

Abstract
RATIONALE
Recent experiments utilizing photodissociation in linear ion traps have enabled significant development of Radical Directed Dissociation (RDD) for the examination of peptides and proteins. The increased mass accuracy and resolution of available in FTICR-MS should enable further progress in this area. Preliminary experiments of photoactivated radicals are reported herein.
METHODS
A 266 nm Nd:YAG laser is coupled to a FTICR or linear ion trap mass spectrometer. Radical peptides and proteins are generated by ultraviolet photodissociation (PD) and further activated by collisions or infrared photons.
RESULTS
A 266 nm UV laser and an IR laser can be simultaneously coupled to a 15 Tesla FTICR mass spectrometer. The ultra-low pressure environment in FTICR-MS makes collisional cooling less competitive, and thus more secondary fragments are generated by UVPD than in linear ion traps. Activation by SORI-CID or IRMPD also yields additional secondary fragmentation relative to CID in an ion trap. Accurate identification of RDD fragments is possible in FTICR-MS.
CONCLUSIONS
Relative to linear ion trap instruments, PD experiments in FTICR-MS are more difficult to execute due to poor ion cloud overlap and the low pressure environment. However, the results can be more easily interpreted due to the increased resolution and mass accuracy.
Keywords: photodissociation, 266 nm, FTICR, IRMPD, radical directed dissociation
INTRODUCTION
Bottom-up proteomics is widely used for protein identification and de novo protein sequencing.[1] Tandem mass spectrometry (MS/MS) can effectively verify the sequence of peptides with high confidence, speed and sensitivity, particularly with the aid of a high performance mass spectrometer. Collision-induced dissociation (CID) is the most widely used dissociation method for peptide/protein characterization due to its high efficiency and ease of implementation.[2] Electron-based dissociation methods, including electron-capture dissociation (ECD) and electron-transfer dissociation (ETD), have also proven to be effective in peptide sequencing and especially for the identification of post-translational modifications (PTMs).[3] The fragmentation types in these electron-based methods are complementary to the vibrational excitation dissociation pathways accessed by CID and thus can potentially provide other useful structural information not available from CID. In contrast to even electron dissociation pathways that dominate in CID, many of the fragments in ECD and ETD are generated by radical chemistries that initiate various homolytic bond cleavages.
In addition to electron-based methods, radicals can also be generated on polypeptides in a more controlled manner through modification with radical precursors. Various radical precursors and methods have been developed to generate hydrogen-deficient radical peptides in the gas phase, including oxidative dissociation of peptide-metal complexes,[4–6] and homolytic bond cleavage in modified groups such as nitrate ester,[7] peroxycarbamate,[8] nitroso,[9] and tempo.[10] All these methods use CID to facilitate activation of radical precursors containing labile bonds that generate hydrogen-deficient radicals upon dissociation. In addition, photodissociation (PD) can be used for radical initiation, which offers the advantage of selective activation of specific chromophores.[11–13] Ultraviolet photodissociation (UVPD) at 266 nm has been used to selectively cleave carbon–iodine (C–I) or carbon–sulfur bonds that are present in chromophore-modified peptides and proteins or native sulfur–sulfur bonds present in disulfide bridges. After generation of radical peptides, further dissociation (either spontaneously or aided by collisional activation) leads to radical directed dissociation (RDD), including backbone fragmentation and side chain loss. One typical feature in RDD is relatively abundant side-chain loss, which allows unambiguous identification of many specific amino acid residues and even distinction of isobaric residues such as leucine and isoleucine.[14]
Both ion trap (linear and quadrupole) and Fourier transform ion cyclotron resonance (FTICR) mass spectrometers have the capability to trap ions for a flexible amount of time, which makes them ideal for coupling with lasers for photodissociation experiments. A variety of lasers with different wavelengths have been implemented on quadrupole ion trap[15–22] and FTICR mass spectrometers[23–26]. Although both instruments are capable of trapping ions, there are also important and obvious differences between them. For example, FTICR instruments are capable of achieving much higher resolution and mass accuracy than ion traps. There are also significant differences in pressure, with FTICR cells operating at 6–7 orders of magnitude lower pressure than ion traps. The lower pressure of the FTICR cell is advantageous for infrared activation of ions because there is minimal cooling.[27] On the other hand, FTICR cells are much larger than ion traps, require more time per scan, and they are located in the center of a large cryogenic magnet, which makes optimization of laser alignment including maximizing laser/ion cloud overlap significantly more challenging than interfacing lasers to linear ion traps.
In the present work, 266 nm Nd:YAG lasers are coupled to both FTICR and linear ion trap (LIT) mass spectrometers. UVPD is performed on trapped ions in both instruments to cleave C–I bonds. The ions are then subjected to further activation by CID or infrared multiphoton dissociation (IRMPD) to investigate their subsequent radical chemistry. The differences in radical directed dissociation between FTICR and LIT instruments are examined.
EXPERIMENTAL
Materials
A synthetic peptide with amino acid sequence RGYALG (residues 21–26 of chicken lysozyme and a synthetic peptide substrate for a cyclic-AMP dependent protein kinase[28] was purchased from American Peptide Company (Sunnyvale, CA, USA). Sodium iodide, chloramine-T, sodium metabisulfite and other organic solvents were purchased from Thermo Fisher Scientific (Waltham, MA USA). Bovine ubiquitin was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Peptide derivatization
Peptides were labeled at the N-terminus with N-hydroxysuccinimide (NHS) activated iodo-benzoyl ester (2x molar ratio) in 1:3 borate buffer (0.2M, pH 8.5): dioxane solution for 30 mins at 40 °C. The side products at the arginine and tyrosine side chains were removed by incubation in 1M hydroxylamine (adjust pH to 8–9 by NaOH) for 4 hours.
Protein iodination
The tyrosine residue in ubiquitin (Y59) was iodinated with sodium iodide and chloramine-T at room temperature. After 10 minutes reaction time, excess sodium metabisulfite was added to quench the reaction. Stoichiometric quantities of reagents (1:2:1:2 molar ratio of peptide/sodium iodide/chloramine-T/sodium metabisulfite) were used to limit the iodination extent and produce mainly mono-iodinated tyrosine. A reversed phase cartridge (Michrom Bioresources, Inc., Auburn, CA, USA) was used for peptide and protein desalting and purification. The samples were washed with 2% acetonitrile with 0.1% trifluoroacetic acid and eluted with 80% acetonitrile with 0.1% trifluoroacetic acid.
Photodissociation MS in linear ion trap instruments
The polypeptides (10 μM) were dissolved in 50:50 H2O:CH3OH with 0.1% acetic acid and electrosprayed into an LTQ linear quadrupole ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). 266 nm photons were generated from the 4th harmonic of a Nd:YAG laser (Continuum, Santa Clara, CA, USA). The back plate of the instrument was modified with a quartz window to transmit UV photons into the linear ion trap. Laser pulses were synchronized to the isolation step via a digital delay generator.
Photodissociation MS in FTICR-MS
The experiments were performed using a 15-Tesla Bruker SolariX FTICR mass spectrometer with an infinity cell. Data were acquired with an approximate resolving power of 400,000 at m/z 400. The ESI capillary voltage was set to 4 kV. The temperature of dry nitrogen was set to 180 ºC and the flow rate was kept at 2.5 L/min. The RF amplitude of the ion-funnels was 200 Vpp, and the applied voltages were 100 V and 6 V for funnels 1 and 2, respectively. The voltage of skimmer 1 was 20 V and the skimmer 2 voltage was kept at 5 V. The RF frequencies used in all ion-transmission regions were multipole 1 (5 MHz), quadrupole (2 MHz), and transfer hexapole (2 MHz). Ions were accumulated for up to 20 s in the hexapole collision cell before being transmitted to the infinity ICR cell. The time-of-flight was set to 1.5 ms. The vacuum pressures for different regions were ~2 mbar for the source region, ~2×10−6 mbar for the quadrupole region, and ~2×10−9 mbar for the UHV-chamber pressure. IRMPD was performed with a Synrad 30 W CO2 laser (Mukilteo, WA, USA). Firing of the laser pulses (266 nm, Continuum Nd:YAG laser) was controlled by custom-programmed digital signals from the FTICR mass spectrometer. To align both UVPD and IRMPD lasers at the back of the infinity cell, a fused silica window (Esco Optics, Oak Ridge, NJ, USA), positioned as a beam splitter, reflected the 10.6 μm beam 90 degrees to the cell while transmitting the UV beam to the infinity cell. The experiment proceeded as follows: ions were filtered by m/z value in the quadrupole (Q1) and accumulated in the hexapole collision cell (h), then submitted to the infinity cell. One to ten UV pulses (10 Hz) homolytically cleaved carbon-iodine bonds. The radical species produced were isolated in the infinity cell (pulse time of 0.1 s and isolation power of 35%) and then subjected to IRMPD or sustained off-resonance irradiation (SORI)-CID. A gas pulse was programmed between the in-cell-isolation and IRMPD events; fragmentation of the radical species was then induced by IRMPD (50%, 0.5 s). For the SORI-CID experiments, the pulsed gas pressure was maintained at 8 mbar and the SORI-CID power was varied up to 5% with a pulse length of 0.2 s and a frequency offset of −500 Hz. Up to 50 scans were averaged for each spectrum, and all spectra were calibrated internally with the precursor ion mass or externally with cesium iodide.
RESULTS AND DISCUSSION
RDD of peptide RGYALG
Figure 1 shows the 266 nm single laser pulse UVPD mass spectra obtained from both the FTICR and the LIT systems for the iodobenzoyl-modified peptide, RGYALG. The peak labeled as “–Iodo” represents a hydrogen-deficient radical [RGYALG]• generated by homolytic C–I bond cleavage. Side chain losses (e.g., −43L, −56L, −106Y) and selective backbone fragmentation (i.e., a3) are secondary fragments that are spontaneously produced. Although the measured spectra from the two instruments show similar features, there are some differences in the fragmentation patterns that can be noted upon closer inspection. For example, the PD yield, which is quantified by the percentage of C–I bond cleavage given from formula (1), is greater in the LIT. The PD yield from the LIT (Fig. 1a) is 97%, which is close to the expected PD yield (~100%) for 4-iodobenzoyl chromophore under the optimal conditions. A marginally lower PD yield, 79%, is observed from the FTICR-MS experiments (Fig. 1b). Identical lasers were used in the two experiments, but we were unable to obtain greater yields using FTICR-MS.
Figure 1.
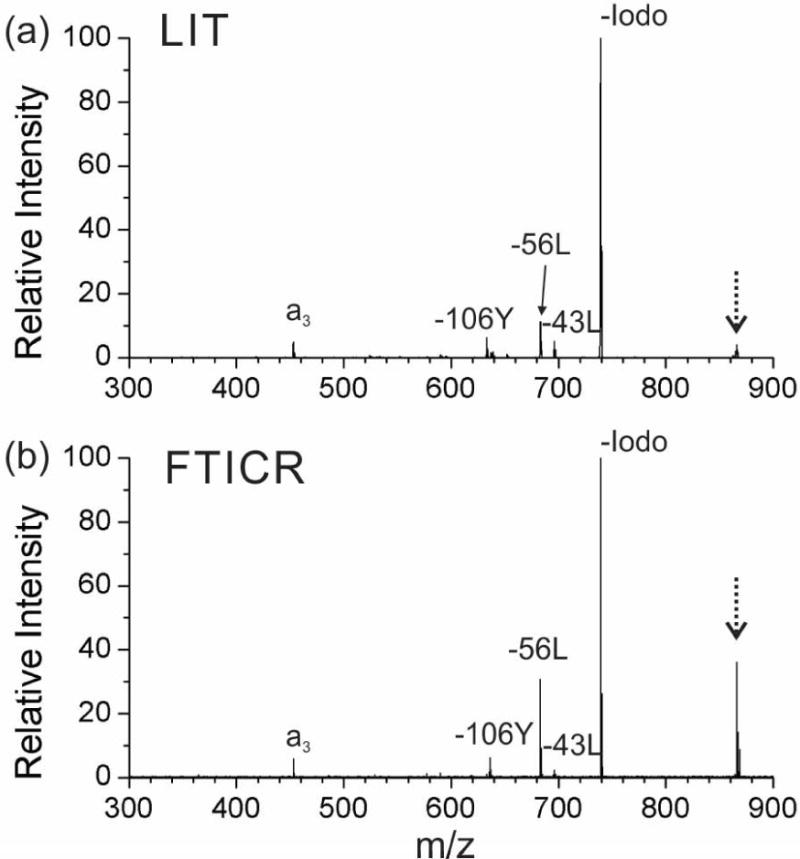
266 nm UV photodissociation (UVPD) spectrum of peptide N-4-iodo-benzoyl-RGYALG (m/z 739.4) in the 1+ charge state with the (a) linear ion trap (LIT) mass spectrometer and (b) FTICR mass spectrometer. (The peak for the precursor ion is noted with a dashed arrow.)
There are several factors that may cause the decreased efficiency in the FTICR cell, including incomplete overlap of the ion cloud and laser beam, or reduced photon flux due to beam divergence as the laser pulse must travel much further to reach the ions. In terms of laser beam-ion cloud overlap, there are several potential causes. The ion cloud may be larger than the laser beam. Alternatively, the ion cloud may not sample the area covered by the laser pulse during the brief time that the laser is firing (~8 ns), i.e., it may be a temporal and spatial overlap problem. Further experiments will explore these possibilities below.
| (1) |
A second difference observed from Fig. 1 is the yield of secondary fragments (e.g., −43L, −56L, −106Y and a3) from RDD as defined by formula (2). The RDD yield from the LIT and the FTICR mass spectrometer are 22% and 35%, respectively. More secondary fragmentation is observed in the FTICR mass spectrometer, which is consistent with the expected trend given the pressure differences between the two instruments. More efficient collisional cooling of the ions is expected in the LIT, which leads to less spontaneous RDD than in FTICR-MS.
| (2) |
The fragmentation characteristics of the radical peptides were investigated further by utilizing additional excitation following re-isolation of the radical product ion (the m/z 739 “-Iodo” peak). Collisional activation in a LIT is shown in Fig. 2a. Most of the observed product ions are the same as those secondary fragments that were measured in the PD experiment (compare with Fig. 1a), although some of the relative intensities differ. For example the loss of tyrosine side chain (−106Y) is much more abundant relative to the leucine side chain losses (−56L, −43L). A few minor secondary fragments are also observed in Fig. 2a, which result from consecutive radical initiated losses of small molecules (i.e., -43L-44CO2 and -56L-44CO2). For all consecutive losses, the radical remains on the peptide and even electron fragments are ejected. In the FTICR cell, SORI-CID was used for additional collisional excitation. Significantly more fragments are observed from SORI-CID-FTICR MS (Fig. 2b) than from CID-LIT MS (Fig. 2a), and most of these fragments correspond to multiple consecutive losses of either side chains from tyrosine/leucine or CO2 from the C‐terminus. The high mass accuracy of FTICR MS allows for confident assignment of these product ions (Table 1). It is clear from the results in Figs. 2a and 2b that SORI imparts significantly more energy into the molecule than collisional activation in the LIT. This is not surprising given that the pressure is much lower in the FTICR cell, necessitating that fewer collisions with higher energy occur to activate the ion. In addition, once the ions are activated, they are not collisionally cooled as occurs in the LIT, which will also favor the observation of sequential fragmentation product ions.
Figure 2.
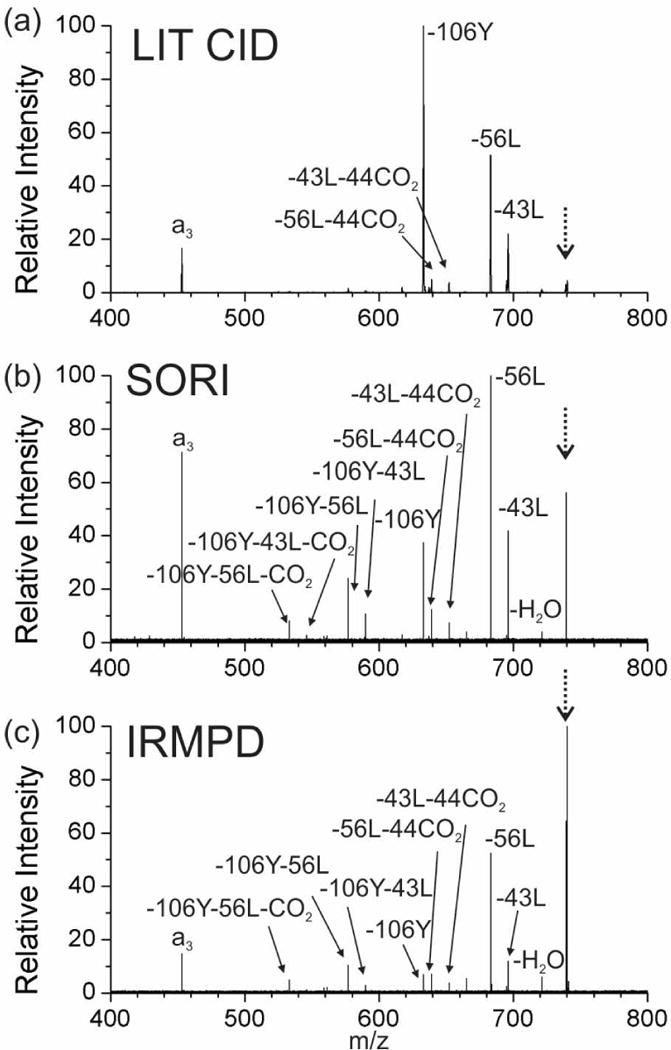
Tandem mass spectrum of radical peptide N-benzoyl-RGYALG in the +1 charge state by (a) CID in linear ion trap, (b) SORI in FTICR-MS and (c) IRMPD in FTICR-MS. (The peak for the precursor ion is noted with a dashed arrow.)
Table 1.
Mass deviations for fragments in Figure 2b.
| Peaks | Theoretical m/z |
Experimental m/z |
p.p.m. relative to theoretical m/z |
|---|---|---|---|
| −H2O | 721.3547 | 721.3552 | 1 |
| −43L | 696.3105 | 696.3113 | 1 |
| −44CO2 | 695.3755 | 695.3763 | 1 |
| −56L | 683.3027 | 683.3037 | 1 |
| −43L-44CO2 | 652.3207 | 652.3222 | 2 |
| −56L-CO2 | 639.3129 | 639.3146 | 3 |
| −106Y | 633.3234 | 633.3252 | 3 |
| −106Y-43L | 590.2687 | 590.271 | 4 |
| −106Y-44CO2 | 589.3336 | 589.3359 | 4 |
| −106Y-56L | 577.2608 | 577.2634 | 4 |
| −106Y-43L-44CO2 | 546.2788 | 546.2816 | 5 |
| −106Y-56L-44CO2 | 533.2710 | 533.2739 | 5 |
IRMPD with FTICR-MS was also tested to provide supplemental activation to the radical peptide generated (Fig. 2c). The relative intensity of the precursor ion remained relatively high, yet many of the fragments have undergone multiple consecutive dissociation events. This suggests that only part of the ion cloud is interacting significantly with the laser, enabling continuous activation of some ions while others experience little or no activation. Importantly, the intensity of the IR laser is significantly reduced in the two laser arrangement relative to a dedicated IRMPD experiment because the silica window that transmits the UV light is not coated for optimal IR reflectivity. The IRMPD and SORI-CID spectra are fairly similar, suggesting that excited product ions were generated by similar pathways in both experiments.
RDD of ubiquitin protein
RDD of ubiquitin was examined on the two instruments and the results for the 9+ charge state are shown in Fig. 3. Collisional activation of the radical protein in the LIT yields very efficient fragmentation, leaving little relative intensity of the precursor ion (Fig. 3a). Although many fragments are easily generated, assigning them is difficult given the low resolving power (~1000) of the LIT. Analogous SORI-CID and IRMPD experiments in the FTICR mass spectrometer are shown in Figs. 3b and 3c, respectively. In general, the fragmentation efficiency was lower in the FTICR mass spectrometer; however, the greatly increased resolution and mass accuracy allow for confident assignment of the product ions. One of the major reasons for the limited fragmentation efficiency for SORI-CID may be that the radical facilitates many low energy dissociation pathways. Numerous attempts to improve the fragmentation efficiency by varying the SORI-CID power, pulsed gas length, or off-set frequency to increase activation led to unacceptable diminution of the overall ion count. The prevalence of products resulting from sequential losses is also minimal for this protein, suggesting that the larger product ions generated from a protein are not sufficiently activated to undergo additional fragmentation.
Figure 3.
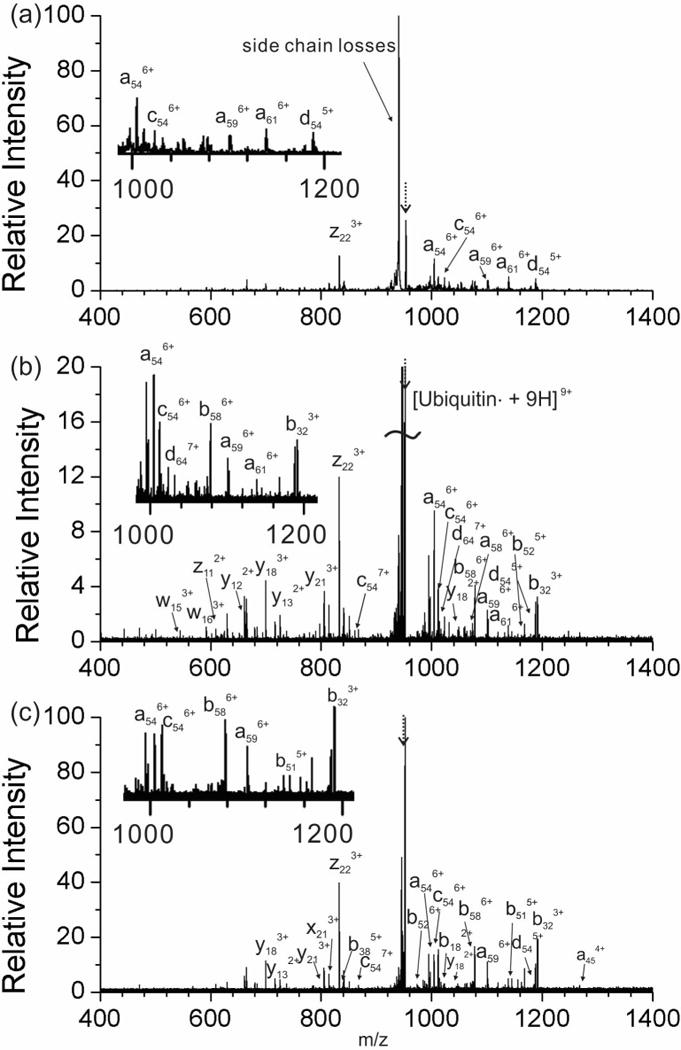
Tandem mass spectrum of radical ubiquitin in the 9+ charge state by (a) CID in linear ion trap, (b) SORI in FTICR-S and (c) IRMPD in FTICR-MS. The average mass measurement accuracy for the FTICR-MS experiments was 1.5 ppm. (The peak for the precursor ion is noted with a dashed arrow.)
Initial IRMPD experiments on radical ubiquitin yielded no product ions at all. Eventually, it was determined that in cell isolation of the radical protein altered the ion cloud sufficiently that there was no overlap with the IR laser. To summarize, quadrupole selected 9+ ubiquitin subjected to IRMPD immediately yielded abundant fragments, but the fragmentation efficiency decreased steeply if the same ions were additionally subjected to in-cell isolation. The degree of the loss of fragmentation efficiency correlated with the power of the in-cell isolation (Fig. S1, Supplementary Information). In order to ameliorate this issue and improve the overlap efficiency between the IR beam and the ion cloud, a short argon gas pulse (0.1–0.2 seconds) was applied after in-cell isolation to attempt to collisionally cool the ions back into the center of the cell. As can be seen in Fig. 3c, a reasonable amount of product ions can be generated by IRMPD following the gas pulse. Another reason for the relatively low fragment intensity in Fig. 3 is that, in the RDD of proteins, side chain fragmentations are very abundant (i.e., the big trailing peak after the radical protein precursor, as shown in Fig. 3) and thus may dilute other backbone fragmentation peaks.
Most fragments from 9+ ubiquitin are multiply charged peaks. Identification of these peaks is easily achievable for high resolution FTICR-MS but challenging in the low resolution LIT. The charge state of individual peaks in Fig. 3a can be assigned using a slow scan mode for a narrow m/z region. However, even this approach is limited to abundant fragments due to constraints imposed by low signal-to-noise. In comparison, significantly more fragments are assigned in high resolution FTICR-MS for both SORI-CID and IRMPD. All the radical initiated backbone fragments in Figs. 3a – 3c (e.g., z22, c54, d54, a59) are close in sequence to Y59, where the radical is initially created. This is consistent with limited radical migration along the protein. In addition, much less secondary fragmentation is observed in Figs. 3b and 3c than with the RDD of peptides in Figs. 2b and 2c. Interestingly, there is no significant difference in fragmentation types among the three methods. As can be seen in Fig. 3, many of the major fragments (e.g., z22, c54, d54, a59) show up in all the three spectra, suggesting that both SORI-CID and IRMPD are appropriate methods for the RDD of proteins.
CONCLUSIONS
The present results illustrate that a 266 nm UV laser can be successfully coupled to a 15-Tesla FTICR mass spectrometer for UVPD and RDD mass spectrometry experiments. The PD yield from a single pulse in an FTICR mass spectrometer is influenced by the alignment efficiency between the UV photons and the ion cloud in the FTICR cell. The ultra-low pressure environment in FTICR cell makes collisional cooling less competitive, and thus more secondary fragments from RDD29 are generated even in the absence of subsequent activation by SORI-CID or IRMPD. The major fragmentation pathways are the same among linear ion trap CID, SORI-CID and IRMPD. However, both peptide precursor ions and fragments are activated by SORI-CID and IRMPD. Therefore, consecutive dissociation is more prevalent in RDD spectra for FTICR-MS. Accurate identification of RDD fragments with FTICR-MS confirms previous experimental results obtained in a lower resolution linear ion trap. Compared with peptides, RDD of small proteins such as ubiquitin is difficult to perform by SORI-CID or IRMPD. For example, UVPD followed by IRMPD suffers from decreased alignment efficiency as a result of altered ion cloud shape, which is caused by in cell isolation. Increasing the pressure by a gas pulse after in-cell isolation can effectively accelerate collisional cooling and improve IRMPD efficiency.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge technical support from Jeremy Wolff (Bruker Daltonics) and funding from the National Institute of Health (R21GM103531 to RRJ and JAL, and R01GM103479 and S10RR028893 to JAL).
References
- 1.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 2.Mcluckey SA. Principles of Collisional Activation in Analytical Mass-Spectrometry. J Am Soc Mass Spectrom. 1992;3:599. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- 3.Ge Y, Rybakova IN, Xu QG, Moss RL. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc Natl Acad Sci USA. 2009;106:12658. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu IK, Rodriquez CF, Lau TC, Hopkinson AC, Siu KWM. Molecular radical cations of oligopeptides. J Phys Chem B. 2000;104:3393. [Google Scholar]
- 5.Barlow CK, McFadyen WD, O’Hair RAJ. Formation of cationic peptide radicals by gas-phase redox reactions with trivalent chromium, manganese, iron, and cobalt complexes. J Am Chem Soc. 2005;127:6109. doi: 10.1021/ja043088f. [DOI] [PubMed] [Google Scholar]
- 6.Laskin J, Yang ZB, Ng CMD, Chu IK. Fragmentation of alpha-Radical Cations of Arginine-Containing Peptides. J Am Soc Mass Spectrom. 2010;21:511. doi: 10.1016/j.jasms.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 7.Headlam HA, Mortimer A, Easton CJ, Davies MJ. beta-scission of C-3 (beta-carbon) alkoxyl radicals on peptides and proteins: A novel pathway which results in the formation of alpha-carbon radicals and the loss of amino acid side chains. Chem Res Toxicol. 2000;13:1087. doi: 10.1021/tx0001171. [DOI] [PubMed] [Google Scholar]
- 8.Masterson DS, Yin HY, Chacon A, Hachey DL, Norris JL, Porter NA. Lysine peroxycarbamates: Free radical-promoted peptide cleavage. J Am Chem Soc. 2004;126:720. doi: 10.1021/ja038615u. [DOI] [PubMed] [Google Scholar]
- 9.Hao G, Gross SS. Electrospray tandem mass spectrometry analysis of S- and N-nitrosopeptides: Facile loss of NO and radical-induced fragmentation. J Am Soc Mass Spectrom. 2006;17:1725. doi: 10.1016/j.jasms.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 10.Lee M, Kang M, Moon B, Oh HB. Gas-phase peptide sequencing by TEMPO-mediated radical generation. Analyst. 2009;134:1706. doi: 10.1039/b904115j. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Julian RR. Photoinitiated intramolecular diradical cross-linking of polyproline peptides in the gas phase. Phys Chem Chem Phys. 2012;14:16243. doi: 10.1039/c2cp42242e. [DOI] [PubMed] [Google Scholar]
- 12.Ly T, Julian RR. Elucidating the Tertiary Structure of Protein Ions in Vacuo with Site Specific Photoinitiated Radical Reactions. J Am Chem Soc. 2010;132:8602. doi: 10.1021/ja910665d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diedrich JK, Julian RR. Site-specific radical directed dissociation of peptides at phosphorylated residues. J Am Chem Soc. 2008;130:12212. doi: 10.1021/ja8023719. [DOI] [PubMed] [Google Scholar]
- 14.Wee S, O’Hair RAJ, McFadyen WD. Side-chain radical losses from radical cations allows distinction of leucine and isoleucine residues in the isomeric peptides Gly-XXX-Arg. Rapid Commun Mass Spectrom. 2002;16:884. doi: 10.1002/rcm.658. [DOI] [PubMed] [Google Scholar]
- 15.Crowe MC, Brodbelt JS. Infrared multiphoton dissociation (IRMPD) and collisionally activated dissociation of peptides in a quadrupole ion trap with selective IRMPD of phosphopeptides. J Am Soc Mass Spectrom. 2004;15:1581. doi: 10.1016/j.jasms.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 16.Remes PM, Glish GL. Mapping the Distribution of Ion Positions as a Function of Quadrupole Ion Trap Mass Spectrometer Operating Parameters to Optimize Infrared Multiphoton Dissociation. J Phys Chem A. 2009;113:3447. doi: 10.1021/jp808955w. [DOI] [PubMed] [Google Scholar]
- 17.Kim TY, Thompson MS, Reilly JP. Peptide photodissociation at 157 nm in a linear ion trap mass spectrometer. Rapid Commun Mass Spectrom. 2005;19:1657. doi: 10.1002/rcm.1969. [DOI] [PubMed] [Google Scholar]
- 18.Madsen JA, Boutz DR, Brodbelt JS. Ultrafast Ultraviolet Photodissociation at 193 nm and its Applicability to Proteomic Workflows. J Proteome Res. 2010;9:4205. doi: 10.1021/pr100515x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun QY, Julian RR, Sun QY, Julian RR. Probing sites of histidine phosphorylation with iodination and tandem mass spectrometry. Rapid Commun Mass Spectrom. 2011;25:2240. doi: 10.1002/rcm.5116. [DOI] [PubMed] [Google Scholar]
- 20.Wilson JJ, Brodbelt JS. MS/MS simplification by 355 nm ultraviolet photodissociation of chromophore-derivatized peptides in 4-3 quadrupole ion trap. Anal Chem. 2007;79:7883. doi: 10.1021/ac071241t. [DOI] [PubMed] [Google Scholar]
- 21.Kalcic CL, Gunaratne TC, Jonest AD, Dantus M, Reid GE. Femtosecond Laser-induced Ionization/Dissociation of Protonated Peptides. J Am Chem Soc. 2009;131:940. doi: 10.1021/ja8089119. [DOI] [PubMed] [Google Scholar]
- 22.Racaud A, Antoine R, Joly L, Mesplet N, Dugourd P, Lemoine J. Wavelength-Tunable Ultraviolet Photodissociation (UVPD) of Heparin-Derived Disaccharides in a Linear Ion Trap. J Am Soc Mass Spectrom. 2009;20:1645. doi: 10.1016/j.jasms.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 23.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Infrared Multiphoton Dissociation of Large Multiply-Charged Ions for Biomolecule Sequencing. Anal Chem. 1994;66:2809. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 24.Flora JW, Muddiman DC. Determination of the relative energies of activation for the dissociation of aromatic versus aliphatic phosphopeptides by ESI-FTICR-MS and IRMPD. J Am Soc Mass Spectrom. 2004;15:121. doi: 10.1016/j.jasms.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Cooper HJ, Case MA, McLendon GL, Marshall AG. Electrospray ionization Fourier transform ion cyclotron resonance mass spectrometric analysis of metal-ion selected dynamic protein libraries. J Am Chem Soc. 2003;125:5331. doi: 10.1021/ja021138f. [DOI] [PubMed] [Google Scholar]
- 26.Dodds ED, German JB, Lebrilla CB. Enabling MALDI-FTICR-MS/MS for high-performance proteomics through combination of infrared and collisional activation. Anal Chem. 2007;79:9547. doi: 10.1021/ac701763t. [DOI] [PubMed] [Google Scholar]
- 27.Hashimoto Y, Hasegawa H, Waki L. High sensitivity and broad dynamic range infrared multiphoton dissociation for a quadrupole ion trap. Rapid Commun Mass Spectrom. 2004;18:2255. doi: 10.1002/rcm.1619. [DOI] [PubMed] [Google Scholar]
- 28.Kemp BE, Benjamini E, Krebs EG. Synthetic Hexapeptide Substrates and Inhibitors of 3′-5′-Cyclic Amp-Dependent Protein-Kinase. Proc Natl Acad Sci USA. 1976;73:1038. doi: 10.1073/pnas.73.4.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leymarie N, Costello CE, O’Connor PB. Electron capture dissociation initiates a free radical reaction cascade. J Am Chem Soc. 2003;125:8949. doi: 10.1021/ja028831n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.