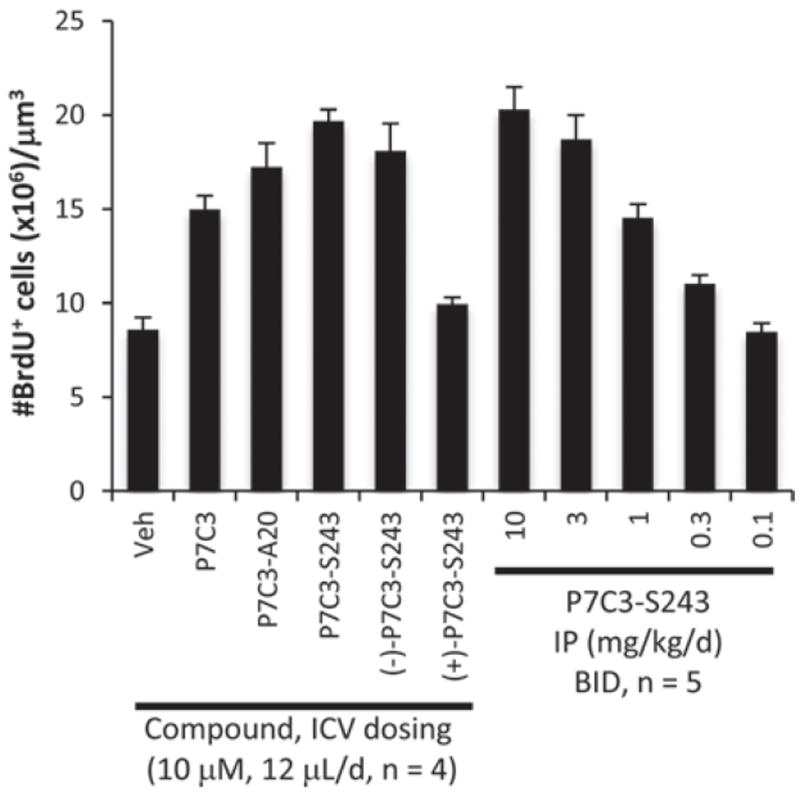
Efficacy of P7C3 derivatives in an in vivo mouse model of neuro- genesis. Compounds were administered as indicated for 7 days along with daily injection of BrdU. Data are expressed as mean ± SEM.
The P7C3 class is broadly neuroprotective
MPTP model of PD
P7C3 was discovered as an agent that could enhance hippocampal neurogenesis, but additional investigation revealed that it and its derivatives increased the number of newly born neurons by virtue of their ability to block neuronal cell death. We therefore hypothesized that the P7C3 class of compounds might also block cell death of mature neurons within various regions of the adult brain. If so, then the P7C3 class might represent general neuroprotective agents that could be used to therapeutic benefit in the treatment of neurodegenerative diseases. A first test of this hypothesis focused on animal models of Parkinson’s disease. PD is a progressive neurological disorder affecting 7–10 million people worldwide. It is incurable and associated with significant morbidity.16 Early stages of the disease are characterized by diminishing motor function, whereas advanced stages feature cognitive and behavioral impairments. Current treatments aim to manage early motor symptoms, but fail to address the underlying cause of the disease: the death of dopaminergic neurons within the substantia nigra pars compacta (SNc).
The selective neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) provides an experimental model of PD.17 It is converted into the toxic metabolite 1-methyl-4-phenylpyridinium cation, MPP+, which is then selectively transported into dopaminergic cells within the SNc.18 These are the same cells that die in PD, so MPP+ -mediated toxicity to dopaminergic neurons recapitulates the neurodegeneration observed in PD.19 In fact, the toxicity of MPTP was originally discovered as the causative agent for the Parkinsonian symptoms displayed by recreational drug users who inadvertently ingested desmethylprodine that was contaminated with MPTP. In mice, a 5 day regimen of MPTP begins to kill dopaminergic neurons in the SNc, and if left untreated nearly half of these cells will have died 21 days later (Fig. 4). The vitality of these cells can be monitored by immunohistochemical staining for tyrosine hydroxylase (TH), which catalyses conversion of L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA), the precursor to dopamine.
Fig. 4.
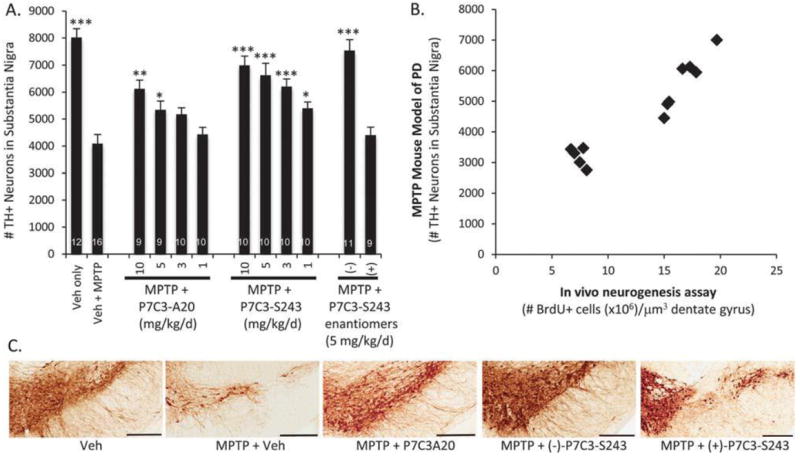
Neuroprotection in the MPTP model of Parkinson’s disease. (A) Mice were administered MPTP (30 mg k−1 d−1) for 5 days. On the 6th day, treatment with drug was initiated at the indicated dose (IP, BID, 21 d). Bars indicate number of tyrosine hydroxylase positive cells detected by immunohisotochemical staining of the substantial nigra. Error bars indicate SEM. Asterisks indicate p < 0.01 (*), p < 0.001 (**) or p < 0.0001 (***) relative to Veh + MPTP group. Numbers of mice in each group are shown within the bars. (B) Correlation between efficacy in the MPTP model of PD and the in vivo neurogenesis assay for analogs of P7C3. (C) Representative immunohistochemical pictures of TH staining in the SNc are shown for 5 mg kg−1 d−1 treatment groups. Bar = 300 mM.
P7C3-A20 and (−)-P7C3-S243 protect TH + cells from toxicity associated with MPTP.11,20 Specifically, compound treatment was initiated a full 24 hours after the final dose of toxin to ensure that any effects reflected genuinely neuroprotective activity rather than simply blocking uptake or metabolism of MPTP. Using this protocol, we observed evidence of efficacy at doses as low as 1 mg kg-1 d-1, and nearly complete rescue of TH + cells at 5 mg kg-1 d-1 (−)-P7C3-S243. The (+)-enantiomer did not protect under the conditions of this experiment. By virtue of preventing MPTP-mediated death of TH + cells in the SNc, treatment with P7C3-A20 or (−)-P7C3-S243 also preserved the integrity of axonal projections of dopaminergic cells. These extensions distribute dopamine to the striata and can also be visualized by immunohistochemical staining for TH. Mice treated with P7C3-A20 or (−)-P7C3-S243 after receiving MPTP maintained nearly all of their dopaminergic axons. Furthermore, P7C3 and 10 analogues have been tested in the MPTP model of PD, and the correlation between this assay and the in vivo neurogenesis assay described above is of high statistical significance.19 Control experiments excluded the trivial possibility that inactive compounds were failing to enter the brain. The concordance between the original hippocampal neuroprotection assay and the PD model indicates that the same mechanistic basis underpins protection of both newly born and mature neurons, and suggests broad utility of P7C3 and its analogues for blocking neuronal cell death.
G93A-SOD1 model of ALS
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease in which spinal cord motor neurons deteriorate and die. The loss of motor neurons causes muscle weakness and ultimately the loss of all voluntary movement. This rare disease strikes without warning in adulthood, with paralysis progressing inexorably such that most patients die from pneumonia or respiratory failure within 2–5 years of diagnosis. ALS is currently untreatable.21 In this context, we were eager to determine whether the neuroprotective effects of the P7C3 class of small molecules would show beneficial activity an animal model of ALS. The G93A-SOD1 mouse model represents the most widely used experimental model of ALS. These mice are hemizygous for a transgene expressing 18 ± 2.6 copies of a mutant form of superoxide dismutase 1 (SOD1). Mutations in this gene are associated with around about 3% of sporadic cases and 20% of inherited cases of ALS.22 The mutant form of SOD1 maintains enzymatic activity, and toxicity thus appears to be a gain of function that may be related to an increased likelihood of disruptive protein aggregation.23 Importantly, this model recapitulates several important characteristics of the human disease.24 At around 100 days of age, G93A-SOD1 mice exhibit paralysis in one or more limbs as a consequence of the death of motor neurons within the ventral horn of the spinal cord. Over the next several weeks, the mice lose all motor function in their limbs, and 50% of the mice die within 7 weeks of disease onset.
We asked three questions concerning the utility of the P7C3 and its derivatives in the G93A-SOD1 model of ALS.25 Could these neuroprotective agents slow or stop the death of motor neurons? Could they improve the motility of the mice after disease onset? And could active variants of P7C3 prolong the lifespan of G93A-SOD1 transgenic mice? To address these questions, we started treating G93A-SOD1 mice at day 80 with P7C3-A20 (20 mg kg-1 d-1), which corresponds to disease onset in these mice. Each test mouse was genotyped and sibling-matched with a vehicle-treated mouse to ensure that we were monitoring the effect of drug and not differences in copy number of the mutant sod1 gene. At days 90, 100, 110, and 120, five mice each from the vehicle and P7C3-A20 treated cohorts were sacrificed, and lumbar spinal sections were stained for choline acetyltransferase, a marker for spinal cord motor neurons (Fig. 5). All sections were counted blinded to treatment group. At every time point, P7C3-A20 treated animals possessed a higher density of spinal motor neurons compared to the vehicle control group (p < 0.003). As we have observed in both the hippocampal neuroprotection assay and the MPTP model of PD, P7C3 proved less effective than P7C3-A20, with a significant difference between treatment and control group being obtained only at days 100 and 110 with P7C3.
Fig. 5.
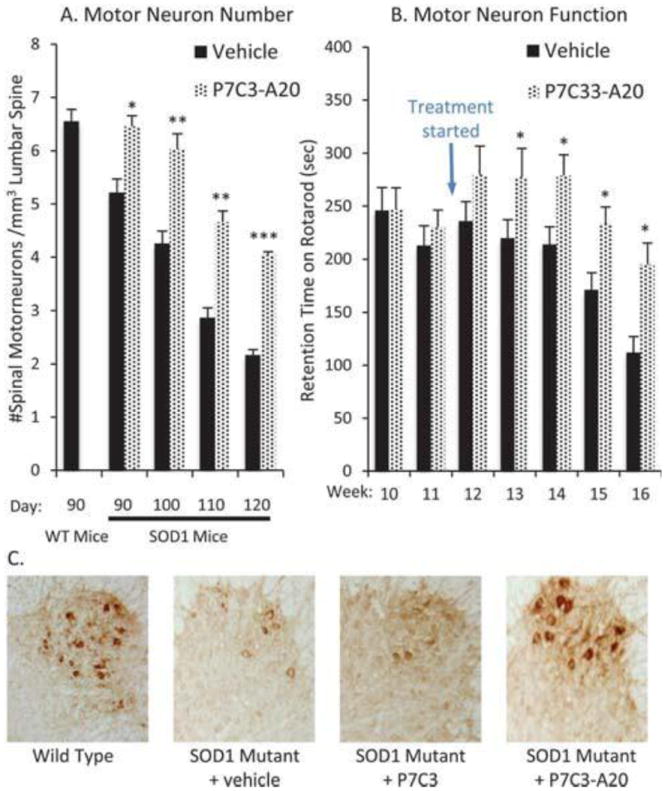
Efficacy of P7C3-A20 in the SOD1 mouse model of ALS. (A) P7C3- A20 blocks motor neuron death in the spinal cord when administered (20 mg kg−1 d−1, ip, bid) starting at the time of disease onset. N = 5 for each timepoint. (B) P7C3-A20 preserves performance in the accelerating rotarod test when administered (20 mg kg−1 d−1, ip, bid) starting at the time of disease onset. N = 20 until week 16 when n = 13. For A and B, asterisks indicate p < 2 × 10−2 (*), p < 2 × 10−4 (**) or p < 2 × 10−6 (***) relative to vehicle. (C) Representative images following immunohisoto- chemical staining with antibodies for choline acetyltransferase
We next investigated whether maintenance of motor neurons translated into improved motility. To this end, we used the accelerating rotarod task. In this trial, mice are placed on an elevated cylinder, which begins rotating at 4 rpm and gradually accelerates to 40 rpm over 10 min. We measured the time mice maintained balance on the rotarod before falling off into soft bedding. When treatment with P7C3-A20 was initiated concurrent with disease onset (day 80), mice showed a slower deterioration of performance in this test. By week 16, the mice in the vehicle control group could stay on the bar only half as long as they could prior to disease onset. By contrast, mice receiving P7C3-A20 decreased their time on the rotarod by only 20% (p = 0.008). Similarly, we observed that P7C3-A20-treated animals took significantly longer front and back strides (p = 0.003, 0.004, respectively) when walking on a flat surface compared to vehicle-treated siblings at the advanced disease state of 132 days. While P7C3 slowed death of the spinal cord neurons, it was not sufficiently active to increase motor neuron function under this testing regime. Neither P7C3 nor P7C3-A20 extended the lifespan of the G93A-SOD1 mice.
The fluid percussion model of TBI. Traumatic brain injury is a serious clinical problem that represents one of the leading causes of death and disability in the United States, with 1.6 million individuals sustaining a TBI every year. Longstanding functional impairment in neurologic function and cognition is associated with focal contusive injury, diffuse axonal damage and neuronal loss after TBI, and there is a profound lack of pharmacologic treatment options for mitigating the damage process. We thus sought to evaluate whether treatment with P7C3-A20 might help aid recovery from neuronal damage and acquired sensorimotor and cognitive deficits in a commonly applied rodent model of TBI: moderate fluid percussion brain injury over the right parietal cortex.26 This form of injury leads to both focal and diffuse brain damage, and faithfully recapitulates symptoms experienced by patients after blunt trauma injury. When treatment with P7C3-A20 was initiated 30 minutes after injury and continued for 7 days thereafter, animals showed a greater than two-fold decrease in contusion volume compared to vehicle treated rats. This protective effect was further associated with increased survival of mature NeuN-positive cortical neurons in the vulnerable pericontusional region.
Treatment with P7C3-A20 also significantly augmented the magnitude of hippocampal neurogenesis after TBI. While hippocampal neural precursor cell proliferation is endogenously enhanced after TBI,27 overall levels of hippocampal neurogenesis decrease as these cells experience an accelerated rate of death after their birth.28 This cell death can be monitored using the expression levels of doublecortin (DCX) in neural precursor cells of the dentate gyrus. DCX is a microtubule-associated protein that serves as a marker of neurogenesis by virtue of transient expression in newly formed neurons immediately prior to their final maturation.29 Treatment with P7C3-A20 after TBI significantly augmented both BrdU and DCX labelling in the hippocampal dentate gyrus, indicating that P7C3-A20 helped overcome the unique vulnerability of maturing hippocampal neurons to higher rates of death after TBI. Subsequent double-labeling in the hippocampus with antibodies to BrdU and NeuN (a marker of mature neurons) five weeks after injury further confirmed that treatment with P7C3-A20 enhanced the net magnitude of hippocampal neurogenesis after TBI. Thus the P7C3 class of molecules protects both mature and maturing neurons after TBI, populations that are otherwise uniquely vulnerable to death following traumatic injury.
Accelerated cell death of both maturing neurons and maturing neural precursor cells is thought to contribute to the constellation of neurological symptoms experienced by patients after TBI. Since treatment with P7C3-A20 was able to effectively block cell death of both classes of neuronal cells, we next tested whether this translated into improved neurological functioning. Specifically, we evaluated animals for contralateral forelimb deficits 1 week after TBI, and for learning in the Morris water maze task 4 weeks after injury. Treatment with P7C3-A20 significantly preserved normal function in both tasks, with vehicle-treated mice showing significant deficits. Thus, P7C3-A20-mediated cellular protection in the brain was correlated with favorable behavioral outcomes. Taken together, these results demonstrate that the P7C3 class of molecules holds promise for developing a new class of neuroprotective drugs for patients suffering from TBI.
Zebrafish model of retinal degeneration
Recently, Waskiewicz, Lehmann and co-workers explored the effects of P7C3 in a zebrafish model of retinal dystrophies.30 These scientists generated zebrafish deficient in the gene for growth differentiation factor 6 (GDF6), a morphogenic protein from the transforming growth factor-b (TGF-β) subfamily of ligands. This growth factor is involved in embryonic development of the retina,31 and mutations in the Dgf6 gene are associated with photoreceptor degeneration and congenital retinal dystrophies. The Gdf6a-/- zebrafish ‘demonstrated profound alterations to the morphology of the individual photoreceptor subtypes’. Retinal degeneration was attributed to increased apoptosis in Gdf6a-/- zebrafish compared to wild-type animals. Given the success of P7C3 in blocking neuronal apoptosis, Waskiewicz and Lehmann treated mutant zebrafish embryos with P7C3 and observed a 70 and 79% reduction in retinal apoptosis at 10 and 100 nM concentrations, respectively (p < 0.001). Moreover, protection of retinal neurons was associated with improved vision. Indeed, compound-treated zebrafish were better able to detect ambient light than DMSO-treated controls. These results suggest that the P7C3 class of neuroprotective agents could find application in the treatment of retinal dystrophies including Leber congenital amaurosis, retinitis pigmentosa, cone-rod dystrophy and other related disorders.
The results in the animal models of neurodegenerative diseases are noteworthy for several reasons. First, we were able to correlate neuroprotective activity with behavioral consequences. The MPTP model of PD lacks a phenotype in mice, but treatment with the P7C3 class of molecules improved motor function in the G93A-SOD1 model of ALS, improved learning and memory in a rat model of TBI, and improved visual perception in the Gdf6a-/- model of retinal degeneration. Second, these results show that P7C3 and related compounds are active outside the brain, since they maintain motor neurons within the spinal column, and also protect retinal neurons in the eye. Third, these results demonstrated that the P7C3 class of compounds are general neuroprotective agents; that is, they function in multiple models of disease in which cell death is initiated through different mechanisms. For these reasons, we are confident that the neuroprotective properties are not an artefact of the individual animal models. Accordingly we conclude that these neuroprotective compounds are blocking a fundamental neuronal cell death pathway and therefore might be used for therapeutic benefit in a variety of settings.
Studies on the mechanism of P7C3
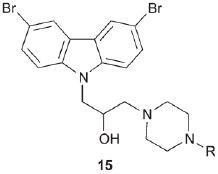
The observation that P7C3 and optimized analogues prevented neuronal cell death in multiple disease settings argued that they were acting at a late and common stage in the apoptotic program. Key steps in apoptosis are the depolarization of the mitochondrial membrane followed by release of cytochrome C. We therefore wondered if the P7C3 derived chemicals might affect mitochondrial integrity or signaling. In this regard, we became intrigued by a class of aminopropyl carbazoles discovered by scientists at Serono Pharmaceutical Research Institute. They described several piperazines of the general form 15 that reportedly inhibited the release of cytochrome C from mitochondria.32 They hypothesized that these carbazoles might inhibit a channel formed by the Bcl-2 family member Bax, although direct binding to Bax or any other protein was not demonstrated. We found that the most potent Serono compound (15, R = H) was active in the hippocampal neuroprotection assay, but less so than P7C3, both in terms of maximum activity and the minimal dose required to observe an effect. Likewise, an inactive Serono analogue (15, R = 4-F-C6H4) did not enhance the number of neural precursor cells. Nonetheless, we have not confirmed that the P7C3 class of chemicals operate through the same mechanism as the Serono chemicals, nor have we confirmed that the molecular target of P7C3 resides in the mitochondria.
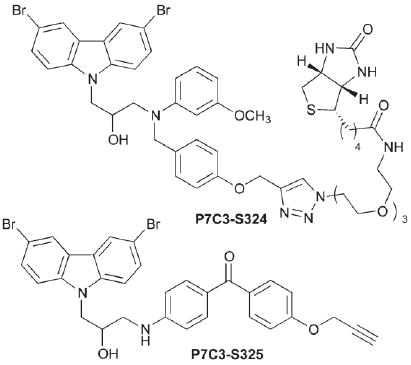
To discover the binding partner for P7C3, we again turned to synthetic chemistry to provide potentially useful reagents. First, as described above, we discovered that individual enantiomers of several derivatives displayed markedly different activity in several assays. We concluded from this observation that the P7C3 class of chemicals interacts with a specific biological macromolecule rather than simply acting within a membrane or as generic antioxidants. Second, through our structure-activity studies, we discovered several regions of the P7C3 scaffold that could tolerate substitution. In particular, P7C3-S324 and P7C3-S325 both approximately double the number of BrdU + cells in the hippocampal neuroprotection assay, an activity equivalent to P7C3 itself. The first of these reagents is decorated with a biotin moiety for use in affinity chromatography; the second features a benzophenone photo-crosslinker and an alkyne for post-crosslinking click conjugation to biotin, a fluorescent dye or a solid support. We are hopeful that these reagents will prove useful in the identification of receptors for P7C3 and the consequent unravelling of the mechanism by which it exerts its neuroprotective effects.
Conclusions
Neurodegenerative diseases represent a profound and growing challenge to the health care system. These disorders cause significant mortality and morbidity and are drivers of increasing health care costs. The large majority of neurodegenerative diseases lack effective therapies even though industrial, academic and governmental laboratories have targeted multiple proteins thought to play a role in disease initiation and/or progression. We adopted an unbiased approach that was agnostic to both target and chemical matter. Our only requirement was that chemicals work safely in live animals. In this way, we identified the P7C3 class of neuroprotective compounds, which were originally discovered as agents that could enhance hippocampal neurogenesis. Over the succeeding five years, we have improved the drug-like properties of lead molecules and demonstrated broad protective properties against mature neurons in various regions of the central nervous system. These studies have revealed the utility of P7C3 and its derivatives in animal models of PD, ALS, TBI, retinal degeneration and age-related decline in cognition. Ongoing studies aim to define their applicability to depression, Alzheimer’s disease and peripheral nerve damage, and will be reported in due course.
Initiating our study with in vivo pharmacology allowed us to focus only on non-toxic, chemically stable and bioavailable drug leads. We therefore circumvented the often lengthy pathway of chemical optimization prior to initial proof-of-concept studies. Additionally, we have been able to simultaneously move forwards – towards drug development – and backwards – towards a fuller mechanistic understanding of P7C3’s activity. While this approach is not common, drug discovery has its origins in unbiased, in vivo screening as far back as Erlich’s identification of the sulfa drugs and continues to dominate the discovery of anti-infectives.33 We suggest that target-agnostic drug discovery may have a valuable role to play in addressing maladies involving multiple cell types such as neurodegenerative diseases. For the sake of patients worldwide, we hope this prediction proves accurate.
Acknowledgments
We gratefully acknowledge the invaluable contributions of Noelle S. Williams and Loraine K. Morlock with regard to ADME/Tox experiments. We are likewise appreciative of the collaborators and scientists within our research groups including Jacinth Naidoo, Karen S. MacMillan, Jue Liang, Lisa Melito, Emanuela Capota, Hector De Jesus-Cortes, Jeremiah Britt, Paula Huntington, Sandi Jo Estill, Ruth Starwalt and Latisha McDaniel. Funding for the work described herein has been provided by the Edward N. and Della C. Thome Memorial Foundation and the Welch Foundation (I-1612) (to JMR), the NIMH (R01 MH087986) to AAP and SLM, funds from The Hartwell Foundation, Ted Nash Long Life Foundation, Brain & Behavior Research Foundation and International Mental Health Research Organization to AAP, and an unrestricted endowment provided to SLM.
Contributor Information
Andrew A. Pieper, University of Iowa Carver College of Medicine, Departments of Psychiatry, Neurology, and Veterans Affairs, 200 Hawkins Ave, Iowa City, Ia 52242, Ph: 319-353-5781 Fax: 319-353-3003, andrew-pieper@uiowa.edu
Steven L. McKnight, University of Texas Southwestern Medical Center, Department of Biochemistry, 5323 Harry Hines Blvd, Dallas, Texas 75390-9038, Ph: 214-648-3342, steven.mcknight@utsouthwestern.edu
Joseph M. Ready, University of Texas Southwestern Medical Center, Department of Biochemistry, 5323 Harry Hines Blvd, Dallas, Texas 75390-9038, Ph: 214-648-0313, joseph.ready@utsouthwestern.edu
Notes and references
- 1.Wokke J. Lancet. 1996;348:795. doi: 10.1016/S0140-6736(96)03181-9. [DOI] [PubMed] [Google Scholar]
- 2.Kumar R, Lozano AM, Kim YJ, Hutchison WD, Sime E, Halket E, Lang AE. Neurology. 1998;51:850. doi: 10.1212/wnl.51.3.850. [DOI] [PubMed] [Google Scholar]
- 3.Ledford H. Nature. 2010;466:1031. doi: 10.1038/4661031a. [DOI] [PubMed] [Google Scholar]
- 4.Hauser RA, Cantillon M, Pourcher E, Micheli F, Mok V, Onofrj M, Huyck S, Wolski K. Lancet Neurol. 2011;10:221. doi: 10.1016/S1474-4422(11)70012-6. [DOI] [PubMed] [Google Scholar]
- 5.(a) Swinney DC, Anthony J. Nat Rev Drug Discovery. 2011;10:507. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]; (b) Lee JA, Uhlik MT, Moxham CM, Tomandl D, Sall DJ. J Med Chem. 2012;55:4527. doi: 10.1021/jm201649s. [DOI] [PubMed] [Google Scholar]
- 6.(a) Altman J. Science. 1962;135:1127. doi: 10.1126/science.135.3509.1127. [DOI] [PubMed] [Google Scholar]; (b) Altman J, Das GD. J Comp Neurol. 1965;124:319. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]; (c) Gross CG. Nat Rev Neurosci. 2000;1:67. doi: 10.1038/35036235. [DOI] [PubMed] [Google Scholar]
- 7.Paton J, Nottebohm F. Science. 1984;225:1046. doi: 10.1126/science.6474166. [DOI] [PubMed] [Google Scholar]
- 8.(a) Encinas JM, Vaahtokari A, Enikolopov G. Proc Natl Acad Sci U S A. 2006;103:8233. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schmidt HD, Duman RS. Behav Pharmacol. 2007;18:391. doi: 10.1097/FBP.0b013e3282ee2aa8. [DOI] [PubMed] [Google Scholar]; (c) Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John MJ, Arango V. Neuropsychopharmacology. 2009;34:2376. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen C-H, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J, De BJ, Ready JM, McKnight SL. Cell. 2010;142:39. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kempermann G, Krebs J, Fabel K. Curr Opin Psychiatry. 2008;21:290. doi: 10.1097/YCO.0b013e3282fad375. [DOI] [PubMed] [Google Scholar]
- 11.(a) MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, Huntington PJ, Estill SJ, Longgood J, Becker GL, McKnight SL, Pieper AA, De Brabander JK, Ready JM. J Am Chem Soc. 2011;133:1428. doi: 10.1021/ja108211m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McKnight SL, Pieper AA, Ready JM, De Brabander J. 8,362,277. U S Pat. 2013
- 12.Naidoo J, Jesus-Cortes HD, Huntington P, Estill S, Morlock LK, Starwalt R, Mangano TJ, Williams NS, Pieper AA, Ready JM. doi: 10.1021/jm401919s. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naidoo J, Bemben CJ, Allwein SR, Liang J, Pieper AA, Ready JM. Tetrahedron Lett. 2013;54:4429. [Google Scholar]
- 14.Klunder JM, Onami T, Sharpless KB. J Org Chem. 1989;54:1295. [Google Scholar]
- 15.Zhao X, Zhuang W, Fang D, Xue X, Zhou J. Synlett. 2009:779. [Google Scholar]
- 16.Lees AJ, Hardy J, Revesz T. Lancet. 2009;373:2055. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 17.Jackson-Lewis V, Przedborski S. Nat Protocols. 2007;2:141. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- 18.(a) Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Proc Natl Acad Sci U S A. 1985;82:2173. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) D’Amato R, Lipman Z, Snyder S. Science. 1986;231:987. doi: 10.1126/science.3080808. [DOI] [PubMed] [Google Scholar]
- 19.Fukuda T. Neuropathology. 2001;21:323. doi: 10.1046/j.1440-1789.2001.00402.x. [DOI] [PubMed] [Google Scholar]
- 20.De Jesus-Cortes H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, Melito LM, Wang G, Williams NS, Ready JM, McKnight SL, Pieper AA. Proc Natl Acad Sci U S A. 2012;109:17010. doi: 10.1073/pnas.1213956109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tandan R, Bradley WG. Ann Neurol. 1985;18:271. doi: 10.1002/ana.410180302. [DOI] [PubMed] [Google Scholar]
- 22.(a) Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Nature. 1993;362:59. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]; (b) Valdmanis P, Daoud H, Dion P, Rouleau G. Curr Neurol Neurosci Rep. 2009;9:198. doi: 10.1007/s11910-009-0030-9. [DOI] [PubMed] [Google Scholar]
- 23.(a) Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Science. 1998;281:1851. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]; (b) Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Hum Mol Genet. 2009;18:3217. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boillée Sv, Vande Velde C, Cleveland DW. Neuron. 2006;52:39. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Gurney M, Pu H, Chiu A, Dal Canto M, Polchow C, Alexander D, Caliendo J, Hentati A, Kwon Y, Deng H, et al. Science. 1994;264:1772. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 25.Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, Starwalt R, Morlock L, Naidoo J, Williams NS, Ready JM, McKnight SL, Pieper AA. Proc Natl Acad Sci U S A. 2012;109:17016. doi: 10.1073/pnas.1213960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaya MO, Bramlett H, Naidoo J, Pieper AA, Dietrich WD., 3rd J Neurotrauma. 2013 doi: 10.1089/neu.2013.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Dash PK, Mach SA, Moore AN. J Neurosci Res. 2001;63:313. doi: 10.1002/1097-4547(20010215)63:4<313::AID-JNR1025>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Chirumamilla S, Sun D, Bullock MR, Colello RJ, Neurotrauma J. 2002;19:693. doi: 10.1089/08977150260139084. [DOI] [PubMed] [Google Scholar]; (c) Urrea C, Castellanos DA, Sagen J, Tsoulfas P, Bramlett HM, Dietrich WD. Restor Neurol Neurosci. 2007;25:65. [PubMed] [Google Scholar]; (d) Gao X, Enikolopov G, Chen J. Exp Neurol. 2009;219:516. doi: 10.1016/j.expneurol.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Chen X-H, Iwata A, Nonaka M, Browne KD, Smith DH. J Neurotrauma. 2003;20:623. doi: 10.1089/089771503322144545. [DOI] [PubMed] [Google Scholar]; (b) Gao X, Deng-Bryant Y, Cho W, Carrico KM, Hall ED, Chen J. J Neurosci Res. 2008;86:2258. doi: 10.1002/jnr.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, Potts MB, Fike JR. Exp Neurol. 2006;202:189. doi: 10.1016/j.expneurol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 29.Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. J Comp Neurol. 2003;467:1. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- 30.Asai-Coakwell M, March L, Dai XH, DuVal M, Lopez I, French CR, Famulski J, De Baere E, Francis PJ, Sundaresan P, Sauve Y, Koenekoop RK, Berry FB, Allison WT, Waskiewicz AJ, Lehmann OJ. Hum Mol Genet. 2013;22:1432. doi: 10.1093/hmg/dds560. [DOI] [PubMed] [Google Scholar]
- 31.(a) Gosse NJ, Baier H. Proc Natl Acad Sci U S A. 2009;106:2236. doi: 10.1073/pnas.0803202106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) French CR, Erickson T, French DV, Pilgrim DB, Waskiewicz AJ. Dev Biol. 2009;333:37. doi: 10.1016/j.ydbio.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 32.(a) Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, Halazy S. J Med Chem. 2003;46:4365. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]; (b) Peixoto PM, Ryu SÄ, Bombrun A, Antonsson B, Kinnally KW. Biochem J. 2009;423:381. doi: 10.1042/BJ20090664. [DOI] [PubMed] [Google Scholar]
- 33.(a) Butera JA. J Med Chem. 2013;56:7715. doi: 10.1021/jm400443k. [DOI] [PubMed] [Google Scholar]; (b) Gilbert IH. J Med Chem. 2013;56:7719. doi: 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sykes ML, Avery VM. J Med Chem. 2013;56:7727. doi: 10.1021/jm4004279. [DOI] [PubMed] [Google Scholar]