

Abstract
A rapid and stability-indicating reversed phase high-performance liquid chromatography (RP-HPLC) method was developed for simultaneous quantification of paracetamol and ibuprofen in their combined dosage form especially to get some more advantages over other methods already developed for this combination. The method was validated according to United States Pharmacopeia (USP) guideline with respect to accuracy, precision, specificity, linearity, solution stability, robustness, sensitivity, and system suitability. Forced degradation study was validated according to International Conference on Harmonisation (ICH). For this, an isocratic condition of mobile phase comprising phosphate buffer (pH 6.8) and acetonitrile in a ratio of 65:35, v/v at a flow rate of 0.7 mL/minute over RP C18 (octadecylsilane (ODS), 150 × 4.6 mm, 5 μm, Phenomenex Inc.) column at ambient temperature was maintained. The method showed excellent linear response with correlation coefficient (R2) values of 0.999 and 1.0 for paracetamol and ibuprofen respectively, which were within the limit of correlation coefficient (R2 > 0.995). The percent recoveries for two drugs were found within the acceptance limit of (97.0–103.0%). Intra-and inter-day precision studies of the new method were less than the maximum allowable limit percentage of relative standard deviation (%RSD) ≤ 2.0. Forced degradation of the drug product was carried out as per the ICH guidelines with a view to establishing the stability-indicating property of this method and providing useful information about the degradation pathways, degradation products, and how the quality of a drug substance and drug product changes with time under the influence of various stressing conditions. The degradation of ibuprofen was within the limit (5–20%, according to the guideline of ICH), while paracetamol showed <20% degradation in oxidation and basic condition.
Keywords: RP-HPLC, stability-indicating, paracetamol, ibuprofen
Introduction
Ibuprofen (IBU) is a non-steroidal anti-inflammatory drug that belongs to arylpropionic acid derivatives1 and is chemically (RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid. Paracetamol or acetaminophen (PARA) is a widely used over-the-counter analgesic (pain reliever) and antipyretic (fever reducer), and it belongs to para-aminophenol derivatives1 and is chemically N-(4-hydroxyphenyl) acetamide as shown in Figure 1.
Figure 1.
Structure of (A) paracetamol and (B) ibuprofen.
Combination of ibuprofen and paracetamol (200/500 mg) tablets is frequently indicated for temporary relief of mild to moderate pain associated with migraine, headache, backache, period pain, dental pain, rheumatic and muscular pain, non-serious arthritis pain, cold and flu symptoms, sore throat, and fever.2 This product is especially suitable for pain that requires stronger analgesia than ibuprofen or paracetamol alone.3 Development of an analytical method for assessment of drugs in pharmaceutical dosage form is of utmost necessity to confirm the quality of tablets or capsules with respect to assay, content uniformity, and dissolution. There are compendial methods for assessment of these two drugs singly in United States Pharmacopeia (USP) and British Pharmacopeia (BP). A number of high-performance liquid chromatography (HPLC) methods have been already stated for determination of ibuprofen and paracetamol in their combination dosage form.4–8 But we found some drawbacks in those methods, which are listed below:
Almost all of those methods have used organic solvents more than 50%, which is not cost effective for routine analysis in pharmaceutical industries.
Mobile phase containing more than 50% organic phase may be detrimental to HPLC column as at that concentration, buffer salts may precipitate.
In some methods, shape and tailing do not seem to be satisfactory.
Stress degradation studies and their chromatograms were not shown properly.
Herein we put an effort to develop a cost-effective, rapid, and robust reversed phase (RP)-HPLC method with enough data of validation parameters. First, pKa of drugs was investigated. pKa of paracetamol and ibuprofen was 9.5 and 4.85, respectively. As a rule of thumb, pH of mobile phase is selected two units above or below the pKa value of drug. If we consider pKa of paracetamol, then we cannot choose the pH above 9.5, which is detrimental to silica beds of column. Therefore, the choice is two units below the pKa of paracetamol. Again, with respect to ibuprofen, we could choose the pH of mobile phase two units below of its pKa (4.8), but at that acidic pH, ibuprofen remains fully undissociated, which results in strong hydrophobic attraction with silica bed and, consequently, longer retention of this drug. Therefore, we thought a pH of around 7.0, which will be about two units far from the pKa of both drugs, and at this pH, both drugs will remain ionized, which makes the retention time much shorter at lower organic concentration. Thus, we tried with different buffers having a pH between 6.5 and 7.5 with different ratios of acetonitrile and methanol in isocratic condition. But we discarded methanol as it took longer time to elute ibuprofen. So our ultimate choice was acetonitrile, and at 35% concentration, the method gave well- resoluted and sharp peaks of paracetamol and ibuprofen. It was found that with increased pH, retention of ibuprofen was decreased. Thus, finally, a pH of 6.8 was chosen so as to get sufficient resolution between the peaks. The method is summarized in Table 1. Typical chromatogram is shown in Figure 2. This study was validated according to the guidelines of International Conference on Harmonisation (ICH) and USP.9,10
Table 1.
Newly developed RP-HPLC method.
| Mobile phase: Phosphate buffer (pH 6.8): Acetonitrile = 65:35, v/v |
| Column: C18 (150 × 4.6 mm, 5 μ, Phenomenex Inc.) |
| Flow rate: 0.7 mL/min |
| Wave length: 222 nm |
| Retention time: Paracetamol: 2.8 ± 0.1 min, Ibuprofen: 4.7 ± 0.1 min |
| Tailing factor: Paracetamol: 1.3, Ibuprofen: 1.1 |
Figure 2.
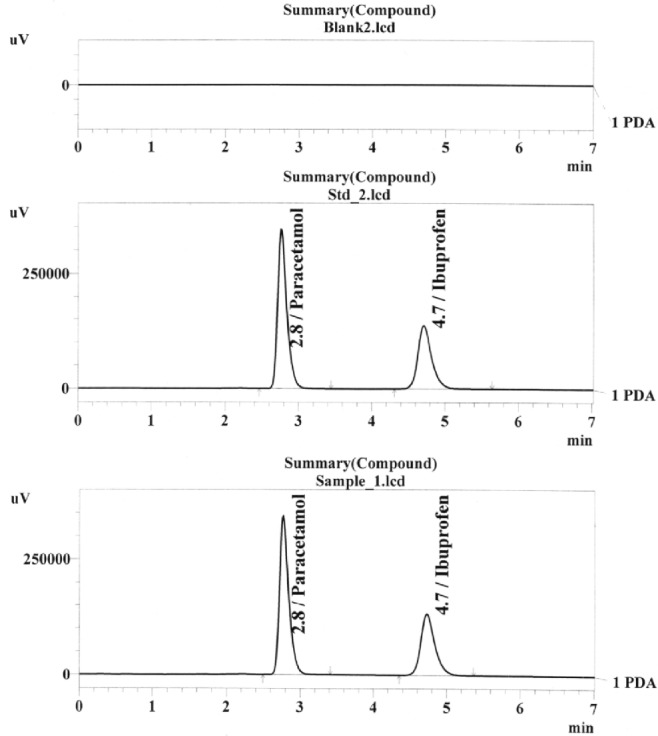
Chromatograms of blank, standard, and sample.
Materials and Methods
Materials
Working standards of paracetamol and ibuprofen were a kind gift of Healthcare Pharmaceuticals Limited, Rajendrapur, Bangladesh. HPLC grade acetonitrile was purchased from Active Fine Chemicals Ltd., Dhaka, Bangladesh.
HPLC system
High-performance liquid chromatographic system (Shimadzu-Prominence, Japan), equipped with an auto sampler (Model—SIL-20AC HT) and UV– visible detector (Model—SPD 20A), was used for the analysis. The data were recorded using LC-solution software. Analytical RP C18 column [octadecylsilane (ODS), 150 × 4.6 mm, 5 μ, Phenomenex Inc., Japan] was used to analyze the standard and samples.
Preparation of mobile phase
A total of 1.75 g of dipotassium hydrogen phosphate salt was dissolved in 900 mL of nanopure water. pH was adjusted to 6.8 ± 0.1 with diluted phosphoric acid or sodium hydroxide, and then volume was made up to 1000 mL with water of the same quality. Then this buffer and HPLC grade acetonitrile were mixed together at a ratio of 65:35, v/v. Finally, it was filtered through a 0.22-μm Millipore filter and sonicated to degas.
Preparation of standard solutions
In all, 50 mg of paracetamol and 20 mg of ibuprofen were taken in a 100-mL volumetric flask, and about 50 mL diluting solution (as mobile phase) was added and sonicated for five minutes to dissolve properly. Then volume was made up to the mark with the same diluent. This was stock solution. A total of 5 mL of this solution was taken into a 50-mL volumetric flask and diluted with diluents up to the mark. Thus, we got the test solution of paracetamol and ibuprofen of a concentration of 50 and 20 μg/mL, respectively. The stock solution was further diluted to get 50–200% of test concentration.
Chromatographic conditions
All analyses were done at ambient temperature under isocratic condition. The mobile phase was run at a flow rate of 0.7 mL/minute for seven minutes. The injection volume was 10 μL for standard and samples. Before analysis, every standard and sample was filtered through 0.2 μm filter tips. The column eluent was monitored with UV detection at 222 nm.
Method validation
The method was validated according to United States of Pharmacopeia (USP) guideline with respect to accuracy, precision, specificity, linearity, solution stability, robustness, sensitivity, and system suitability.10 Forced degradation was validated according to ICH.9,11,12
Specificity
The specificity of the LC method was evaluated to ensure that there was no interference from the degradation products, excipients, or other impurities in the region of actives. The specificity was studied by injecting the unstressed and stressed standard solutions, samples, and blanks.
Solution stability
Stability of drugs in diluting solvent and mobile phase was checked by rendering the test solutions in tightly capped vials at room temperature and in refrigerator at 5°C for three days. The solutions were analyzed by HPLC for three consecutive days.
Forced degradation
Forced degradation studies are undertaken to degrade the active drug deliberately. These studies are used to evaluate an analytical method’s ability to measure an active ingredient and its degradation products without interference. Samples or drug product (spiked placebo) and drug substance are exposed to acid, base, oxidizing agent, reducing agent, and water. The degraded samples were then analyzed using the method to determine if there are interferences with the active. Thus, stability-indicating property was evaluated.
Linearity and range
Linearity was checked on seven different concentrations within 50–200% of the nominal standard concentration. The linearity of the proposed method was evaluated by using calibration curve to calculate coefficient of correlation, slope, and intercept values.
Accuracy
The accuracy of an analytical method expresses the nearness between the expected value and the value found. In the present study, successive analysis (n = 3) for three different concentrations of standard mixture (80, 100, and 120% of nominal concentration) was carried out to determine the accuracy of the proposed method.
Precision
The precision of the assay was assessed with respect to repeatability and reproducibility. The precision of the proposed method was checked by intra- and inter-day repeatability of responses on different columns and different HPLC machines after replicate injections and expressed as %RSD among responses using the formula [%RSD = (standard deviation/mean) × 100%].
Robustness
Robustness is an indication of reliability of the analytical method during normal usage. The effect of the following deliberate changes in chromatographic conditions was monitored: flow rate ±50%, solvent ratio ±15%, pH of buffer solution ±0.2, temperature ±10°C, and detector wavelength ±3.
System suitability
The purpose of the system suitability test is to ensure that the complete testing system, including instruments, reagents, columns, analysts etc., is adequate for the intended analysis. The following parameters are usually determined: theoretical plate count, tailing factors, resolution, and reproducibility.
Limit of detection (LOD) and limit of quantitation (LOQ)
LOD is the lowest amount of analyte in a sample that can be detected but not necessarily quantitated under the stated experimental conditions. On the other hand, LOQ is the lowest amount of analyte in a sample that may be determined with acceptable accuracy and precision.
Results and Discussion
Specificity
The specificity was studied by injecting the unstressed and stressed standard solutions, blank, and pharmaceutical preparation of these drugs combination, several times on several days. It was revealed that there was no interference of peak in the region of paracetamol and ibuprofen in chromatogram for the stressed sample, placebo, and active. Specificity was further confirmed by peak purity index. Hence, the method was considered specific for the product. Representative chromatograms of blank, standard, and sample are shown in Figure 2, and peak purity index is shown in Figure 3.
Figure 3.
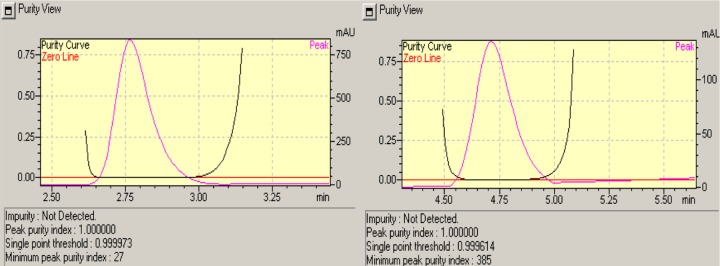
Peak purity of paracetamol and ibuprofen.
Solution stability
Two vials were prepared containing the standard mixture in diluting solution. One vial was kept in room temperature and another in refrigerator. Area change was investigated up to three consecutive days. %RSD of areas for all drugs were well between the limit (1.3%), which demonstrated that the drugs were fairly stable in the diluting solution and in the mobile phase. Results are shown in Table 2.
Table 2.
Results of solution stability.
| DAY | ROOM TEMPERATURE | REFRIGERATOR | ||
|---|---|---|---|---|
| PARA | IBU | PARA | IBU | |
| Day 1 | 1548146 | 782314 | 1548146 | 782314 |
| Day 2 | 1540987 | 70193 | 1542538 | 779734 |
| Day 3 | 1538988 | 765330 | 1541885 | 779556 |
| %RSD | 0.312 | 1.13 | 0.223 | 0.198 |
Forced degradation
Standard mixture was exposed to water, acid (1.0N HCl), base (1.0N NaOH), oxidizing agent (10% H2O2), and reducing agent (10% sodium bisulfate) for 24 hours. Results are summarized in Table 3. Degradation of drug substances between 5 and 20% has been accepted for validation of chromatographic assays.11,12 From the results, it was found that paracetamol was degraded significantly by oxidation. It also exhibited degradation in basic condition. One very notable finding is that ibuprofen remained completely stable in acid media. ibuprofen showed degradation in strong basic condition, but it was within the limit. In case of water hydrolysis, degradation was below the limit for both drugs. That is why we chose the pH of buffer in acidic region and close to neutral pH. Chromatograms are shown in Figure 4.
Table 3.
Summary of stress degradation study.
| DRUG | % OF DEGRADATION AGAINST FRESHLY PREPARED STANDARDa | ||||
|---|---|---|---|---|---|
| ACID | BASE | WATER | OXIDATION | REDUCTION | |
| PARA | 19.9 | 32.5 | 2.02 | 53.4 | 2.83 |
| IBU | 0 | 15.9 | 1.97 | 7.38 | 1.62 |
Notes:
Mean of three runs.
Figure 4.

Chromatograms of stressed condition, clockwise from top-left corner: acid, base, reduction, and oxidation. Peaks of paracetamol and ibuprofen were identified by their retention time, and rest of the peaks was of unknown degradants.
Linearity, accuracy, and precision
Linearity of the standard mixture was examined on 50–200% of nominal concentration. According to USP, the correlation coefficient (R2) for a calibration curve must be >0.995.10 The correlation coefficient was found to be more than 0.995 for both drugs indicating good linearity of calibration curve. The linearity curves are shown in Figure 5.
Figure 5.
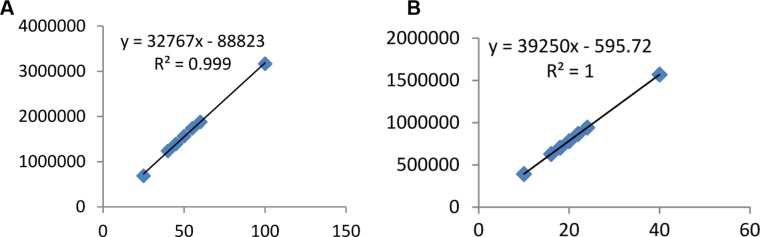
Linearity curve of (A) paracetamol and (B) ibuprofen.
For paracetamol, percent recovery is found to be 101.2, 100.5, and 99.9% for 40, 45, and 50 μg/mL, respectively, and for ibuprofen, percent recovery is found to be 101.1, 100.4, and 100.2% for 16, 18, and 20 μg/mL, respectively, shown in Table 4. All experimental results are in the range of the acceptability for accuracy (97.0–103.0%).10
Table 4.
Accuracy (% recovery) of paracetamol and ibuprofen.
| INJECTED CONC. OF PARACETAMOL (μg/mL) | MEAN RECOVERED CONC. OF PARACETAMOL (μg/mL) | % RECOVERY OF PARACETAMOL | INJECTED CONC. OF IBUPROFEN (μg/mL) | MEAN RECOVERED CONC. OF IBUPROFEN (μg/mL) | % RECOVERY OF IBUPROFEN |
|---|---|---|---|---|---|
| 40 | 40.48 | 101.2 | 16 | 16.17 | 101.1 |
| 45 | 45.23 | 100.5 | 18 | 18.07 | 100.4 |
| 50 | 49.95 | 99.9 | 20 | 20.04 | 100.2 |
The %RSD values found in precision study depicted in Table 5 showed that the proposed method provides acceptable intra- and inter-day variations for paracetamol and ibuprofen in their simultaneous determination. Results are summarized in Table 5 where %RSD was within the limit ≤2.10
Table 5.
Results of method validation parameters.
| PARAMETERS | LIMITS | PARACETAMOL | IBUPROFEN |
|---|---|---|---|
| Linear equation | y = 32767x–88823 | y = 32950x–595 | |
| Correlation coefficient (R2) | ≥0.995 | 0.999 | 1.00 |
| Linearity range | 25–100 μg/mL | 10–40 μg/mL | |
| Precision(intra-day)a: | %RSD < 2% | ||
| Day 1, Analyst 1, Column 1 | 0.956% | 0.821% | |
| Day 2, Analyst 2, Column 2 | 1.344% | 1.937% | |
| Day 3, Analyst 3, Column 3 | 1.987% | 1.124% | |
| Precision(inter-day): | %RSD < 3% | 2.11% | 2.034% |
| LOD | S/N = 3:1 | 2.3 μg/mL | 0.213 μg/mL |
| LOQ | S/N = 10:1 | 7.9 μg/mL | 0.711 μg/mL |
Note:
Mean of six runs.
System suitability
All system suitability parameters, including peak area, theoretical plate, tailing factor, retention time, and resolution, met the compendium acceptance limits.10 Results are summarized in Table 6.
Table 6.
System suitability parameters.
| PARAMETER | PARACETAMOL | IBUPROFEN | USP LIMIT |
|---|---|---|---|
| %RSD of tRa | 0.225 | 0.114 | ≤2% |
| %RSD of areaa | 0.223 | 0.198 | ≤2% |
| Tailing factora | 1.3 | 1.1 | ≤2.0 |
| Resolutionb | – | 6.5 | ≥2.0% |
| Theoretical plate | 2100 | 3743 | ≥2000 |
| Peak purity index | 1.00 | 1.00 | ≥0.98 |
Notes:
Mean of six runs.
Resolution with respect to former peak.
Robustness
Predetermined variations were performed under the experimental conditions to assess their robustness. We changed pH ±0.4, flow rate ±0.3, wave length ±10, and organic concentration ±3.5 units. Results are shown in Table 7.
Table 7.
Summary of robustness.
| PARAMETER CHANGES | ACTUAL VALUES | RETENTION TIME | TAILING FACTOR | PEAK PURITY | |||
|---|---|---|---|---|---|---|---|
| PARA | IBU | PARA | IBU | PARA | IBU | ||
| ph ±0.4 | 7.2 | 2.9 | 4.1 | 1.2 | 1.1 | 1.00 | 1.00 |
| 6.2 | 2.4 | 5.4 | 1.3 | 1.2 | 1.00 | 1.00 | |
| Flow rate ±0.3 | 1.0 | 1.6 | 3.1 | 1.1 | 1.2 | 0.99 | 0.99 |
| 0.4 | 3.8 | 6.3 | 1.5 | 1.3 | 1.00 | 1.00 | |
| Wave length ±10 | 212 | 2.8 | 4.7 | 1.3 | 1.1 | 1.00 | 1.00 |
| 232 | 2.7 | 4.7 | 1.3 | 1.1 | 1.00 | 1.00 | |
| Organic concentration ±3.5 | 38.5% | 2.1 | 4.2 | 1.2 | 1.1 | 0.99 | 0.99 |
| 31.5% | 3.0 | 12.4 | 1.3 | 1.2 | 1.00 | 0.99 | |
Conclusion
Hereby a new and robust method was developed for simultaneous quantification of paracetamol and ibuprofen in their solid dosage form. We validated it strictly maintaining the guidelines of ICH, USP, and Food and Drug Administration (FDA). The method was primarily designed for assay of paracetamol and ibuprofen in tablet or capsule. Moreover, the content of degradation of paracetamol and ibuprofen in various conditions, such as alkaline, acidic, oxidation, reduction and water hydrolysis, were observed and quantitatively analyzed by this HPLC method. The information, thus, obtained will facilitate pharmaceutical development in areas such as formulation development, manufacturing, and packaging, where knowledge of chemical behavior can be used to improve the quality of drug product.
Acknowledgments
The authors extend their appreciation to the Centre for Advanced Research in Sciences, University of Dhaka and Healthcare Pharmaceuticals Limited, Rajendrapur, Bangladesh.
Footnotes
Author Contributions
Conceived and designed the experiments: AR. Analyzed the data: RK. Wrote the first draft of the manuscript: MSJ. Contributed to the writing of the manuscript: MJI. Agree with manuscript results and conclusions: MSJ, MJI, RB, RK, AR. Jointly developed the structure and arguments for the paper: RB and MSJ. Made critical revisions and approved final version: AR. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: Gabor Patonay, Editor in Chief
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
REFERENCES
- 1.Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics. 10th ed. New York, USA: McGraw-Hill; 2001. [Google Scholar]
- 2.Ibuprofen and Paracetamol 200 mg/500 mg tablets–Summary of Product Characteristics. Reckitt Benckiser Healthcare (UK) Ltd; http://www.medicines.org.uk/emc/medicine/23871/SPC0000. [Google Scholar]
- 3.Hay AD, Redmond NM, Montgomery AA, Fletcher M, Hollinghurst S, Peters TJ. Paracetamol plus ibuprofen for the treatment of fever in children (PITCH): randomised controlled trial. BMJ. 2008;337:729. doi: 10.1136/bmj.a1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brune K, Hinz B. Paracetamol, ibuprofen, or a combination of both drugs against knee pain: an excellent new randomised clinical trial answers old questions and suggests new therapeutic recommendations. Ann Rheum Dis. 2011;70:1521–1522. doi: 10.1136/annrheumdis-2011-200242. [DOI] [PubMed] [Google Scholar]
- 5.Battu P, Reddy M. RP-HPLC method for simultaneous estimation of paracetamol and ibuprofen in tablets. Asian J Res Chem. 2009;2:70–72. [Google Scholar]
- 6.Lakka NS, Goswami N, Balakrishna P. Development and validation of a RP-HPLC for simultaneous determination of Ibuprofen and Paracetamol in solid dosage forms: application to dissolution studies. Int J Res Pharm Sci. 2011;2:331–337. [Google Scholar]
- 7.Gnana RM, Geetha G, Sangaranarayanan A. Simultaneous, stability indicating method development and validation for related compounds of ibuprofen and paracetamol tablets by RP-HPLC method. J Chromatogr Sep Tech. 2012;3:3–8. [Google Scholar]
- 8.Tsvetkova BG, Pencheva IP, Zlatkov AB, Peikov PT. Development and validation of RP-HPLC method for simultaneous determination of paracetamol and ibuprofen in fixed dose combinations. Int J Pharm Sci Rev Res. 2012;16:13–16. [Google Scholar]
- 9.Harmonised Tripartite Guideline: Validation of Analytical Procedures: Methodology (Q2B) 2005. International Conference on Harmonisation (ICH), 2005. [Google Scholar]
- 10.United States Pharmacopeia (USP) 34 (NF 29), Chapter <621>, Edition 2011.
- 11.Szepesi G. Selection of high-performance liquid chromatographic methods in pharmaceutical analysis. III. Method validation. J Chromatogr. 1989;464:265–278. doi: 10.1016/s0021-9673(00)94245-6. [DOI] [PubMed] [Google Scholar]
- 12.Carr GP, Wahlich JC. A practical approach to method validation in pharmaceutical analysis. J Pharm Biomed Anal. 1990;86:613–618. doi: 10.1016/0731-7085(90)80090-c. [DOI] [PubMed] [Google Scholar]