

Abstract
Nature is providing a bountiful pool of valuable secondary metabolites, many of which possess therapeutic properties. However, the discovery of new bioactive secondary metabolites is slowing down, at a time when the rise of multidrug-resistant pathogens and the realization of acute and long-term side effects of widely used drugs lead to an urgent need for new therapeutic agents. Approaches such as synthetic biology are promising to deliver a much-needed boost to secondary metabolite drug development through plug-and-play optimized hosts and refactoring novel or cryptic bacterial gene clusters. Here, we discuss this prospect focusing on one comprehensively studied class of clinically relevant bioactive molecules, the polyketides. Extensive efforts towards optimization and derivatization of compounds via combinatorial biosynthesis and classical engineering have elucidated the modularity, flexibility and promiscuity of polyketide biosynthetic enzymes. Hence, a synthetic biology approach can build upon a solid basis of guidelines and principles, while providing a new perspective towards the discovery and generation of novel and new-to-nature compounds. We discuss the lessons learned from the classical engineering of polyketide synthases and indicate their importance when attempting to engineer biosynthetic pathways using synthetic biology approaches for the introduction of novelty and overexpression of products in a controllable manner.
Keywords: refactoring, plug-and-play biology, combinatorial biosynthesis, secondary metabolites, drug discovery
Polyketides: magnificently modular
Polyketides represent an important class of compounds that are extremely diverse in structure and function. Natural screening strategies have brought more than 20 drugs to market, including the immunosuppressants FK506 and rapamycin (Park et al., 2010; Goranovic et al., 2012); hypocholesterolemics, such as lovastatin (Ma & Tang, 2007); anticancer agents, such as doxorubicin (Vasanthakumar et al., 2013); and a host of antimicrobials, including tetracycline and erythromycin (Weissman & Leadlay, 2005; Lesnik et al., 2009). Furthermore, it is predicted that more than 1% of polyketides described have potential drug activity (Koskinen & Karisalmi, 2005), and as a result of this, polyketides have received tremendous attention in efforts to unearth new compounds with bioactive properties. Interest in the discovery of novel acting polyketides has been renewed with the recent surge in microbial genome sequences and the availability of accurate genome mining software to detect a previously unexpected abundance of uncharacterized secondary metabolite biosynthesis gene clusters (BGCs). Advances in sequence analysis software such as antiSMASH are providing a facility to screen for BGCs in an automated computational fashion (Medema et al., 2011a, b2011b; Blin et al., 2013). They are particularly powerful in detecting polyketide BGCs, as these are defined by the presence of highly characteristic signature genes and motifs. Identification of putative BGCs using sequence-based analysis is also enabling the discovery of compounds that are cryptic, which are not expressed under laboratory conditions.
In addition to the clinical relevance and abundance of polyketides, there is one other reason behind the particular interest in polyketides as promising targets for synthetic biology: the highly modular architecture of both the BGCs and the constituent polyketide synthases (PKS) presents an ideal starting point from which to engineer chemical novelty in polyketides.
The biosynthesis of polyketides is modular at many levels. First, the genes responsible for polyketide biosynthesis are typically clustered in the genome (Chen et al., 2006), forming a BGC. Each BGC encodes the PKS responsible for the formation of the carbon backbone, together with the tailoring enzymes required for primary tailoring events, for example cyclization and dimerization of the β-keto-acyl carbon chain, subsequent tailoring events to form the final polyketide structure as well as genes encoding the regulation of the BGC and resistance to the end product if applicable, for example in the case of antibiotic end products. Once transcribed and translated, the PKS enzymes themselves are also modular in nature. The best-characterized PKS, 6-deoxyerythronolide B (6-dEB) synthase from Saccharopolyspora erythraea (Mironov et al., 2004), represents a good example of this. 6-dEB synthase consists of three megasynthases encoded by three ORFs, DEBS1–3 (Fig.1a). Each of these megasynthases comprises a series of modules responsible for the extension of the polyketide carbon backbone through addition and selective reduction of one acyl-CoA monomer to form a β-keto-acyl intermediate. In addition to this, each module can be dissected further still into a series of domains. Each of these domains is unequivocally linked with one specific catalytic function required for chain extension. Some domains are obligatory for recruitment of the acyl-CoA monomer and chain extension, for example acetyltransferase, acyl carrier protein (ACP) and ketosynthase, while others are accessory domains involved in the selective reduction of the β-keto-acyl intermediate to the corresponding alcohol, olefin or methylene group catalysed by ketoreductase, dehydratase and enoyl reductase activity, respectively. Importantly, all of the modules encoded within DEBS1–3 are required for successful synthesis of 6-dEB and act in succession, like a giant molecular assembly line (Weissman & Leadlay, 2005). Because each domain is unequivocally linked with one specific catalytic function and polyketides are synthesized in a collinear fashion, addition, removal and/or substitution of these domains or modules will theoretically result in defined alterations of the end product. Furthermore, the collinear architecture of these domains, and motifs within, can allow prediction of the structure of the polyketide and important elements of its stereochemistry from analysis of its coding sequence (Caffrey, 2003; Reid et al., 2003; Anand & Mohanty, 2012). With these rules in mind, theoretically, we have the potential to engineer rationally a desirable predefined polyketide end product if domains or modules can be stitched together like molecular lego bricks (Weber et al., 2003; Kennedy, 2008).
Figure 1.
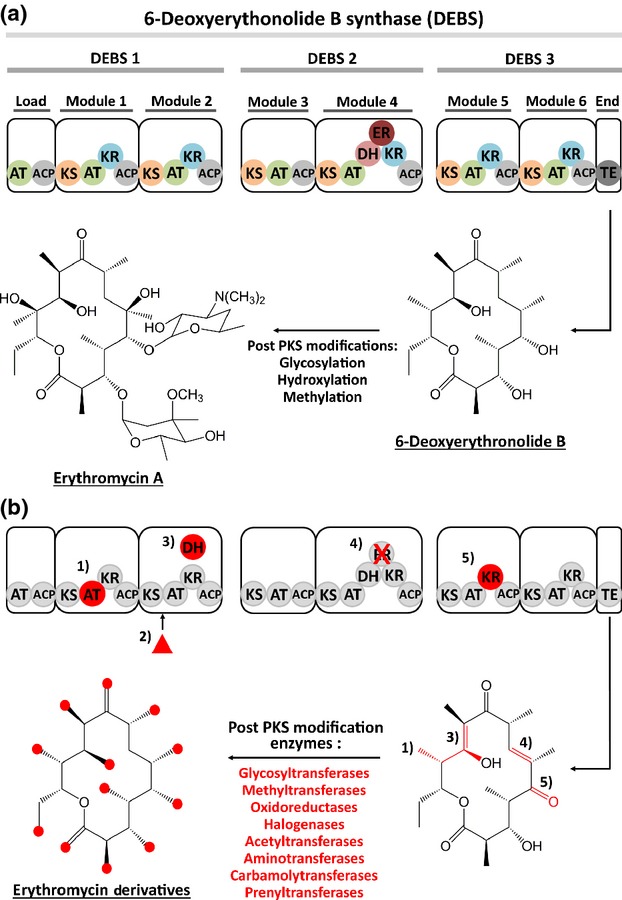
Pictorial illustration of 6-DEBS synthase, a modular type I PKS and successful attempts at engineering this megasynthase. (a) The native biosynthetic gene cluster and end product. (b) Summary of engineered cluster variants and their products; alterations are indicated in red. Manipulation of the polyketide scaffold includes: (1) substitution of domains (Oliynyk et al., 1996); (2) feeding with noncanonical substrates (Jacobsen et al., 1997); (3) domain insertion (McDaniel et al., 1997); (4) inactivation of domains (Donadio et al., 1993); and (5) domain deletions (Donadio et al., 1991). The effects of modifications 1–5 to the 6-dEB scaffold are also indicated in red, as are the positions at which engineered post-PKS tailoring modifications can occur. AT, acetyltransferase domain; ACP, acyl carrier protein domain; KS, ketosynthase domain; ER, enoyl reductase domain; DH, dehydratase domain.
With the increase in the number of characterized PKSs, it is becoming apparent that the collinear relationship between gene structure and chemical end product is not absolute (Piel, 2010); however, as a general rule, collinearity presents an ideal template for engineering the polyketide biosynthetic machinery. Consequently, manipulating polyketide assembly through domain alteration has been one major avenue that classical engineering has explored in order to derivatize known polyketides even before the current era of synthetic biology (Fig.1b; Donadio et al., 1991, 1993; Kao et al., 1994, 1995; Oliynyk et al., 1996; McDaniel et al., 1997; Ranganathan et al., 1999; Rowe et al., 2001). However, not all PKSs show the ‘one domain–one reaction step’ modular organization that is seen in type I PKSs. Chain extension can also occur iteratively through a recursive approach, where domains that are part of a single polypeptide are used repeatedly. This is the case for type II and type III PKSs, as well as for some type I fungal PKS, for example LovB and LovC in lovastatin biosynthesis (Campbell & Vederas, 2010). Although differences occur in enzymatic organization of PKSs, the underlying chemistry behind chain extension, through successive decarboxylative Claisen condensation of acyl-CoA monomers to form a β-keto-acyl intermediate and modification in cis or trans, remains the same for all (Lai et al., 2006). As such, all PKSs are in principle amenable to engineering (Kantola et al., 2003; Yu et al., 2012). Detailed reviews of the underlying biochemistry are available (Lai et al., 2006; Hertweck, 2009; Meier & Burkart, 2009; Walsh & Fischbach, 2010; Williams, 2013).
The immense diversity in the chemical structures of polyketides is the result of continuous evolutionary pressure for the development of chemical novelty facilitated by the modular nature of the PKS. On an evolutionary scale, diversity is introduced into polyketides through both simple mutations within domains and frequent horizontal co-transfer of genes between clusters (Donadio et al., 2005). Evolutionary analysis reveals conserved synteny between gene clusters responsible for the biosynthesis of homologous products, as well as products of considerable structural difference and those in between. Transfer of gene units between BGCs, permitted by their inherent modularity and collinearity, generates a continuous interspecific flow of compounds with novel physicochemical properties, not only as polyketides, but also for the generation of hybrid products containing additional nonribosomal peptide moieties. The recently described BGC encoding the biosynthesis of three zeamine-related antibiotics in the Serratia plymuthica RVH1 genome provides a good example of the plasticity of BGCs and their ability to co-transfer between organisms. This BGC comprises genes for five PKSs, three nonribosomal peptide synthetases and one mixed fatty acid synthase/PKS enzyme, which are required for the synthesis of the hybrid product backbones, as well as additional tailoring genes (Masschelein et al., 2013). Hybrid products, such as these, elucidate the tolerance of synthases to integrate noncanonical substrates from different biosynthetic systems into the growing carbon backbone successfully and are naturally occurring versions of domain alteration attempts paralleled in the laboratory-based engineering of PKSs.
Generation of novelty through exchange of domains between BGCs polished under evolutionary selection pressures, as above, invariably results in successful product assembly – as millions of failed ‘experiments’ are rapidly discarded by natural selection. This process cannot be replicated easily in vitro, and simple domain substitutions between BGCs commonly result in the failure of product release and maturation (Xu et al., 2013). Failure of product biosynthesis is regularly the result of the inflexibility of downstream enzymes to tolerate novel substrates. Without additional engineering, most domains incorporate only one substrate into the growing polyketide backbone and show little flexibility to introduce noncanonical substrates. Lessons learned from reprogramming PKSs using classical molecular biology approaches, detailed below, are supporting this general observation, but also, more interestingly, are revealing exceptions. This has provided an instruction manual that exemplifies the scope and limitations of plasticity of PKS to tolerate the integration of exogenous extenders into the growing β-keto-acyl chain.
Diversification of polyketides can occur at four steps throughout biosynthesis resulting from: (1) the choice of building blocks and chain length, (2) the extent of reduction and stereochemistry of β-keto intermediates (Reid et al., 2003), primary cyclization, alkylation and branching, (3) rearrangements and secondary cyclization and (4) postpolyketide tailoring: glycosylation, oxygenation, etc. In the following discussion, we focus on these events as two main phases of polyketide synthesis: core scaffold biosynthesis (steps 1–3) and subsequent or concurrent tailoring events (step 4; Fig.2).
Figure 2.
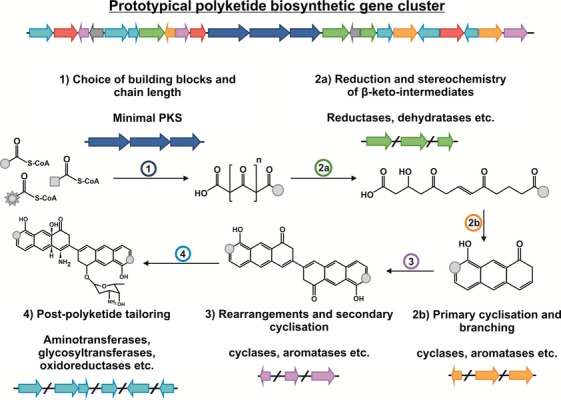
Schematic illustration of the four steps of polyketide biosynthesis encoded by a prototypical polyketide biosynthetic gene cluster. Each of these steps offers the potential for end product diversification by evolution or engineering as described in the text.
Modularity of scaffold biosynthesis
Initiation of biosynthesis
The composition of the polyketide backbone, or scaffold, structure is governed by the stringency of acetyltransferase domains to load a specific acyl-CoA substrate, but also through substrate stereochemistry and redox pattern (Sundermann et al., 2013): each PKS assembles an individual product through the choice of acyl-CoA units, their level of reduction and subsequent tailoring. Initiation of scaffold biosynthesis requires selection and recruitment of a starter unit onto a didomain, comprising an acetyltransferase and an ACP, collectively termed the loading module. The resulting initial starter unit serves as the first substrate in the growth of the final β-keto-acyl chain. Generation of diversity through the promiscuity of acetyltransferase domains to load multiple different starter units, termed polyspecificity, is more commonly observed than by polyspecificity of extender modules later in biosynthesis (McDaniel et al., 1999; Yuzawa et al., 2012). Introduction of diversity during initiation of biosynthesis also commonly occurs through the multiple different priming mechanisms used by the array of loading modules available (Moore & Hertweck, 2002; Hertweck, 2009). Due to the mechanistic promiscuity of the starter domains, combinatorial biosynthesis attempts to manipulate PKS modules often start with here. For example, the acetyltransferase and ACP loading module of DEBS1 naturally recruit a propionate starter unit. Substitution with loading modules from tylosin and oleandomycin type I megasynthases from Streptomyces fradiae and Streptomyces antibioticus, respectively, resulted in controlled integration of propionate or acetate as a starter unit (Long et al., 2002). Similarly, the replacement of the isobutyryl-CoA-specific loading module initiating avermectin biosynthesis in Streptomyces avermitilis M1 by the unique phoslactomycin polyketide cyclohexanecarboxylic unit loading module from Streptomyces platensis resulted in production of the veterinary antiparasitic doramectin (Wang et al., 2011). Alteration of loading modules for the initiation of biosynthesis is therefore one step showing promise for the generation of novel polyketides.
Chain extension
After initiation, continued assembly of the polyketide scaffold requires loading of extender units onto the acetyltransferase and ACP and incorporation into the β-keto-acyl intermediate by the ketosynthase. At this stage, diversity can be introduced through the installation of noncanonical extender units resulting from the polyspecificity of loading domains, domain substitutions or by the iterative action of an otherwise modular PKS (Kapur et al., 2012). The collection of commonly used extender units nature provides is modest: Canonical extender units comprise malonyl- and methylmalonyl-CoA. Substitution for domains loading other, less commonly used, extender units will allow introduction of a broadened chemistry into the polyketide backbone. For example, reductive carboxylation of α,β-unsaturated acyl-CoA precursors via crotonyl-CoA reductase/carboxylase homologues facilitates inclusion of hexyl-, propyl-, chloroethyl- and isobutylmalonyl-CoA into the polyketide scaffold (Eustaquio et al., 2009; Liu et al., 2009; Wilson et al., 2011). Alternatively, functionalization of extender units on stand-alone ACPs allows the incorporation of allyl- (Mo et al., 2011), amino-, hydroxyl- (Chan et al., 2006) and methoxymalonyl-ACP (Wu et al., 2000) extender units into the polyketide scaffold.
Predicting the polyspecificity of extender modules to introduce these rarer extender units is not straightforward. The mechanical processing and discrimination between acyl-CoA extender units by loading domains in type I modular PKSs is currently little understood. Investigations to elucidate why particular substrates are preferred or chosen are presenting a growing body of evidence suggesting that PKSs may be able to tolerate and incorporate exogenous natural and non-natural extender units into the β-keto-acyl chain. For example, analysis of the acyl-CoA substrate selectivity of PikAIV, a pikromycin synthase from Streptomyces venezuelae, elucidated the polyspecificity of extender modules towards substrates not readily present in the producer. PikAIV successfully loaded malonyl-, propionyl-, ethyl- and native methylmalonyl-CoA to the ACP. In the case of malonyl- and propionyl-CoA, active site occupancy was low at 3% and 19%, respectively. More interestingly, the rare extender ethylmalonyl-CoA showed acetyltransferase loading of 90% and low levels of hydrolytic release indicating its potential for incorporation during assembly; the native substrate methylmalonyl-CoA showed 100% acetyltransferase saturation (Bonnett et al., 2011). In the case of PikAIV, all acyl-CoA substrates were loaded; however, incorporation into the carbon chain depended upon the rate of subsequent hydrolytic release. These findings suggest that extender modules may show a greater tolerance to incorporate exogenous precursors lacking evolved selectivity, consistent with findings previously reported (Pohl et al., 2001). Substituting extender domains for the addition of novel extender units showing limited hydrolytic release could therefore result in the generation of novel polyketides; however, no concrete rules defining what properties extender modules require to do so have been elucidated, despite observed discrimination between sizes of extender units and incorporation.
Product release
Manipulating starter and extender modules of PKSs may permit the introduction of novel acyl-CoA substrates into the polyketide scaffold. However, for these to show activity, they must be released from the PKS. For successful release, it is important to identify which catalytic domains act as decision gates, thereby permitting continuation of downstream biosynthesis of altered β-keto-acyl intermediates. Elucidating such points will significantly aid success when engineering BGCs. Yeh et al. (2013) experimentally indicated that the phylogeny between nonreducing iterative PKS (nrPKS) modules is a good predictor of successful polyketide assembly and release from engineered BGCs. Increased phylogenetic proximity between gene units translated to improved domain–domain interactions and as a result, improved the release of the polyketide end product. In contrast, Xu et al. (2013) show for type II nrPKSs that the best predictors of thioesterase acceptance, and therefore release, are the shape and size of the polyketide substrate and consequently indicate the stringency of thioesterase domains in carrying out discriminative decision gate functions. For example, if the native substrate of a thioesterase was a nonaketide, but the engineered assembly line presented it with a heptaketide, the rate of release was almost zero (Vagstad et al., 2013). Substituting thioesterase domains often resulted in abolished product formation, despite the presence of an abundance of β-keto-acyl intermediates produced by the upstream domains, whereas judicious choices of thioesterase substitution resulted in the successful production of an unnatural polyketide product, radilarin (Xu et al., 2013). Successful polyketide release from thioesterase in the case of resorcyclic acid lactones and dihydroxyphenylacetate acid lactones may be dependent upon substrate size (Xu et al., 2013). However, contrastingly, truncation of the DEBS1–3 megasynthase through relocation of the thioesterase domains downstream of the modular DEBS1 resulted in assembly and successful release of a much shortened triketide lactone (Kao et al., 1995; Pfeifer et al., 2001). These contrasting results indicated the complex nature of the thioesterase and show the requirement for further work to build rules to predict thioesterase domain tolerance for substrates. Currently, for successful incorporation of novel starter and extender units, and successful product release, analysis of domains must be carried out on a case-by-case basis.
Modularity of tailoring reactions
Introducing diversity within the polyketide scaffold provides the ability to diversify the backbone structure. Further tailoring of these structures generates an additional level of complexity, and pathway engineering over the past decade has generated new-to-nature products through novel glycosylation, acyltransfer, hydroxylation, epoxidation, alkylation, transamination and desaturation reactions acting on naturally occurring products (Rix et al., 2002; Olano et al., 2010).
Tailoring enzymes can introduce chemical groups that often are more relevant to engineer for the alteration of specific activity of the polyketide than the backbone construct. 6-dEB is a precursor in the biosynthesis of the macrolide antibiotic erythromycin. Biosynthesis of erythromycin requires the action of tailoring enzymes encoded by ORFs located within the BGC encoding DEBS1–3. Without the required glycosylation, hydroxylation and methylation reactions catalysed by tailoring enzymes, 6-dEB cannot become active as erythromycin (Weissman & Leadlay, 2005). This is similarly the case for a group of type II aromatic polyketides with anticancer activities, the anthracyclines. The mechanisms of action of anthracyclines such as doxorubicin are mediated through DNA damage caused by the inhibition of DNA topoisomerase II, DNA binding and subsequent alkylation and intercalation within DNA of the target cells (Minotti et al., 2004). While the basic aglycone structures comprise 7,8,9,10-tetrahydro-5,12-naphthacene quinones, the observed anticancer activities of anthracyclines are heavily dependent on the attached sugars (Weymouth-Wilson, 1997). Furthermore, alteration of the attached sugars can modify not only activity, but also other parameters, such as toxicity. The clinical applications of doxorubicin are limited by dose-dependent cardiotoxic side effects. Epirubicin, an analogue of doxorubicin, with opposing configuration of a C-4 hydroxyl group on the deoxysugar, shows significantly less cardiotoxicity, while maintaining comparable antitumor properties (Hurteloup & Ganzina, 1986). Therefore, exchanging the sugars attached to the aglycone scaffold can tune the overall properties of therapeutic polyketides. Derivatization in this manner could be achieved through the addition of glycosyltransferases into BGCs, as opposed to introducing the sugar moieties semi-synthetically. First steps towards this have been undertaken (Han et al., 2011) and are revealing the substrate tolerance of individual tailoring enzymes.
In a parallel to the promiscuity of the scaffold biosynthesis genes, a case study of glycosyltransferases showcases the ability of tailoring enzymes to accept a broader range of substrates and a tolerance to modifying foreign acceptors molecules. ElmGT from Streptomyces olivaceus involved in elloramycin biosynthesis (Ramos et al., 2008) can glycosylate 8-demethyltetracenomycin C with D-mycarose, D-olivose, L-olivose, L-rhodinose, L-rhamnose and a disaccharide comprising two D-olivose moieties, showing extensive tolerance to glycosylate scaffolds with multiple sugars. Other glycosylases show tolerance to introduce a more defined range of sugars to a wider number of acceptor scaffolds. For example, the L-olivosyl glycosyltransferase OleG2 from S. antibioticus involved in oleandomycin production has promiscuity for NDP-L-mycarose, NDP-L-rhamnose and the foreign acceptor erythronolide B. The activities of ElmGT and OleG2 show a high tolerance to introduce noncanonical substrates onto aglycone scaffolds. This level of promiscuity for novel substrates and scaffolds is also consistent for EryCIII and EryBV from S. erythraea endogenous to the erythromycin BGC and the heterologously expressed UrdGT2 from S. fradiae Tü2717 involved in urdamycin A production, for the generation of novel C-glycosylated compounds (Wohlert et al., 1998; Doumith et al., 1999; Aguirrezabalaga et al., 2000; Gaisser et al., 2000; Rodriguez et al., 2000; Tang & McDaniel, 2001; Yoon et al., 2002).
Such flexibility of tailoring modifications may be a result of frequent co-transfer of tailoring genes between clusters, or a result of multifunctional tailoring of a series of different polyketides within the host; however, in both cases, the ability to introduce a variety of scaffolds opens up promising prospects for further diversification by synthetic biology. Full characterization and cataloguing of tailoring enzymes in a standardized ‘biobrick’ fashion would ultimately allow a user to pick and choose which modifications are desirable (Knight, 2003). Integration of tailoring enzymes into plug-and-play hosts as well as in standardized constructs would facilitate the rapid derivatization of novel compounds. Tailoring in this fashion shows potential to speed up rational design of polyketides at the increased rate that will be necessary, for example, to overcome the rapid and unavoidable emergence of resistance against them in pathogenic bacteria.
Synthetic biology: the future of combinatorial biosynthesis
Combinatorial biosynthesis has successfully exploited the functional collinearity of PKS domains, their structural modularity across many levels and their enzymatic flexibility and promiscuity for noncanonical substrates, to expand the accessible part of the polyketide universe. Progress, however, has been slower than expected. This is predominantly a result of our inability to predict the tolerance of enzymes to facilitate the downstream biosynthesis or incorporation of novel substrates. The inherent complexity of PKSs may exceed our capacity to define a set of pre-established rules that when followed ensure a judicious choice of modules or domains incorporated into a reengineered BGC for the successful generation of novel products. To overcome this limitation, high-throughput approaches, on a scale comparable to the working of evolutionary recombination, will be necessary. This is where synthetic biology's recent advances of writing genetic code at an unprecedented scale and complexity will usefully complement the repertoire of classical genetic engineering methodologies.
The first and most obvious application of synthetic biology will be for the production of novel compounds discovered by genome mining and metagenomics. It has been generally observed that the majority of BGCs in newly sequenced genomes are cryptic or silent, and the corresponding products are not produced at detectable levels in normal culture conditions. This is usually due to strict repressive control of gene expression by global regulation embedded within the coding sequence in the form of a complex combination of promoters, 5′-UTRs (Breaker, 2008; Ishihama, 2010), feed-forward and feed-back loops (Vasanthakumar et al., 2013), pause sites and small noncoding RNA (Georg & Hess, 2011; Guell et al., 2011). The intertwined nature of the control circuitry makes it difficult to circumvent native regulation and force expression. ‘Refactoring’, a synthetic biology methodology derived from software engineering, aims at decoupling this endogenous regulation through a comprehensive rewriting of BGCs. The resulting DNA sequence of a refactored BGC is as dissimilar as possible from the wild-type DNA sequence, yet still encoding the same amino acid end products. Rewriting the BGC in this fashion will remove all internal redundancy and regulation, including those regulatory elements that are currently undiscovered (Temme et al., 2012). Once a gene cluster has been refactored, we are able to introduce new, controllable and desired regulation to allow biosynthesis and characterization of the end compound, for example using orthogonal T7-based promoter libraries (Alper et al., 2005; Shis & Bennett, 2013).
Refactoring BGCs aims to by-pass the classical discovery limitations, decoupling desired product expression from the complex endogenous regulatory cascade. But the resulting engineered clusters can also be designed in such a way that they facilitate further reengineering through additions, deletions, substitutions, domain swapping or other modifications (Fig.1b). Of course, the same limitations for successful product assembly and release apply as they do in combinatorial biosynthesis. This also implies that the generation of novel products cannot reliably be achieved using random modularization of gene units from a multitude of different sources. Refactoring, however, is providing the methodology required to generate vast libraries of BGCs for the prospective biosynthesis of novel PKSs, and by its highly parallel approach, it may enable the elucidation of more informative assembly rules for the engineering of chemical novelty.
Refactoring approaches to synthetic biology do not need to be restricted to the nucleotide level. Ultimately, the aim would also be to optimize the encoded protein sequences to enhance their modularity and thus increase the engineering potential of PKSs. Advances towards this are already being made (Lockless & Muir, 2009); however, as a general strategy, such protein-level refactoring is currently still unrealistic. Nonetheless, the deluge of new genome sequence data is providing increasingly detailed insights into the rules that govern domain compatibility during the natural evolution of polyketide diversity (Chen et al., 2007; Thattai et al., 2007; Yuzawa et al., 2012), and multiplexed genome engineering strategies (Wang & Church, 2011) can be used to systematically explore these rules in the context of specific biosynthetic pathways.
In conclusion, the engineering-inspired approach of synthetic biology raises the dissection, standardization and decoupling of distinct catalytic units from highly integrated cellular processes to new levels of ambition. By learning from the pioneering efforts of combinatorial biosynthesis, as described above, these emergent technologies will soon yield the raw materials required to construct rationally designed biosynthetic machinery and regulatory circuits from first principles on a scale and, at a speed, far superseding our current capacity. Ultimately, the success of the next generation of polyketide bioprospecting for drug discovery will depend on an intimate interaction between protein chemistry, evolutionary genomics and synthetic biology.
Acknowledgments
We thank Jason Micklefield and Ashley Chessher for their helpful comments on the manuscript. M.C. was funded by a BBSRC studentship (BB/J014478/1).
References
- Aguirrezabalaga I, Olano C, Allende N, Rodriguez L, Brana AF, Mendez C. Salas JA. Identification and expression of genes involved in biosynthesis of L-oleandrose and its intermediate L-olivose in the oleandomycin producer Streptomyces antibioticus. Antimicrob Agents Chemother. 2000;44:1266–1275. doi: 10.1128/aac.44.5.1266-1275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper H, Fischer C, Nevoigt E. Stephanopoulos G. Tuning genetic control through promoter engineering. P Natl Acad Sci USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand S. Mohanty D. Modeling holo-ACP:DH and holo-ACP:KR complexes of modular polyketide synthases: a docking and molecular dynamics study. BMC Struct Biol. 2012;12:21. doi: 10.1186/1472-6807-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E. Weber T. antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnett SA, Rath CM, Shareef AR, Joels JR, Chemler JA, Hakansson K, Reynolds K. Sherman DH. Acyl-CoA subunit selectivity in the pikromycin polyketide synthase PikAIV: steady-state kinetics and active-site occupancy analysis by FTICR-MS. Chem Biol. 2011;18:1075–1081. doi: 10.1016/j.chembiol.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker RR. Gene regulation by riboswitches. FASEB J. 2008;22:1. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- Caffrey P. Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- Campbell CD. Vederas JC. Biosynthesis of lovastatin and related metabolites formed by fungal iterative PKS enzymes. Biopolymers. 2010;93:755–763. doi: 10.1002/bip.21428. [DOI] [PubMed] [Google Scholar]
- Chan YA, Boyne MT, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL. Thomas MG. Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units. P Natl Acad Sci USA. 2006;103:14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Vater J, Piel J, et al. Structural and functional characterization of three polyketide synthase gene clusters in Bacillus amyloliquefaciens FZB 42. J Bacteriol. 2006;188:4024–4036. doi: 10.1128/JB.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, Cane DE. Khosla C. Structure-based dissociation of a type I polyketide synthase module. Chem Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadio S, Staver MJ, McAlpine JB, Swanson SJ. Katz L. Modular organization of genes required for complex polyketide biosynthesis. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- Donadio S, McAlpine JB, Sheldon PJ, Jackson M. Katz L. An erythromycin analog produced by reprogramming of polyketide synthesis. P Natl Acad Sci USA. 1993;90:7119–7123. doi: 10.1073/pnas.90.15.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadio S, Sosio M, Stegmann E, Weber T. Wohlleben W. Comparative analysis and insights into the evolution of gene clusters for glycopeptide antibiotic biosynthesis. Mol Genet Genomics. 2005;274:40–50. doi: 10.1007/s00438-005-1156-3. [DOI] [PubMed] [Google Scholar]
- Doumith M, Legrand R, Lang C, Salas JA. Raynal MC. Interspecies complementation in Saccharopolyspora erythraea: elucidation of the function of oleP1, oleG1 and oleG2 from the oleandomycin biosynthetic gene cluster of Streptomyces antibioticus and generation of new erythromycin derivatives. Mol Microbiol. 1999;34:1039–1048. doi: 10.1046/j.1365-2958.1999.01666.x. [DOI] [PubMed] [Google Scholar]
- Eustaquio AS, McGlinchey RP, Liu Y, et al. Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-L-methionine. P Natl Acad Sci USA. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisser S, Reather J, Wirtz G, Kellenberger L, Staunton JU. Leadlay PF. A defined system for hybrid macrolide biosynthesis in Saccharopolyspora erythraea. Mol Microbiol. 2000;36:391–401. doi: 10.1046/j.1365-2958.2000.01856.x. [DOI] [PubMed] [Google Scholar]
- Georg J. Hess WR. cis-Antisense RNA, Another Level of Gene Regulation in Bacteria. Microbiol Mol Biol Rev. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goranovic D, Blazic M, Magdevska V, et al. FK506 biosynthesis is regulated by two positive regulatory elements in Streptomyces tsukubaensis. BMC Microbiol. 2012;12:238. doi: 10.1186/1471-2180-12-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guell M, Yus E, Lluch-Senar M. Serrano L. Bacterial transcriptomics: what is beyond the RNA horiz-ome? Nat Rev Microbiol. 2011;9:658–669. doi: 10.1038/nrmicro2620. [DOI] [PubMed] [Google Scholar]
- Han AR, Park JW, Lee MK, Ban YH, Yoo YJ, Kim EJ, Kim E, Kim BG, Sohng JK. Yoon YJ. Development of a Streptomyces venezuelae-based combinatorial biosynthetic system for the production of glycosylated derivatives of doxorubicin and its biosynthetic intermediates. Appl Environ Microbiol. 2011;77:4912–4923. doi: 10.1128/AEM.02527-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- Hurteloup P. Ganzina F. Clinical studies with new anthracyclines epirubicin, idarubicin, esorubicin. Drugs Exp Clin Res. 1986;12:233–246. [PubMed] [Google Scholar]
- Ishihama A. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol Rev. 2010;34:628–645. doi: 10.1111/j.1574-6976.2010.00227.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen JR, Hutchinson CR, Cane DE. Khosla C. Precursor-directed biosynthesis of erythromycin analogs by an engineered polyketide synthase. Science. 1997;277:367–369. doi: 10.1126/science.277.5324.367. [DOI] [PubMed] [Google Scholar]
- Kantola J, Kunnari T, Mantsala P. Ylihonko K. Expanding the scope of aromatic polyketides by combinatorial biosynthesis. Comb Chem High Throughput Screening. 2003;6:501–512. doi: 10.2174/138620703106298680. [DOI] [PubMed] [Google Scholar]
- Kao CM, Luo GL, Katz L, Cane DE. Khosla C. Engineered biosynthesis of a triketide lactone from an incomplete modular polyketide synthase. J Am Chem Soc. 1994;116:11612–11613. [Google Scholar]
- Kao CM, Luo GL, Katz L, Cane DE. Khosla C. Manipulation of macrolide ring size by directed mutagenesis of a modular polyketide synthase. J Am Chem Soc. 1995;117:9105–9106. [Google Scholar]
- Kapur S, Lowry B, Yuzawa S, Kenthirapalan S, Chen AY, Cane DE. Khosla C. Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation. P Natl Acad Sci USA. 2012;109:4110–4115. doi: 10.1073/pnas.1118734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J. Mutasynthesis, chemobiosynthesis, and back to semi-synthesis: combining synthetic chemistry and biosynthetic engineering for diversifying natural products. Nat Prod Rep. 2008;25:25–34. doi: 10.1039/b707678a. [DOI] [PubMed] [Google Scholar]
- Knight TF. MIT Synthetic Biology Working Group Technical Reports. 2003. Idempotent vector design for standard assembly of BioBricks.
- Koskinen AMP. Karisalmi K. Polyketide stereotetrads in natural products. Chem Soc Rev. 2005;34:677–690. doi: 10.1039/b417466f. [DOI] [PubMed] [Google Scholar]
- Lai JR, Koglin A. Walsh CT. Carrier protein structure and recognition in polyketide and nonribosomal peptide biosynthesis. Biochemistry. 2006;45:14869–14879. doi: 10.1021/bi061979p. [DOI] [PubMed] [Google Scholar]
- Lesnik U, Gormand A, Magdevska V, Fujs S, Raspor P, Hunter I. Petkovic H. Regulatory elements in tetracycline-encoding gene clusters: the otcG gene positively regulates the production of oxytetracycline in Streptomyces rimosus. Food Technol Biotechnol. 2009;47:323–330. [Google Scholar]
- Liu Y, Hazzard C, Eustaquio AS, Reynolds KA. Moore BS. Biosynthesis of salinosporamides from alpha, beta-unsaturated fatty acids: implications for extending polyketide synthase diversity. J Am Chem Soc. 2009;131:10376–10377. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless SW. Muir TW. Traceless protein splicing utilizing evolved split inteins. P Natl Acad Sci USA. 2009;106:10999–11004. doi: 10.1073/pnas.0902964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long PF, Wilkinson CJ, Bisang CP, et al. Engineering specificity of starter unit selection by the erythromycin-producing polyketide synthase. Mol Microbiol. 2002;43:1215–1225. doi: 10.1046/j.1365-2958.2002.02815.x. [DOI] [PubMed] [Google Scholar]
- Ma SM. Tang Y. Biochemical characterization of the minimal polyketide synthase domains in the lovastatin nonaketide synthase LovB. FEBS J. 2007;274:2854–2864. doi: 10.1111/j.1742-4658.2007.05818.x. [DOI] [PubMed] [Google Scholar]
- Masschelein J, Mattheus W, Gao LJ, et al. A PKS/NRPS/FAS hybrid gene cluster from Serratia plymuthica RVH1 encoding the biosynthesis of three broad spectrum, zeamine-related antibiotics. PLoS One. 2013;8:1. doi: 10.1371/journal.pone.0054143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel R, Kao CM, Fu H, Hevezi P, Gustafsson C, Betlach M, Ashley G, Cane DE. Khosla C. Gain-of-function mutagenesis of a modular polyketide synthase. J Am Chem Soc. 1997;119:4309–4310. [Google Scholar]
- McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M. Ashley G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. P Natl Acad Sci USA. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Alam MT, Breitling R. Takano E. The future of industrial antibiotic production: from random mutagenesis to synthetic biology. Bioeng Bugs. 2011a;2:230–233. doi: 10.1111/j.1751-7915.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E. Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011b;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JL. Burkart MD. The chemical biology of modular biosynthetic enzymes. Chem Soc Rev. 2009;38:2012–2045. doi: 10.1039/b805115c. [DOI] [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G. Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- Mironov VA, Sergienko OV, Nastasyak IN. Danilenko VN. Biogenesis and regulation of biosynthesis of erythromycins in Saccharopolyspora erythraea. Appl Biochem Microbiol. 2004;40:531–541. [PubMed] [Google Scholar]
- Mo S, Kim DH, Lee JH, et al. Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J Am Chem Soc. 2011;133:976–985. doi: 10.1021/ja108399b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BS. Hertweck C. Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat Prod Rep. 2002;19:70–99. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- Olano C, Mendez C. Salas JA. Post-PKS tailoring steps in natural product-producing actinomycetes from the perspective of combinatorial biosynthesis. Nat Prod Rep. 2010;27:571–616. doi: 10.1039/b911956f. [DOI] [PubMed] [Google Scholar]
- Oliynyk M, Brown MJB, Cortes J, Staunton J. Leadlay PF. A hybrid modular polyketide synthase obtained by domain swapping. Chem Biol. 1996;3:833–839. doi: 10.1016/s1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]
- Park SR, Yoo YJ, Ban YH. Yoon YJ. Biosynthesis of rapamycin and its regulation: past achievements and recent progress. J Antibiot. 2010;63:434–441. doi: 10.1038/ja.2010.71. [DOI] [PubMed] [Google Scholar]
- Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE. Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E-coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2010;27:996–1047. doi: 10.1039/b816430b. [DOI] [PubMed] [Google Scholar]
- Pohl NL, Hans M, Lee HY, Kim YS, Cane DE. Khosla C. Remarkably broad substrate tolerance of Malonyl-CoA synthetase, an enzyme capable of intracellular synthesis of polyketide precursors. J Am Chem Soc. 2001;123:5822–5823. doi: 10.1021/ja0028368. [DOI] [PubMed] [Google Scholar]
- Ramos A, Lombo F, Brana AF, Rohr J, Mendez C. Salas JA. Biosynthesis of elloramycin in Streptomyces olivaceus requires glycosylation by enzymes encoded outside the aglycon cluster. Microbiology. 2008;154:781–788. doi: 10.1099/mic.0.2007/014035-0. [DOI] [PubMed] [Google Scholar]
- Ranganathan A, Timoney M, Bycroft M, et al. Knowledge-based design of bimodular and trimodular polyketide synthases based on domain and module swaps: a route to simple statin analogues. Chem Biol. 1999;6:731–741. doi: 10.1016/s1074-5521(00)80020-4. [DOI] [PubMed] [Google Scholar]
- Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR. McDaniel R. A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- Rix U, Fischer C, Remsing LL. Rohr J. Modification of post-PKS tailoring steps through combinatorial biosynthesis. Nat Prod Rep. 2002;19:542–580. doi: 10.1039/b103920m. [DOI] [PubMed] [Google Scholar]
- Rodriguez L, Oelkers C, Aguirrezabalaga I, Brana AF, Rohr J, Mendez C. Salas JA. Generation of hybrid elloramycin analogs by combinatorial biosynthesis using genes from anthracycline-type and macrolide biosynthetic pathways. J Mol Microbiol Biotechnol. 2000;2:271–276. [PubMed] [Google Scholar]
- Rowe CJ, Bohm IU, Thomas IP, et al. Engineering a polyketide with a longer chain by insertion of an extra module into the erythromycin-producing polyketide synthase. Chem Biol. 2001;8:475–485. doi: 10.1016/s1074-5521(01)00024-2. [DOI] [PubMed] [Google Scholar]
- Shis DL. Bennett MR. Library of synthetic transcriptional AND gates built with split T7 RNA polymerase mutants. P Natl Acad Sci USA. 2013;110:5028–5033. doi: 10.1073/pnas.1220157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E. Schulz F. Enzyme-directed mutasynthesis: a combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem Biol. 2013;8:443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- Tang L. McDaniel R. Construction of desosamine containing polyketide libraries using a glycosyltransferase with broad substrate specificity. Chem Biol. 2001;8:547–555. doi: 10.1016/s1074-5521(01)00032-1. [DOI] [PubMed] [Google Scholar]
- Temme K, Zhao DH. Voigt CA. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. P Natl Acad Sci USA. 2012;109:7085–7090. doi: 10.1073/pnas.1120788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thattai M, Burak Y. Shraiman BI. The origins of specificity in polyketide synthase protein interactions. PLoS Comput Biol. 2007;3:1827–1835. doi: 10.1371/journal.pcbi.0030186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagstad AL, Newman AG, Storm PA, Belecki K, Crawford JM. Townsend CA. Combinatorial domain swaps provide insights into the rules of fungal polyketide synthase programming and the rational synthesis of non-native aromatic products. Angew Chem Int Ed Engl. 2013;52:1718–1721. doi: 10.1002/anie.201208550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanthakumar A, Kattusamy K. Prasad R. Regulation of daunorubicin biosynthesis in Streptomyces peucetius – feed forward and feedback transcriptional control. J Basic Microbiol. 2013;53:636–644. doi: 10.1002/jobm.201200302. [DOI] [PubMed] [Google Scholar]
- Walsh CT. Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132:2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HH. Church GM. Multiplexed genome engineering and genotyping methods: applications for synthetic biology and metabolic engineering. In: Voigt C, editor; Synthetic Biology, Pt B: Computer Aided Design and DNA Assembly. San Diego, CA: Elsevier Academic Press Inc; 2011. pp. 409–426. Vol. 498 (, ed.) &. [DOI] [PubMed] [Google Scholar]
- Wang JB, Pan HX. Tang GL. Production of doramectin by rational engineering of the avermectin biosynthetic pathway. Bioorg Med Chem Lett. 2011;21:3320–3323. doi: 10.1016/j.bmcl.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Weber T, Welzel K, Pelzer S, Vente A. Wohlleben W. Exploiting the genetic potential of polyketide producing streptomycetes. J Biotechnol. 2003;106:221–232. doi: 10.1016/j.jbiotec.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Weissman KJ. Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- Weymouth-Wilson AC. The role of carbohydrates in biologically active natural products. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- Williams GJ. Engineering polyketide synthases and nonribosomal peptide synthetases. Curr Opin Struct Biol. 2013;23:603–612. doi: 10.1016/j.sbi.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MC, Nam SJ, Gulder TAM, Kauffman CA, Jensen PR, Fenical W. Moore BS. Structure and biosynthesis of the marine streptomycete ansamycin ansalactam A and its distinctive branched chain polyketide extender unit. J Am Chem Soc. 2011;133:1971–1977. doi: 10.1021/ja109226s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlert SE, Blanco G, Lombó F, et al. Novel hybrid tetracenomycins through combinatorial biosynthesis using a glycosyltransferase encoded by the elm genes in cosmid 16F4 and which shows a broad sugar substrate specificity. J Am Chem Soc. 1998;120:10596–10601. [Google Scholar]
- Wu K, Chung L, Revill WP, Katz L. Reeves CD. The FK520 gene cluster of Streptomyces hygroscopicus var. ascomyceticus (ATCC 14891) contains genes for biosynthesis of unusual polyketide extender units. Gene. 2000;251:81–90. doi: 10.1016/s0378-1119(00)00171-2. [DOI] [PubMed] [Google Scholar]
- Xu YQ, Zhou T, Zhang SW, Xuan LJ, Zhan JX. Molnar I. Thioesterase domains of fungal nonreducing polyketide synthases act as decision gates during combinatorial biosynthesis. J Am Chem Soc. 2013;135:10783–10791. doi: 10.1021/ja4041362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HH, Chang SL, Chiang YM, Bruno KS, Oakley BR, Wu TK. Wang CC. Engineering fungal nonreducing polyketide synthase by heterologous expression and domain swapping. Org Lett. 2013;15:756–759. doi: 10.1021/ol303328t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YJ, Beck BJ, Kim BS, Kang H-Y, Reynolds KA. Sherman DH. Generation of multiple bioactive macrolides by hybrid modular polyketide synthases in Streptomyces venezuelae. Chem Biol. 2002;9:203–214. doi: 10.1016/s1074-5521(02)00095-9. [DOI] [PubMed] [Google Scholar]
- Yu DY, Xu FC, Zeng J. Zhan JX. Type III polyketide synthases in natural product biosynthesis. LUBMB Life. 2012;64:285–295. doi: 10.1002/iub.1005. [DOI] [PubMed] [Google Scholar]
- Yuzawa S, Kapur S, Cane DE. Khosla C. Role of a conserved arginine residue in linkers between the ketosynthase and acyltransferase domains of multimodular polyketide synthases. Biochemistry. 2012;51:3708–3710. doi: 10.1021/bi300399u. [DOI] [PMC free article] [PubMed] [Google Scholar]