

Abstract
Microglia are resident antigen-presenting cells in the central nervous system (CNS) that either suppress or promote disease depending on their activation phenotype and the microenvironment. Multiple sclerosis (MS) is a chronic inflammatory disease causing demyelination and nerve loss in the CNS, and experimental autoimmune encephalomyelitis (EAE) is an animal model of MS that is widely used to investigate pathogenic mechanisms and therapeutic effects. We isolated and cultured microglia from adult mouse brains and exposed them to specific combinations of stimulatory molecules and cytokines, the combination of IL-4, IL-10, and TGF-β yielding the optimal regime for induction of an immunosuppressive phenotype (M2). M2 microglia were characterized by decreased expression or production of CD86, PD-L1, nitric oxide, and IL-6, increased expression of PD-L2, and having a potent capacity to retain their phenotype on secondary proinflammatory stimulation. M2 microglia induced regulatory T cells, suppressed T-cell proliferation, and downmodulated M1-associated receptor expression in M1 macrophages. Myelin oligodendrocyte glycoprotein (MOG)-induced EAE was induced in DBA/1 mice and at different time points (0, 5, 12, or 15 days postimmunization) 3 × 105 M2 microglia were transferred intranasally. A single transfer of M2 microglia attenuated the severity of established EAE, which was particularly obvious when the cells were injected at 15 days postimmunization. M2 microglia-treated mice had reduced inflammatory responses and less demyelination in the CNS. Our findings demonstrate that adult M2 microglia therapy represents a novel intervention that alleviated established EAE and that this therapeutic principle may have relevance for treatment of MS patients.
Keywords: EAE, microglia, macrophages, phenotype, cell therapy
Introduction
Microglia belong to the family of mononuclear phagocytes, are considered to be resident macrophages in the central nervous system (CNS), and derive from yolk-sac progenitors (Ransohoff and Cardona, 2010; Saijo and Glass, 2011). Macrophages are an abundant immune-competent cell type that play a crucial role in immune responses to pathogens, and as such are significant mediators of inflammatory processes. However, inflammation is associated with deleterious effects for the tissue environment, and must thus be repressed to allow complete healing. Macrophages also play a major role in the resolution of inflammation by producing anti-inflammatory cytokines and chemokines and by eliminating tissue debris (Murray and Wynn, 2011; Weisser et al., 2013). Microglia/macrophages can exhibit both proinflammatory and anti-inflammatory properties, depending on the disease stage and the signals they receive, i.e., the inflammatory balance in the microenvironment. Different activation states of microglia/macrophages have been described in both rodents and man, generally assigned into two groups: M1 classically activated or type I microglia/macrophages, which are proinflammatory effectors, and M2 alternatively activated or type II microglia/macrophages that exhibit anti-inflammatory properties (Crain et al., 2013; Martinez et al., 2008). A more modern view reflects a spectrum of activation states between these extremes (Mosser and Edwards, 2008) and a genetic influence on activation grades (Andersson et al., 2004).
Multiple sclerosis (MS) is a chronic inflammatory demyelinating autoimmune disease of the CNS. It is the most frequent neurological disease in young adults and affects over 2 million people worldwide. Most types of immune cell have been associated with MS pathogenesis, demonstrating the importance of both innate and adaptive immune mechanisms. Infiltrating macrophages and activated CNS-resident microglia can cause and mediate destruction of myelin and axons through both direct damage and production of proinflammatory molecules, thereby increasing the disease severity and giving a poor prognosis (Almolda et al., 2011). Experimental autoimmune encephalomyelitis (EAE), the animal model of MS, is often used to study neuroinflammatory disease mechanisms and potential therapies. The similarities between MS and EAE are established in many aspects, such as disease course, pathological observations, and inflammatory mechanisms (Owens and Sriram, 1995). We have previously established a MOG-EAE model in DBA/1 mice that has a very severe disease course, and have characterized its immunological basis as well as using it to test therapeutic interventions (Abdul-Majid et al., 2003; Wallberg and Harris, 2005; Wallberg et al., 2003). As the degree of infiltration of monocytes into the CNS has been determined to correlate with progression to paralytic EAE (Ajami et al., 2011) and the numbers of CNS myeloid cells contracts during remissions and expands during relapses (King et al., 2009), the importance of myeloid cells in EAE pathogenesis is clear. Importantly, the imbalance of monocyte activation profiles and impaired M2 expression are key factors in relapse development (Mikita et al., 2011), and sustained proinflammatory microglial activation prevents repair (Rasmussen et al., 2011).
The primary aims of MS therapy are reinstating physiological function after a disease episode, preventing new attacks and long-term disability. Existing medications are modestly effective at decreasing the number of attacks in relapsing-remitting MS (RRMS) and in reducing the accumulation of brain lesions. Treatment of secondary progressive MS (SPMS) and primary progressive MS (PPMS) is problematic and more difficult than RRMS as many patients do not respond to any available therapy (Anlar, 2009; Jones and Coles, 2010; Pozzilli and Prosperini, 2008). As current treatments are generally only partly beneficial, MS remains a significant health burden for both individuals and society.
There is increasing interest in the use of cell-based therapies as a promising strategy for a variety of autoimmune diseases, including MS. Mesenchymal stem cells (MSCs) display stromal features and exert bystander immunomodulatory and neuroprotective activities, and it has been reported that transplanted MSCs promote functional recovery and myelin repair in different MS animal models (Auletta et al., 2012; Burman et al., 2013). However, these cells have been determined to have various adverse effects, especially in the context of their direct and indirect involvement in cancer. It has also been demonstrated that regulatory T cells (Tregs) play a critical role in the protection and recovery of EAE (Wallberg et al., 2003).
In recent years, mononuclear phagocytes have been studied in the treatment of inflammatory disorders. Following our initial report (Wallberg and Harris, 2005) the administration of ex vivo activated M2 macrophages/monocytes in suppressing ongoing disease has been repeated and extended by other groups, indicating a clear and immunomodulatory potential of this strategy in EAE (Mikita et al., 2011). We have additionally demonstrated proof-of-concept that adoptive transfer of specifically activated anti-inflammatory macrophages could prevent development of chronic autoimmune inflammatory disease in a setting of type 1 diabetes (T1D) (Parsa et al., 2012).
Considering that microglia are in their natural environment in the CNS, we hypothesized that the adoptive transfer of specifically activated anti-inflammatory microglia during EAE could extend this therapeutic concept. Although most microglial studies use cells derived from neonatal animals, we recently reported significant differences in neonatal and adult microglia (Scheffel et al., 2012). We thus used adult M2 microglia in the severe MOG-EAE DBA/1 model to assess their therapeutic potential.
Materials and Methods
Animals
Female DBA/1 mice were purchased from Taconic (Lille Skensved, Denmark) and maintained in our animal facility at Karolinska Hospital. All mice were between 9 and 11 weeks of age, weighing 20–23 g, were pathogen-free, and had access to chow and water ad libitum. All experiments performed were in accordance with the local ethical committee in Stockholm North.
EAE Induction and Clinical Evaluation
Recombinant protein corresponding to the N-terminal sequence of mouse myelin oligodendrocyte glycoprotein (MOG) (amino acids 1–125) was expressed in Escherichia coli and purified to homogeneity by chelate chromatography as previously described (Amor et al., 1994). Purified MOG dissolved in 6 M urea was dialyzed against sodium acetate buffer (10 mM, pH 3.0) to obtain a soluble preparation that was stored frozen at −20°C. Mice were anesthetized with isoflurane (Forane; Abbott Laboratories, Abbot Park, IL) and injected subcutaneously at the dorsal tail base with 100 μL inoculum containing 50 μg of MOG in phosphate-buffered saline (PBS) emulsified in Complete Freund's Adjuvant (CFA) containing 100 μg heat-killed Mycobacterium tuberculosis H37Ra (Difco Laboratory, Detroit, MI). Mice were weighed and scored daily for clinical signs of EAE using a 6-point scale as follows: 0, no clinical signs of EAE; 1, tail weakness or tail paralysis; 2, hindlimb paraparesis or hemiparesis; 3, hindlimb paralysis or hemiparalysis; 4, tetraplegia or moribund; and 5, death.
Primary Culture of Adult Microglia and Bone Marrow-Derived Macrophages
Primary glial cells from adult mice were cultured as described previously with modifications (Moussaud and Draheim, 2010). Briefly, 10-week-old female DBA/1 mice were deeply anesthetized and perfused in the left ventricle with ice-cold PBS supplemented with 2 U/mL heparin (LEO Pharma AB, Malmö, Sweden). After 5-min perfusion the brain was aseptically removed and freed from the meninges. The brain of one animal was then transferred to a Petri dish containing 5 mL of enzymatic solution and finely minced. The enzymatic solution contained 116 mM NaCl, 5.4 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 1.5 mM CaCl2, 1 mM MgSO4, 0.5 mM EDTA, 25 mM glucose, 1 mM cysteine, and 20 U/mL papain (all from Sigma, Stockholm, Sweden). The tissue was then incubated at 37°C, 5% CO2 and continuously stirred. After a 90-min incubation the digested brain was transferred to a 50-mL conical tube and the enzymatic reaction was stopped by the addition of 20 mL of 20% heat-inactivated fetal bovine serum (FBS) in Hank's balanced salt solution (HBSS) (Invitrogen, Sweden). After centrifugation at 200g for 7min at room temperature (RT), the pellet was resuspended in 2 mL of 0.5 mg/mL DNase I (Roche, Sweden) in HBSS and incubated for 5 min at RT. Finally, the homogenate was filtered through a 40-μm cell strainer (Becton Dickinson, Sweden) and centrifuged again at 200g for 7 min at RT. A discontinuous gradient density centrifugation step using Percoll (Sigma) was then performed; the cells were resuspended in 20 mL of 20% stock isotonic Percoll in HBSS. The cell suspension was carefully overlain with 20 mL pure HBSS and centrifuged at 200g for 20 min with slow acceleration and no brake. The pellet that contained mixed glial cells was washed once in HBSS before being resuspended in Dulbecco's modified Eagle's/F12 medium with GlutaMAX™I (DMEM/F12) supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine (all reagents from Life Technologies, Paisley, Scotland), and 30% M-CSF-conditioned L929 cell line supernatant. The cell suspension from one mouse was plated in one T75-cell culture flask (Invitrogen, Carlsbad, CA) coated with poly-l-lysine (Sigma) and incubated at 37°C and 5% CO2 in a humidified incubator. The medium was changed twice a week until the cells became confluent (after ∼14 days). When full confluence of the cell layer was reached, mixed glial cells were harvested using prewarmed trypsin (Gibco, Grand Island, NYA).
To separate microglia and astrocytes from the mixed glial cells, magnetic separation was performed according to the manufacturer's instructions. In brief, the mixed cell suspension was centrifuged at 300g for 10 min, the cell pellet was resuspended in MACS buffer (Miltenyi Biotec GmbH, Germany), and thereafter subjected to AutoMACS magnetic positive separation using anti-CD11b MicroBeads (Miltenyi Biotec GmbH). The resulting microglia were seeded in 48-well plates (2 × 105 cells per well) or temperature-sensitive Petri dishes (Thermo Scientific, Sweden) for stimulation before adoptive transfer.
Bone marrow-derived macrophages were cultured as described previously (Weischenfeldt and Porse, 2008). In brief, femoral bone marrow cells were collected, single-cell suspensions were prepared and cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 20% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine, and 20% M-CSF-conditioned L929 cell line supernatant for 10 days. Cells were harvested using 2 mM EDTA (Sigma).
Microglia/macrophages were then either left (a) unstimulated, (b) stimulated with lipopolysaccharide (LPS)/IFN-γ (50 ng/mL; 20 ng/mL), or (c) with 20 ng/mL recombinant mouse IL-4, IL-10, and recombinant human TGF-β1 (R&D Systems, MN), either alone or in combination. Cells were analyzed using flow cytometry. To check the morphology of activated microglia/macrophages, the cells were labeled with lectin (Vector Laboratories, Stockholm, Sweden) and counterstained with DAPI.
Co-Culture of Microglia with T Lymphocytes or M1 Macrophages
Splenocytes were obtained by mechanically disrupting spleens through a 40-μm nylon strainer. Erythrocytes were lysed with ACK buffer (Gibco) for 5 min on ice and washed with DPBS. T lymphocytes were isolated from splenocytes using the Pan T Cell Isolation Kit II, an LS Column, and a MidiMACS™ Separator (Miltenyi Biotec GmbH). Microglia and T lymphocytes were co-cultured in either 24- or 96-well flat-bottom plates (Corning, NY) for 48 h. T lymphocytes were stimulated with 0.5 μg/mL anti-CD3 and anti-CD28 molecular complex (BD) for 2 h before the co-culture. Proliferation was measured using [methyl-3H]thymidine (Amersham, Aylesbury, UK) at a concentration of 1 µCi/well. Flow cytometry was used to assess induction of Tregs.
To assess if M2 microglia could directly modulate an M1 macrophage phenotype, M2 microglia or untreated microglia (as control) and M1 macrophages were co-cultured at different ratios (M2:M1 = 1:2, 1:1, or 2:1) for 24 h. To identify the polarized microglia/macrophages after mixing them, M1 macrophages were prelabeled with 2 μg/mL XenoLight DiD (Invitrogen) before co-culture. The modulated phenotype of M1 macrophages was then evaluated by examining surface marker expression using flow cytometric analysis.
Intranasal Adoptive Transfer of Microglia to EAE Mice
Microglia stimulated with IL-4/IL-10/TGF-β for 24 h or untreated microglia (cultured in medium alone) were injected intranasally (i.n) with PBS as vehicle into EAE mice at different time points [days 0, 5, 12, or 15 postimmunization (p.i)]. Intranasal cell or vehicle (PBS) application was performed during a short isoflurane anesthesia. Before treatment all animals received 100 U hyaluronidase (Sigma) i.n dissolved in 12 µL of sterile PBS. Mice received twice in each nostril alternate applications (left–right) of 3 μL drops containing cell suspension/hyaluronidase or PBS (the second set of right–left applications was performed 2 min after the first set). One hour after i.n pretreatment with hyaluronidase a microglia suspension (3 × 105 in 12 μL of sterile PBS) or vehicle (PBS) was applied using the same procedure. In separate experiments, IL-4/IL-10/TGF-β polarized macrophages were injected intravenously into EAE mice at two different time points (days 0 or 15 p.i).
Histological Analysis
Day 30 p.i (15 days after adoptive microglia transfer) mice were anesthetized and perfused via the aorta with 30 mL of PBS and 20 mL of 4% paraformaldehyde. Histological evaluation was performed on 14-µm frozen sections of brains and spinal cords stained with hematoxylin–eosin and Luxol fast blue to assess inflammation and demyelination, respectively. In adjacent serial sections immunohistochemistry was performed with antibodies against macrophages/activated microglia (Iba1; Wako, Osaka, Japan) and astrocytes (GFAP; AbCam, Cambridge, UK). Bound antibody was visualized using HRP-labeled secondary antibody and DAB reagent and control sections were incubated in the absence of primary antibody. Inflammatory cell infiltration was assessed blindly in a semiquantitative fashion, from − (no infiltration) to +++ (severe infiltration).
Flow Cytometric Analysis
Stimulated primary cultured microglia were analyzed by flow cytometry for their activation phenotypes. Microglia were detached with 2 mM EDTA (Sigma) for 30 min at 37°C. Antibodies against CD86 (eBioscience), PD-L1 (MIH5, eBioscience), and PD-L2 (TY25, eBioscience) were used for analysis.
In addition, day 30 p.i (day 15 after adoptive microglia transfer) mice were anesthetized and transcardically perfused with PBS. Microglia and infiltrating cells were isolated from brains and spinal cords and stained with antibodies specific for CD11b (Biolegend), CD3 (17A2, Biolegend), CD4 (GK1.5, Beckman Coulter), CD8α (53-6.7, Beckman Coulter), CD62L (MEL-14, Beckman Coulter), CD44 (IM7, BD), IFNγ (BD), IL-17 (BD), and isotype controls rat IgG2a (eBioscience) or rat IgG2b (eBioscience). Samples were run in a Gallios flow cytometer (Beckman Coulter, Brea, CA) and analyzed using Kaluza v1.1 software (Beckman Coulter).
Tracking of Adoptive Transferred Microglia
For tracking of transferred cells, IL-4/IL-10/TGF-β-stimulated microglia were incubated with 2 μg/mL XenoLight DiI (Invitrogen) for 5 min at 37°C and then quickly transferred to 4°C for 15 min before i.n injection. Twenty-four hours or 72 h after microglia delivery, MOG-EAE mice were sacrificed. Frozen sections of whole CNS tissue (including the olfactory bulb) were prepared as described for histological analysis, and single-cell suspensions from deep cervical lymph nodes, superficial cervical lymph nodes, as well as inguinal lymph nodes were prepared and stained for flow cytometric analyses.
Cytokine Analysis and Nitric Oxide Detection
ELISA kits for detection of secreted IL-6 and IL-10 in cell culture supernatants were purchased from Biolegend (San Diego, US) and used according to the manufacturer's instructions. In brief, supernatants were collected from microglia cultures, run in duplicate wells, and absorbances were measured at 450 nm and subtracted with the absorbance at 562 nm using a microplate reader (LabSystems, Basingstoke, UK).
The measurement of nitric oxide (NO) in the microglia cell culture supernatants was performed using the modified Griess reagent (Sigma). Samples were plated in duplicate wells of flat 96-well plates (Corning), mixed with equal volumes of freshly prepared Griess reagent, and incubated for 15 min at room temperature. Absorbance was measured at 540 nm using a microplate reader (LabSystems).
Statistical Analysis
Statistical significance was determined by Mann–Whitney U-test. A P-value of <0.05 (*) was considered significant, and error bars are presented as SEM. Statistical analysis was conducted using GraphPad software v6 (San Diego, CA).
Results
Microglial Stimulation with IL-4/IL-10/TGF-β Induces a Robust M2 Phenotype
Cytokines induce a multitude of effects on microglia, directing receptor expression, intracellular signaling, phenotype stability, and biological properties. We have previously defined optimal protocols for reliable and stable induction of an anti-inflammatory macrophage phenotype using combined stimulation with IL-4/IL-10/TGFβ (Parsa et al., 2012). In this study, we modified a novel protocol for culturing adult microglia. We stimulated these microglia using the same cytokine stimulatory protocol and assessed their phenotype.
Similar to macrophages, distinct M1 and M2 phenotypes of microglia were confirmed by their different morphologies (Fig. 1). After 24 h of stimulation, IFNγ/LPS-treated microglia (M1 phenotype) expressed higher levels of CD86 and PD-L1, and lower levels of PD-L2. Conversely, IL-4/IL-10/TGFβ-treated microglia (M2 phenotype) had decreased expression of CD86, mild expression of PD-L1, and strong expression of PD-L2 (Fig. 2A–C). IL-4 seemed to be the key cytokine in this context, because IL-4 alone could induce high expression of PD-L2 in microglia (Fig. 2C). Additionally, IFNγ/LPS-stimulated microglia secreted high levels of IL-6 and NO, whereas these were almost immeasurable in IL-4/IL-10/TGFβ-stimulated microglia, either alone or in combination (Fig. 2D,E).
Figure 1.
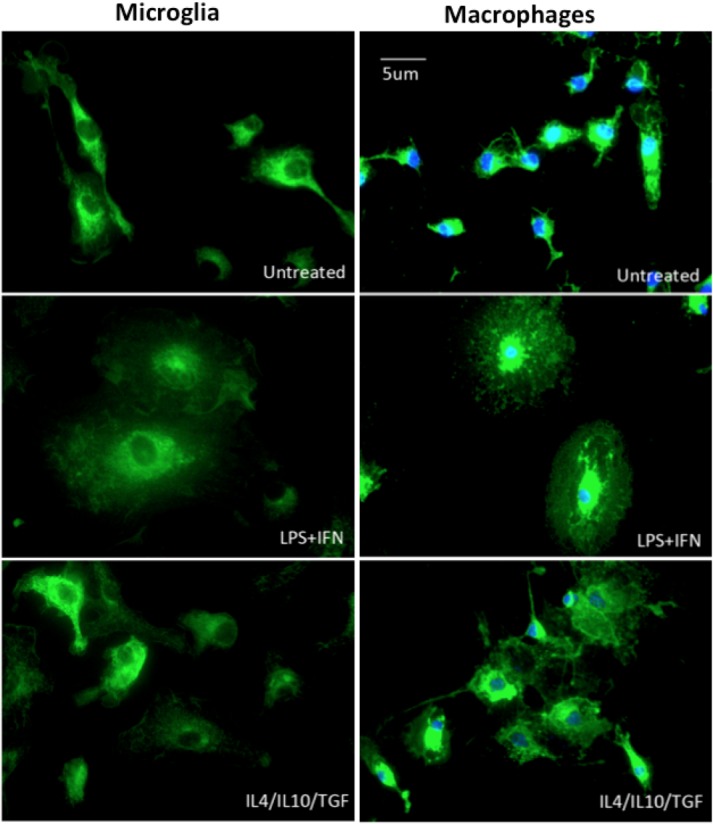
Morphology of M1 and M2 of microglia/macrophage phenotypes. Cultured microglia/macrophages from adult DBA/1 mice were stimulated for 24 h with IFNγ/LPS (M1 phenotype) or IL-4/IL-10/TGFβ (M2 phenotype). Microglia/macrophages were labeled with lectin (green) and nuclei stained with DAPI (blue). Scale bar: 5 µm.
Figure 2.

Microglial stimulation with IL-4/IL-10/TGF-β induces an M2 phenotype. Cultured microglia from adult DBA/1 mice were stimulated for 24 h with IFNγ/LPS or IL-4/IL-10/TGFβ, either alone or in combination. Expression of: CD86 (A); PD-L1 (B); PD-L2 (C); IL-6 (D); nitric oxide (NO) (E); and IL-10 (F). Secondary IFNγ/LPS proinflammatory stimulation following initial M2 stimulation: levels of IL-6 (G); NO (H); and IL-10 (I). *P < 0.05; **P < 0.01; ***P < 0.001. Data represent at least three independent experiments.
To determine if the induced M2 phenotype would convert to an M1 phenotype following secondary proinflammatory stimulation, microglia were first M2-induced for 24 h, supernatants were washed away, and fresh medium containing LPS/IFN-γ was added for another 24 h. Resultant proinflammatory IL-6 and NO levels were significantly lower, while anti-inflammatory cytokine IL-10 levels were higher from these secondary proinflammatory activated IL-4/IL-10/TGF-β-induced M2 cells, indicating that a relatively stable M2 macrophage phenotype had been induced by these cytokines (Fig. 2G–I). Moreover, TGF-β was observed to be crucial for phenotype retention (Fig. 2G–I).
Adoptive Transfer of M2 Microglia Attenuates the Clinical Symptoms of Established EAE
Similar to our previous findings with bone marrow-derived macrophages, our in vitro microglia stimulation studies indicated that a combination of IL-4/IL-10/TGF-β induced an optimal suppressive regulatory microglia phenotype (M2) that was dominantly anti-inflammatory. The in vivo immunoregulatory activity of these cells in a setting of EAE was next investigated through their adoptive transfer into EAE mice. M2 microglia were injected i.n into MOG-EAE DBA/1 mice at different time points (days 0, 5, 12, or 15 p.i). When we transferred unstimulated microglia to EAE mice we did not observe any treatment effects, with no difference in clinical disease development compared with the vehicle-treated group (data not included), and thus the latter control group was used subsequently. Strikingly, a single M2 microglia treatment alleviated the clinical symptoms of ongoing EAE at most time points except at day 0 p.i (Fig. 3A, day 0; Fig. 3B, day 5; Fig. 3C, day 12; and Fig. 3D, day 15). Taken together, these results indicate the potent ability of M2 microglia to attenuate the clinical symptoms of ongoing EAE, with the most benefit being recorded at later clinical time points (during chronic disease). When cytokine-induced immunomodulatory macrophages were injected intravenously at day 15 p.i into EAE mice, similar beneficial effects were observed (Fig. 4).
Figure 3.
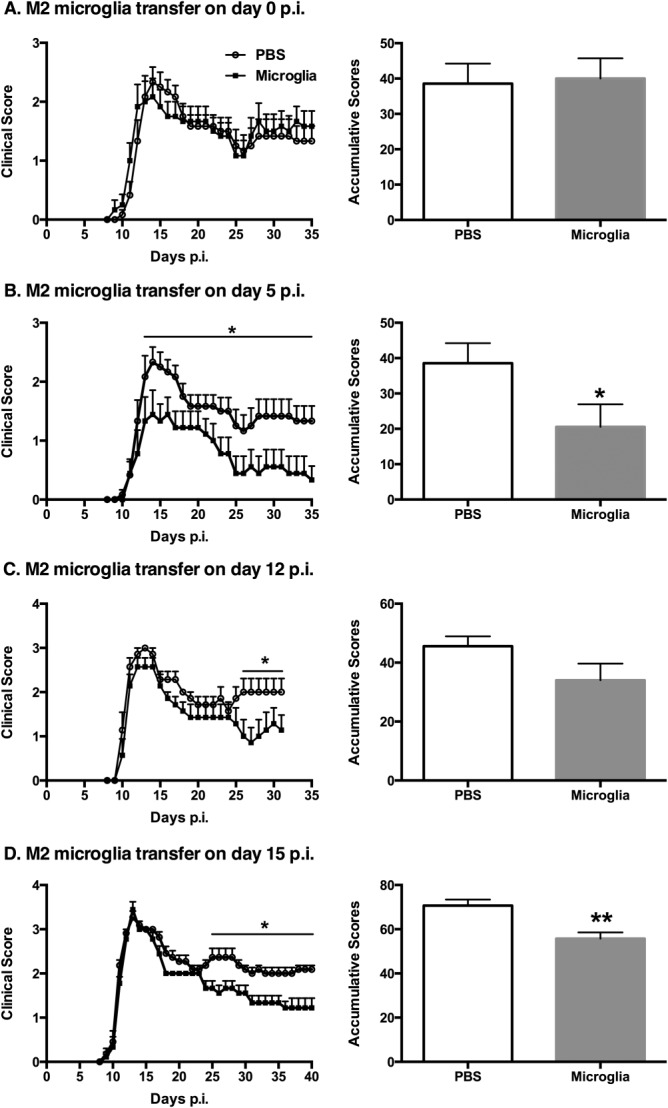
Adoptive transfer of M2 microglia attenuates the clinical symptoms of established EAE. M2 microglia were injected intranasally into MOG-EAE DBA/1 mice at different time points postimmunization (p.i): (A) day 0; (B) day 5; (C) day 12; and (D) day 15. Comparison of accumulative clinical scores between PBS-treated EAE mice and M2 microglia-treated EAE mice is presented for each time point. *P < 0.05; **P < 0.01. Data represent two independent experiments.
Figure 4.

Adoptive transfer of M2 macrophages at day 15 postimmunization (p.i) diminishes the severity of established EAE. M2 macrophages were injected intravenously into MOG-EAE DBA/1 mice at two time points p.i: (A) day 0 and (B) day 15. Comparison of accumulative clinical scores between PBS-treated EAE mice and M2 macrophage-treated EAE mice is presented for each time point. *P < 0.05. Data represent two independent experiments.
EAE Mice Treated with M2 Microglia Have Reduced Inflammatory Responses and Less Demyelination in the CNS
The typical pathological changes of EAE consist of CNS inflammatory cell infiltration, demyelination, and axonal loss during severe disease. To assess inflammation and demyelination, CNS tissues from EAE mice were divided into 10 segments as depicted in Fig. 5A and stained with hematoxylin–eosin, luxol fast blue, and antibodies specific for Iba1 and GFAP, with inflammatory cell infiltration being assessed blindly in a semiquantitative fashion. At day 30 p.i (15 days after adoptive microglia transfer) PBS-treated mice clearly demonstrated inflammatory infiltration, microglial and astrocyte activation in the brain stem, cerebellum, and the whole spinal cord. It was also obvious that demyelination occurred throughout the spinal cord in PBS-treated mice. Moreover, the infiltration and demyelination were especially strong in the lumbar spinal cord (Fig. 5C). Conversely, a lower degree of infiltration and demyelination was detected in mice treated with M2 microglia. The infiltration scores indicated that transfer of M2 microglia led to significantly reduced spinal cord destruction (Fig. 5B). Smaller infiltrates were detectable in the forebrain of approximately half of the animals, and although there was a trend toward less infiltration in the brain of M2 microglia-treated mice, this did not reach statistical significance.
Figure 5.
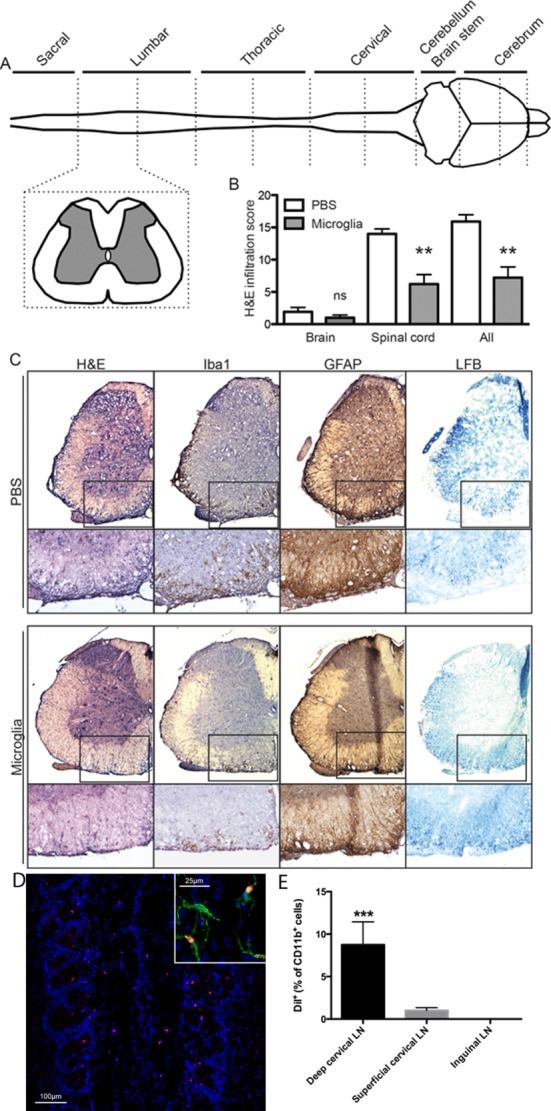
EAE mice treated with M2 microglia have reduced inflammatory responses and less demyelination in the CNS at day 30 postimmunization (day 15 after adoptive microglia transfer) as assessed by immunohistochemistry. (A) Schematic figure illustrated that CNS tissues from EAE mice were divided into 10 segments and stained with hematoxylin–eosin, luxol fast blue, and antibodies against Iba1 and GFAP, with inflammatory cell infiltration being assessed blindly in a semiquantitative fashion, from − (no infiltration) to +++ (severe infiltration). (B) The infiltration scores indicated that transfer of M2 microglia led to diminished spinal cord destruction. (C) Representative slices from lumbar spinal cord showed reduced degree of inflammation and demyelination in mice treated with M2 microglia. (D) Fluorescent DiI-labeled microglia (red) were detected in the olfactory bulb 24 and 72 h after delivery. DiI-labeled cells were designated as microglia by staining with Iba1 (green) and DAPI (blue). (E) DiI-positive cells were detected in the brain-draining deep cervical lymph nodes (LN) 72 h after delivery. **P < 0.01; ***P < 0.001. Data represent two independent experiments.
The fate of the transferred cells is a central issue and using fluorescent DiI labeling we were able to detect transferred microglia in the olfactory bulb 24 and 72 h after delivery (Fig. 5D). Moreover, after 72 h the microglia were also detected in the brain-draining deep cervical lymph nodes (Fig. 5E). At this time point we could not detect labeled cells in the rest of the brain or spinal cord.
Adoptive Transfer of M2 Microglia Suppresses T-Cell Activation and Th17 Production in the CNS of EAE Mice
Given the impressive therapeutic effect of M2 microglia therapy in EAE mice, CNS tissues (brain and spinal cord) from M2 microglia- and PBS-treated mice were more closely examined for immune activities by flow cytometry on day 30 p.i (day 15 after adoptive microglia transfer). Consistent with the histological findings, compared with PBS-treated mice M2 microglia-treated mice had reduced macrophage/microglia (CD11b+) and T-cell (CD3+) infiltration (Fig. 6A,B). Even though there was no difference in the percentages of CD4 and CD8 cells (Fig. 6C,D), the PBS-treated group had a higher number of activated CD4 T cells (CD62L−, CD44high) than the M2 microglia-treated group (Fig. 6F). Moreover, the PBS- and M2 microglia- treated mice had similar numbers of IFN-γ+ CD4 T cells in the CNS tissues (Fig. 6G). However, fewer IL-17+ CD4 T cells were evident in the CNS tissue following M2 microglia treatment (Fig. 6H).
Figure 6.

Adoptive transfer of M2 microglia suppresses T-cell activation and Th17 production in the CNS of EAE mice. CNS tissues from M2 microglia- and PBS-treated mice were examined for immune activities by flow cytometry on day 30 postimmunization (day 15 after adoptive microglia transfer). (A) Macrophages/microglia; (B) T-cell infiltration; percentage of CD4 (C) and CD8 cells (D); percentage of naïve CD4 T cells (E) and activated CD4 T cells (F); percentage of IFN-γ+ (G) and IL-17+ (H) CD4 T cells. *P < 0.05. Data represent two independent experiments.
The Immunomodulatory Properties of M2 Microglia Act on Both Innate and Adaptive Immune Cells
In an effort to try and further discern the observed mechanism of therapeutic action of M2 microglia we assessed their effects on different aspects of innate and adaptive immunity. The expansion of effector T cells is important in driving experimental autoimmune disease during both initiation and perpetuation (locally) of disease. We included M2-activated microglia in an in vitro T-cell proliferation assay and demonstrated a significant reduction in the T-cell proliferative capacity (Fig. 7A). We next assessed the capacity of M2 microglia to induce de novo Tregs by incubating them together with naïve T cells. The observed induction of a CD4+Foxp3+ phenotype indicated that M2 microglia were potent inducers of Tregs in the absence of antigen loading (Fig. 7B). Considering that proinflammatory M1 macrophages/microglia are the major cause of tissue damage during EAE we next addressed what effect co-incubation of M2 microglia would have on the phenotype of M1-activated macrophages. We assessed both expression of CD86 and PD-L1, both of which are highly expressed on M1 myeloid cells. We thus co-incubated M2 microglia with fluorescently labeled M1 macrophages in order to be able to subsequently immunophenotype the M1 cells. The data presented in Fig. 7C,D demonstrate that titration of M2 against M1 resulted in a reduced M1 phenotype as a direct result of the action of M2 microglia.
Figure 7.
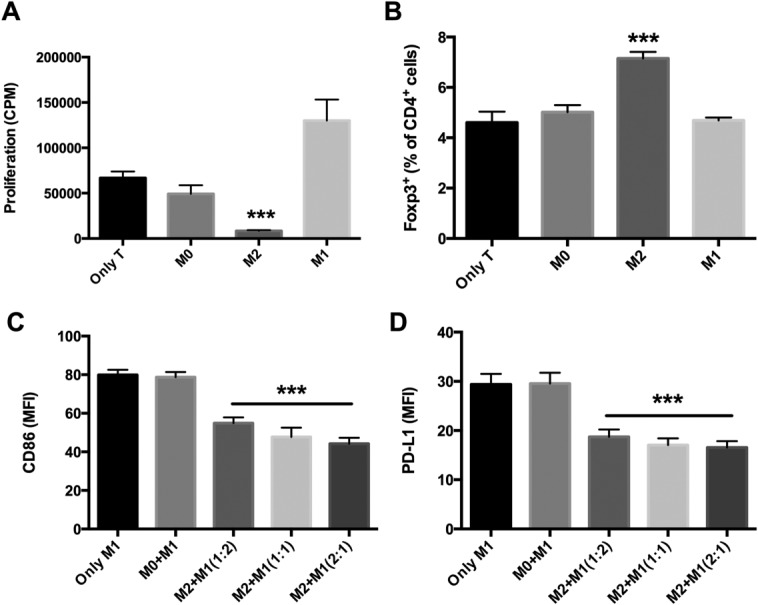
The immunomodulatory properties of M2 microglia were evaluated by co-culture with T lymphocytes or M1 macrophages. (A) M2-activated microglia demonstrated a significant reduction in in vitro T-cell proliferative capacity. (B) M2 microglia had the capacity to induce Tregs (CD4+Foxp3+ cells) de novo by incubating them together with naïve T cells. (C and D) Co-incubation of M2 microglia could modulate the phenotype of M1-activated macrophages by decreasing the expression of CD86 and PD-L1, respectively. ***P < 0.001. Data represent two independent experiments.
It is thus clear that M2 microglia have many potential immunomodulatory modes of action that could be expected to be significant in downregulating disease-promoting immune reactions occurring in the cervical lymph nodes or even within the afflicted CNS.
Discussion
We have previously demonstrated that adoptive transfer of specifically activated anti-inflammatory macrophages could prevent development of T1D in a mouse model (Parsa et al., 2012). In this study, we expanded this therapeutic concept through adoptive transfer of anti-inflammatory microglia into mice with ongoing EAE. The novel aspects of our study are (a) the use of adult microglia; (b) the use of a novel intranasal route of administration; and (c) the use of a late disease course time point in a severe EAE model. Our major finding is that a single adoptive transfer of in vitro-induced immunosuppressive M2 microglia can effectively alleviate the disease severity of established MOG-EAE in DBA1 mice.
MS is the most common neurological disease of young adults in Western societies, having a higher prevalence among females than males (2:1). In most patients the disease starts as RRMS and subsequently develops into SPMS, although 10% of patients debut with PPMS (Huijbregts et al., 2004; Quintana et al., 2008). The most commonly used therapies today, including injections of IFNβ, polypeptide glatiramer acetate, and the other approved disease-modifying drugs, are all restricted to use in RRMS, but fail to slow disease progression in both PPMS and SPMS (Anlar, 2009; Jones and Coles, 2010; Pozzilli and Prosperini, 2008). Additional treatments, in particularly targeting the significant SPMS group, are therefore a currently unmet medical need.
During recent years the use of specific immunosuppressive innate myeloid cells in therapy has been tested in a variety of experimental inflammatory settings including MS (Mikita et al., 2011), kidney inflammation (Ranganathan et al., 2013), and spinal cord injury (Hino et al., 2009). The first human trials using immunosuppressive macrophages have been conducted in a setting of spinal cord injury (Lammertse et al., 2012), and dendritic cell vaccination is common in cancer settings (Mantia-Smaldone and Chu, 2013). However, despite promising effects in animal models, translation into a human setting has been less efficacious in both settings. We advocate the use of stringently optimized stimulation protocols to ensure maximal retention of functional capacity post-transfer, and consider this as a flaw of the currently tested human therapies.
As microglia are primary immune effector cells of the CNS, serving in the surveillance and maintenance, protection, and restoration of the CNS homeostasis, we reasoned that they might have superior immunosuppressive capacity than M2 macrophages within the CNS. Although microglia are functionally equivalent to other populations of tissue-resident macrophages, by origin as well as functional characteristics (Hanisch, 2013; Saijo and Glass, 2011), it is currently definitively unproven as to what the relative contributions of microglia and systemic infiltrating macrophages are as effector cells in EAE and MS. Likewise, it is similarly unknown what the relative abilities of microglia and macrophages are as immunosuppressive cells (Ajami et al., 2011; Heppner et al., 2005; Ponomarev et al., 2005). In our in vitro immunological characterization studies we observed comparable activation states of M1 and M2 macrophages/microglia for all phenotypic markers studied. In vivo, the adoptive transfer of one dose of immunosuppressive M2 microglia alleviated the symptoms of EAE, accompanied by approved pathological changes, reduced T-cell infiltration and activation. Considering that the MOG-EAE DBA/1 model develops a particularly severe clinical disease, this therapeutic effect is significant.
There are several potential modes of action of the therapeutic M2 microglia. We demonstrate immunomodulatory effects on T-cell proliferation and on M1 activation phenotype, as well as specific induction of Tregs in vitro. Whether this latter response could be improved by loading of M2-activated cells with specific antigen before adoptive transfer remains to be determined, although there is no requirement per se for antigen preloading in induction of Tregs in vitro. However, there is a possibility for the uptake of antigen in the draining lymph nodes where we do find transferred M2 microglia, and it is in the nature of M2-stimulated macrophages and microglia to be highly phagocytic. The capacity of alternatively activated macrophages to reciprocally interact with Tregs has been previously reported (Mahnke et al., 2008; Tiemessen et al., 2007), and this capacity warrants more attention in the field of myeloid cell immunotherapy. We believe that the production of the anti-inflammatory cytokines IL-10 and TGF-β by the M2 microglia is critical for their ability to reduce the activities of proinflammatory T cells and macrophages, and the use of appropriate gene-deficient mice will aid to elucidate the relative contributions of these cytokines in the therapeutic effect in future studies. An additional potential mode of action of the transferred M2 microglia could be in stimulating the natural healing processes of the damaged tissue, and exploring this possibility is a current focus of our research efforts.
Critical consideration of efficiency of cell transfer is a vital aspect of myeloid cell therapy strategies. As most intravenously injected cells end up in the lung and direct intrathecal administration is associated with local tissue damage, intranasal cell transplantation into the CNS was used in this study. Intranasal administration can deliver peptides, chemical drugs, metals, viruses, plasmid, siRNA, and bacterial phages directly into the brain through the olfactory nerve pathway or vascular pathway (Danielyan et al., 2011; Jiang et al., 2011). We have previously used this strategy to deliver Tregs to the CNS (Fransson et al., 2012). In this study, we could detect labeled M2 cells in the deep cervical lymph nodes, indicating this as the main focus for immunomodulation, which is supported by the lower number of CD4+ T cells in the CNS of treated mice. However, we consider that determining the exact routes that adoptively transferred microglia navigate post-transfer requires further investigation using advanced imaging techniques such as IVIS, PET, and MRI scanning. Suffice to say that treatment effectiveness will be largely determined by how many immunomodulatory M2 cells access the appropriate compartments, and efficiency of route of administration will be a key issue for eventual translation into the clinic.
Considering the crucial role of infiltrating autoreactive T cells within the target organ in the development of EAE and MS, the deactivation of T cells could be one of the mechanisms explaining the therapeutic effects of adoptive microglial transfer to established EAE. Our histological and flow cytometric data supported this notion. Moreover, M2 microglia-transferred EAE mice exhibited fewer IL-17+ CD4 T cells but equivalent numbers of IFN-γ+ CD4 T cells in their CNS tissue. Additionally, we noted that M2 microglia-transferred EAE mice maintained better balance posture compared with PBS-treated EAE mice, a clinical symptom previously associated with Th17 cells (Stromnes et al., 2008). Early infiltration of Th17 or Th1/Th17 cells into the CNS is an important event in microglial activation during the early disease process (Murphy et al., 2010).
Although we and other groups are developing myeloid cell therapy platforms for translation into the clinic, the obvious question of the feasibility of a microglial therapy arises. A protocol for deriving microglia from mouse embryonic stem (ES) cells has been described (Napoli et al., 2009), and new protocols using iPS cells are currently being developed (Czepiel et al., 2011). The potential use of personalized microglia is thus a future clinical possibility. When cytokine-induced immunomodulatory macrophages were injected at day 15 p.i into the EAE mice, although somewhat milder, similar beneficial therapeutic effects were observed, suggesting a potential alternative option of using macrophages instead of microglia in clinical settings.
In conclusion, EAE could be alleviated by a single dose transfer of adult microglia in DBA/1 mice, representing a novel intervention of established EAE. That the best therapeutic effect was obvious at a more advanced stage of disease indicates that this approach might be of particular relevance as a treatment of SPMS patients.
Acknowledgments
Grant sponsors: Swedish Research Council, David and Astrid Hagelén Foundation, Tore Nilsons Foundation for Medical Research, and Karolinska Institutet.
References
- Abdul-Majid KB, Wefer J, Stadelmann C, Stefferl A, Lassmann H, Olsson T, Harris RA. Comparing the pathogenesis of experimental autoimmune encephalomyelitis in CD4-/- and CD8-/- DBA/1 mice defines qualitative roles of different T cell subsets. J Neuroimmunol. 2003;141:10–19. doi: 10.1016/s0165-5728(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- Almolda B, Gonzalez B, Castellano B. Antigen presentation in EAE: Role of microglia, macrophages and dendritic cells. Front Biosci. 2011;16:1157–1171. doi: 10.2741/3781. [DOI] [PubMed] [Google Scholar]
- Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, Matthieu JM, Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153:4349–4356. [PubMed] [Google Scholar]
- Andersson A, Kokkola R, Wefer J, Erlandsson-Harris H, Harris RA. Differential macrophage expression of IL-12 and IL-23 upon innate immune activation defines rat autoimmune susceptibility. J Leukoc Biol. 2004;76:1118–1124. doi: 10.1189/jlb.0704385. [DOI] [PubMed] [Google Scholar]
- Anlar O. Treatment of multiple sclerosis. CNS Neurol Disord Drug Targets. 2009;8:167–174. doi: 10.2174/187152709788680670. [DOI] [PubMed] [Google Scholar]
- Auletta JJ, Bartholomew AM, Maziarz RT, Deans RJ, Miller RH, Lazarus HM, Cohen JA. The potential of mesenchymal stromal cells as a novel cellular therapy for multiple sclerosis. Immunotherapy. 2012;4:529–547. doi: 10.2217/imt.12.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman J, Fransson M, Totterman TH, Fagius J, Mangsbo SM, Loskog AS. T cell responses after hematopoietic stem cell transplantation for aggressive relapsing-remitting multiple sclerosis. Immunology. 2013;140:211–219. doi: 10.1111/imm.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res. 2013;91:1143–1151. doi: 10.1002/jnr.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czepiel M, Balasubramaniyan V, Schaafsma W, Stancic M, Mikkers H, Huisman C, Boddeke E, Copray S. Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia. 2011;59:882–892. doi: 10.1002/glia.21159. [DOI] [PubMed] [Google Scholar]
- Danielyan L, Schafer R, von Ameln-Mayerhofer A, Bernhard F, Verleysdonk S, Buadze M, Lourhmati A, Klopfer T, Schaumann F, Schmid B, Koehle C, Proksch B, Weissert R, Reichardt HM, van den Brandt J, Buniatian GH, Schwab M, Gleiter CH, Frey IIWH. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res. 2011;14:3–16. doi: 10.1089/rej.2010.1130. [DOI] [PubMed] [Google Scholar]
- Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, Loskog AS. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9:112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK. Functional diversity of microglia—How heterogeneous are they to begin with? Front Cell Neurosci. 2013;7:65. doi: 10.3389/fncel.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Hino M, Ogata T, Morino T, Horiuchi H, Yamamoto H. Intrathecal transplantation of autologous macrophages genetically modified to secrete proenkephalin ameliorated hyperalgesia and allodynia following peripheral nerve injury in rats. Neurosci Res. 2009;64:56–62. doi: 10.1016/j.neures.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Huijbregts SC, Kalkers NF, de Sonneville LM, de Groot V, Reuling IE, Polman CH. Differences in cognitive impairment of relapsing remitting, secondary, and primary progressive MS. Neurology. 2004;63:335–339. doi: 10.1212/01.wnl.0000129828.03714.90. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Zhu J, Xu G, Liu X. Intranasal delivery of stem cells to the brain. Expert Opin Drug Deliv. 2011;8:623–632. doi: 10.1517/17425247.2011.566267. [DOI] [PubMed] [Google Scholar]
- Jones JL, Coles AJ. New treatment strategies in multiple sclerosis. Exp Neurol. 2010;225:34–39. doi: 10.1016/j.expneurol.2010.06.003. [DOI] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammertse DP, Jones LA, Charlifue SB, Kirshblum SC, Apple DF, Ragnarsson KT, Falci SP, Heary RF, Choudhri TF, Jenkins AL, Betz RR, Poonian D, Cuthbert JP, Jha A, Snyder DA, Knoller N. Autologous incubated macrophage therapy in acute, complete spinal cord injury: Results of the phase 2 randomized controlled multicenter trial. Spinal Cord. 2012;50:661–671. doi: 10.1038/sc.2012.39. [DOI] [PubMed] [Google Scholar]
- Mahnke K, Ring S, Bedke T, Karakhanova S, Enk AH. Interaction of regulatory T cells with antigen-presenting cells in health and disease. Chem Immunol Allergy. 2008;94:29–39. doi: 10.1159/000154854. [DOI] [PubMed] [Google Scholar]
- Mantia-Smaldone GM, Chu CS. A review of dendritic cell therapy for cancer: Progress and challenges. BioDrugs. 2013;27:453–468. doi: 10.1007/s40259-013-0030-9. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, Brochet B, Canron MH, Franconi JM, Boiziau C, Petry KG. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. 2011;17:2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaud S, Draheim HJ. A new method to isolate microglia from adult mice and culture them for an extended period of time. J Neurosci Methods. 2010;187:243–253. doi: 10.1016/j.jneumeth.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Kierdorf K, Neumann H. Microglial precursors derived from mouse embryonic stem cells. Glia. 2009;57:1660–1671. doi: 10.1002/glia.20878. [DOI] [PubMed] [Google Scholar]
- Owens T, Sriram S. The immunology of multiple sclerosis and its animal model, experimental allergic encephalomyelitis. Neurol Clin. 1995;13:51–73. [PubMed] [Google Scholar]
- Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, Holmberg D, Harris RA. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes. 2012;61:2881–2892. doi: 10.2337/db11-1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- Pozzilli C, Prosperini L. Clinical markers of therapeutic response to disease modifying drugs. Neurol Sci 29 (Suppl. 2008;2):S211–S213. doi: 10.1007/s10072-008-0939-9. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Farez MF, Viglietta V, Iglesias AH, Merbl Y, Izquierdo G, Lucas M, Basso AS, Khoury SJ, Lucchinetti CF, Cohen IR, Weiner HL. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci USA. 2008;105:18889–18894. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan PV, Jayakumar C, Ramesh G. Netrin-1-treated macrophages protect the kidney against ischemia-reperfusion injury and suppress inflammation by inducing M2 polarization. Am J Physiol Renal Physiol. 2013;304:F948–F957. doi: 10.1152/ajprenal.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Rasmussen S, Imitola J, Ayuso-Sacido A, Wang Y, Starossom SC, Kivisakk P, Zhu B, Meyer M, Bronson RT, Garcia-Verdugo JM, Khoury SJ. Reversible neural stem cell niche dysfunction in a model of multiple sclerosis. Ann Neurol. 2011;69:878–891. doi: 10.1002/ana.22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Scheffel J, Regen T, Van Rossum D, Seifert S, Ribes S, Nau R, Parsa R, Harris RA, Boddeke HW, Chuang HN, Pukrop T, Wessels JT, Jürgens T, Merkler D, Brück W, Schnaars M, Simons M, Kettenmann H, Hanisch UK. Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia. 2012;60:1930–1943. doi: 10.1002/glia.22409. [DOI] [PubMed] [Google Scholar]
- Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg M, Harris RA. Co-infection with Trypanosoma brucei brucei prevents experimental autoimmune encephalomyelitis in DBA/1 mice through induction of suppressor APCs. Int Immunol. 2005;17:721–728. doi: 10.1093/intimm/dxh253. [DOI] [PubMed] [Google Scholar]
- Wallberg M, Wefer J, Harris RA. Vaccination with myelin oligodendrocyte glycoprotein adsorbed to alum effectively protects DBA/1 mice from experimental autoimmune encephalomyelitis. Eur J Immunol. 2003;33:1539–1547. doi: 10.1002/eji.200323772. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc. 2008;2008:pdb prot5080. doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- Weisser SB, McLarren KW, Kuroda E, Sly LM. Generation and characterization of murine alternatively activated macrophages. Methods Mol Biol. 2013;946:225–239. doi: 10.1007/978-1-62703-128-8_14. [DOI] [PubMed] [Google Scholar]