

Abstract
Sensitivity to FVIII inhibitors of the native plasma-derived (pd) FVIII/VWF complex vs. the complexes formed after exogenous FVIII infusion in the haemophilic patient has not been thoroughly studied. The role of VWF in the interaction of FVIII with inhibitors was studied in vitro using different combinations of VWF and FVIII concentrates. Normal plasma, pdFVIII/VWF and isolated FVIII (recombinant FVIII, B-domain deleted and pdFVIII) were used. Titre (BU) was kinetically determined (up to 2 h) in serial dilutions of inhibitor IgG (purified from a pool of plasmas with inhibitors) mixed with VWF and then incubated with the different FVIII. Inhibitor was also added to previously mixed VWF+FVIII. Residual FVIII:C was determined. TGA assays were performed with FVIII-deficient plasma spiked with the FVIII-VWF mixtures with/without an ESH-8 antibody. Inhibitor titres for plasma and pdFVIII/VWF were comparable at all time points. Titres for all concentrates of isolated FVIII were significantly higher than those for plasma or pdFVIII/VWF (1.4–1.9 fold) even after preincubation with VWF. At t = 0 h, titres for plasma or pdFVIII/VWF were unquantifiable, but were detectable for isolated FVIII (0.6–1.6 BU). In contrast to pdFVIII/VWF, the decrease in thrombin generation parameters by isolated FVIII in the presence of ESH-8 was significant (P < 0.01) even when previously combined with VWF. In conclusion, VWF protection against FVIII inhibitor activity might be higher with native pdFVIII/VWF complex than with the corresponding compound formed from the isolated proteins. Bethesda assay titration using different FVIII concentrates would be advisable to guide the treatment of inhibitor patients.
Keywords: Bethesda assay, factor VIII, FVIII inhibitors, FVIII/VWF complex, thrombin generation assay, von Willebrand factor
Introduction
The formation of inhibitors to factor VIII (FVIII) is currently the most common and challenging complication of haemophilia treatment 1. Both environmental as well as treatment-related risk factors are associated with inhibitor formation 2,3.
In the circulation, FVIII is tightly bound to von Willebrand factor (VWF), forming a complex that plays a significant role in FVIII protection from degradation by plasma proteinases 4,5 and interfering with antigen recognition 4. Some clinical studies have shown that VWF-containing FVIII (pdFVIII/VWF) concentrates result in lower immunogenicity (inhibitor formation) than isolated FVIII products in patients with haemophilia A 6–8. Data from clinical studies reporting the incidence of inhibitors in previously untreated patients have suggested that recombinant FVIII (rFVIII) has more than twice the immunogenicity of plasma-derived FVIII (pdFVIII; 27.0 vs. 10.8%) 9,10, while additional meta-analyses have shown that the risk of inhibitor development with rFVIII is approximately twice that of pdFVIII (27.4 vs. 14.3%) 9. However, this issue is far from settled 11.
pdFVIII/VWF concentrates have been shown to generate more thrombin in the presence of inhibitors than monoclonal-purified pdFVIII and rFVIII concentrates, which is critical for coagulation 12. Shi, et al. have shown, both in vitro and in mice, that VWF has a dose-dependent protective effect on FVIII and reduces inhibitor inactivation of FVIII 13. VWF is known to mask FVIII epitopes within the A2, A3 and C2 domains, which may reduce the formation of inhibitors by partially masking FVIII epitopes 14,15.
When infused into a haemophilic patient, isolated FVIII spontaneously binds to circulating VWF, with an apparent stoichiometric ratio of 1 IU FVIII:1 IU VWF 16. However, the precise in vivo molecular mechanisms of the FVIII-VWF interactions are not well known. The recognition of FVIII by inhibitors is also not well understood. When the Bethesda assay is used with different commercial FVIII concentrates, a wide range of inhibitor titres is obtained 17,18. Performing concentrate-based assays for direct evaluation of inhibitor reactivity has previously been proposed 18.
The recognition of FVIII by inhibitors and the potential differential characteristics of the native pdFVIII/VWF complex vs. the compound formed after exogenous FVIII infusion in the haemophilic patient warrant further investigation. For this study, we used a series of in vitro assays to test inhibitor reactivity in different combinations of VWF, FVIII concentrates (plasma-derived and recombinant) and inhibitors. Our results highlight the differential sensitivity to inhibitors of the native pdFVIII/VWF complex vs. the combination of purified, isolated FVIII and VWF proteins.
Material and Methods
Objectives and experimental design
The role of VWF in the interaction of FVIII with inhibitors was studied in vitro following two approaches:
In the first approach, the inhibitor reactivity (from a pool of haemophilic plasma with inhibitors) against FVIII from concentrates of different origins was investigated kinetically using the Bethesda assay, in comparison to normal human plasma. Two experimental models were tested: (i) FVIII added to previously mixed VWF+inhibitor (the haemophilia-mimic case), which theoretically models what occurs when FVIII is infused into a patient's blood already containing VWF and inhibitors; and (ii) inhibitor added to previously mixed VWF and FVIII (the factors-mixture case), in which the formation of a VWF+FVIII compound can occur prior to the interaction with the inhibitor.
In the second approach, the reactivity of inhibitors was analysed by the thrombin generation assay (using an antibody against FVIII C2 domain), comparing the native pdFVIII/VWF complex and the VWF+FVIII compound resulting from the combination of the isolated FVIII (of plasma or recombinant origin) and VWF proteins.
Biologicals
The native VWF-complexed FVIII concentrates of plasma origin (pdFVIII/VWF) used in the study were Fanhdi® (Grifols, Barcelona, Spain) and Alphanate® (Grifols, Los Angeles, CA, USA). Since both products share an identical purification process, for assessments they were considered the same concentrate type. Both products contain an approximate 1:1 ratio between FVIII:C and VWF:RCo activities. The pdFVIII was a monoclonally purified product containing no, or very little, VWF 19. The FVIII concentrates produced by a recombinant DNA technique (containing no VWF) were: a third generation full-length rFVIII, and a B-domain deleted rFVIII (BDD-rFVIII). The VWF was a commercially available plasma-derived VWF concentrate. The FVIII-deficient plasma (containing VWF) and normal pooled plasma were purchased from Diagnostic Grifols (Barcelona, Spain).
Inhibitor human IgG was purified from a commercial pool of haemophilic plasmas with inhibitors (Technoclone, Vienna, Austria) using protein G Sepharose chromatography (GE Healthcare, Uppsala, Sweden). Characterization of the pool performed in our laboratory showed the presence of antibodies against both light and heavy (A1–A2) chains. The Mab ESH-8, human anti-FVIII C2 domain antibody was obtained from American Diagnostica GbmH (Pfungstadt, Germany).
FVIII activity assays
The (modified) chromogenic method was performed with the Coamatic FVIII kit (Chromogenix, Bedford, USA). Briefly, 50 μL samples were added to 96 well microtitre plates in duplicates and warmed during 2 min at 37°C. Fifty (50) μL of assay components, including bovine factor IXa, factor X and thrombin co-lyophilized with CaCl2 and phospholipid were added to each well, and then the plates were incubated 10s with shaking and 167s without shaking at 37°C. Subsequently, 50 μL of the chromogenic FXa substrate mixture S-2765/I-2581 was added to each plate and the plate was incubated 10s with shaking at 37°C and 1 min without shaking at 37°C. Finally, 50 μL of acetic acid 20% was added to each well to stop the reaction and the plate was transferred to the microplate reader (Model ELX808, BioTek Instruments, Inc. Winooski VT, USA) preset at 37°C. The absorbance was read at 405 nm. A standard curve was plotted using a secondary standard calibrated against human coagulation FVIII concentrate Pharmacopea Europea BRP Batch 4.
For comparison to the chromogenic method results, residual FVIII:C of experimental samples at the time point t = 2 h of the inhibitor assay was determined by the one-stage clotting assay 20 with a coagulometre (Amelung, Lieme, Germany).
Inhibitor assay
In the haemophilia-mimic case, serial dilutions of inhibitor IgG were mixed with an equal volume of 2 IU mL−1 VWF and later incubated with an equal volume of 1 IU mL−1 of FVIII of different origin (pdFVIII/VWF, pdFVIII, rFVIII, BDD-rFVIII) and normal human plasma. In the factors-mixture case 2 IU mL−1 of each VWF and FVIII from different sources were mixed and then serial dilutions of inhibitor IgG were added in an equal volume.
In both cases samples were incubated at 37°C at different times (0, 15, 30, 60 and 120 min). The inhibitor titre of the experimental samples was determined following the Bethesda assay design. After incubation, residual FVIII:C was assayed by a chromogenic assay. Bethesda Units (BU) were defined by dilution of the inhibitory antibodies until 50% of the initial FVIII:C was neutralized. The inhibitor assay was repeated at least three times for each experimental condition. Buffer controls with no added inhibitors were assayed in parallel.
A series of controls with one of the VWF-devoid FVIII concentrates was performed to verify that inhibitor reactivity against the mixture of FVIII and VWF (factors-mixture case) was not affected by the preincubation time prior to addition of inhibitor IgG. Hence, the BDD-rFVIII+VWF mixture was incubated for 1 h at 37°C. Then, the titre in BU was obtained immediately (0 h) and after 2 h (representing the extreme points of the kinetic study). Results were compared with those of the FVIII+VWF mixture without the prior incubation during 1 h at 37°C.
Thrombin generation assay
FVIII-deficient plasma was spiked with FVIII concentrate from the different sources up to 1 IU FVIII mL−1. In parallel, FVIII-deficient plasma was spiked with each FVIII concentrate and mixed 1:1 with ESH-8 antibody to final concentrations of 1 IU FVIII mL−1 and 1 μg ESH-8 mL−1 (0.73 BU as determined in our laboratory) respectively. Additionally, 1:1 (IU) mixtures of isolated VWF and FVIII concentrates (rFVIII, BDD-rFVIII, pdFVIII) were prepared in vitro to be assayed in the same manner, in parallel to pdFVIII/VWF.
All samples were placed on a 96-well plate and assayed by TGA 21. Reagents for TGA including thrombin fluorogenic substrate, tissue factor/phospholipid solution RC low and thrombin calibrator were purchased from Technoclone (Vienna, Austria). Fluorescence was read for 60 min (microplate fluorescence reader FLX800 BIO-TEK Instruments, Inc.). The TGA was performed at least three times for each experimental condition and all data were standardized with the value of FVIII-deficient plasma. The parameters analysed include Endogenous Thrombin Potential (ETP), peak thrombin concentration, and velocity. Data were analysed using the evaluation software provided by the manufacturer.
Statistical analyses
Mean, median, standard deviation (SD), minimum–maximum and frequency in percentage were used where appropriate. The inhibitor titre data were analysed using Student's t test and thrombin generation data were analysed using an anova test. Brown–Forsythe's test for homogeneity of variances was performed to ensure that the results from Student's t test and the anova had been found appropriately. All P values were two-sided. Statistical significance was set at P < 0.05. GraphPad Prism® Statistics 5.0 (GraphPad Software Inc.; La Jolla, CA, USA) was used for analysis and charting.
Results
Kinetics of FVIII-inhibitor interactions
The inhibitor titre for the pdFVIII/VWF complex concentrates was comparable to the titre in normal plasma at all time points studied in both the haemophilia-mimic case (in which FVIII is added to previously mixed VWF+inhibitor) and the factors-mixture case (in which the inhibitor is added to previously mixed VWF+FVIII). At time t = 0 titres were unquantifiable while at 2 h, titres ranged from 11.6 ± 2.7 to 12.6 ± 1.8 BU [P < 0.001; Fig.1(a)].
Figure 1.
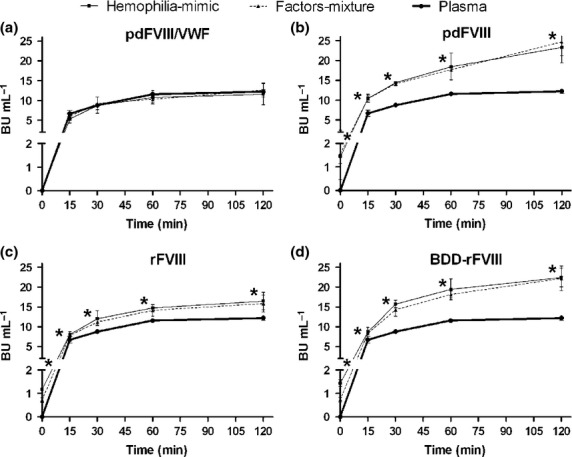
Kinetics of inhibitor reactivity, in Bethesda Units, against FVIII concentrates compared with normal plasma (pdFVIII/VWF: plasma-derived FVIII/VWF complex; pdFVIII: plasma-derived FVIII; rFVIII: full-length recombinant FVIII; BDD-rFVIII: B-domain deleted recombinant FVIII). In the haemophilia-mimic case FVIII containing products are added to previously mixed VWF+inhibitor, while in the factors-mixture case the inhibitor is added to previously mixed VWF+FVIII (n = 3 for plasma, n = 8–9 for pdFVIII/VWF; n = 3–5 for pdFVIII; n = 3 for rFVIII and n = 3 for BDD-rFVIII). *P < 0.05 to P < 0.001 with respect to plasma.
Conversely, the inhibitor titres obtained with all isolated FVIII products (pdFVIII, rFVIII and BDD-rFVIII) in the haemophilia-mimic case were significantly higher than in normal plasma and the pdFVIII/VWF complex concentrates at all the time points (P < 0.05–0.001), with ratios reaching 1.4–1.9 fold, respectively, after 2 h of incubation (titres ranging from 16.5 ± 2.1 to 23.3 ± 3.9 BU). This pattern did not change when the inhibitor was added to the FVIII+VWF mixture (factors-mixture case). Titres were even detectable at t = 0 (1.2–1.5 BU mL−1 in the haemophilia-mimic case and 0.6–1.6 BU in the factors-mixture case). Kinetic profiles of pdFVIII, rFVIII and BDD-rFVIII are plotted in Figs.1(b–d), respectively.
Figure2 shows detailed results of residual FVIII:C at t = 0 of the haemophilia-mimic case with three inhibitor dilutions. Only pdFVIII/VWF complex concentrates values remained similar to those of plasma at all the dilutions, whereas in the three VWF-devoid FVIII products (plasma-derived or recombinant) percentages of FVIII:C were always below those of plasma. At dilution ½, values were, respectively, 59–68% vs. 84% (P < 0.001).
Figure 2.
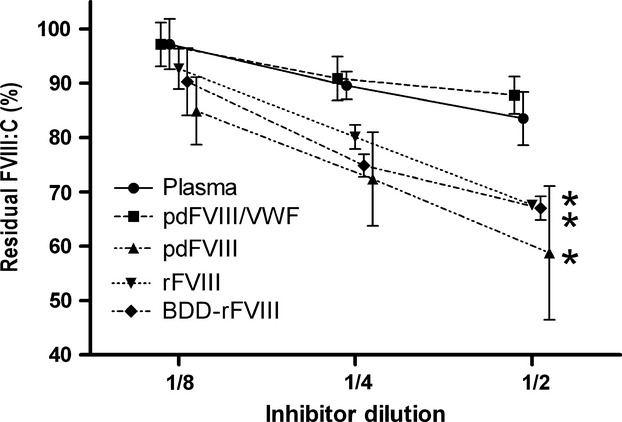
Residual FVIII activity (FVIII:C) of the FVIII concentrates with three inhibitor dilutions in the haemophilia-mimic case (pdFVIII/VWF: plasma-derived FVIII/VWF complex; pdFVIII: plasma-derived FVIII; rFVIII: full-length recombinant FVIII; BDD-rFVIII: B-domain deleted recombinant FVIII) (n = 3-6). *P < 0.001 with respect to plasma.
Controls in which the BDD-rFVIII+VWF mixture was preincubated for 1 h prior to inhibitor addition showed very similar titres as those found in the mixture without preincubation (0.66 ± 0.20 and 0.80 ± 0.20 BU mL−1, respectively, at t = 0 h, n = 3; 23.3 ± 5.2 and 24.4 ± 4.3 BU mL−1, respectively, at t = 2 h, n = 3).
There were no discrepancies between the chromogenic and the one-stage clotting assay results at the 2 h time point of the inhibitory assay in the haemophilia-mimic case. The respective ratios of BU in FVIII product vs. plasma were 0.95–1.04 for pdFVIII/VWF, 1.91–1.94 for pdFVIII, 1.35–1.30 for rFVIII and 1.84–1.73 for BDD-rFVIII.
Inhibitor reactivity in native pdFVIII/VWF vs. FVIII+VWF mixture
The pdFVIII/VWF complex concentrates consistently showed higher residual FVIII activity (i.e. less sensitivity to inhibition) in comparison to the mixture from isolated FVIII and VWF (1.5-fold, approximately), independent of the type of isolated FVIII assayed (pdFVIII, rFVIII and BDD-rFVIII). This observation was consistent over the time course of the assay. Furthermore, a 1 h preincubation of the isolated FVIII and VWF prior to addition of inhibitor had no effect on the results.
The decrease of ETP mediated by the anti-C2 antibody (ESH-8) in the presence of pdFVIII/VWF complex concentrates was very mild and not statistically significant (down to 88% of the ETP value of the product without inhibitor). The decrease of peak thrombin concentration and velocity index were moderate although significant (down to 78% and 63%, of the value of the product without inhibitor, respectively; P < 0.01). However, in the presence of isolated FVIII products the decreases were markedly higher and statistically significant in all cases (43–60% peak thrombin concentration and 32–46% velocity index; P < 0.01). Detailed results are summarized in Fig.3(a). The differences in inhibitor reactivity between pdFVIII/VWF complex concentrates and the VWF-devoid products persisted even when VWF was added to the FVIII products before measuring thrombin generation [Fig.3(b)].
Figure 3.
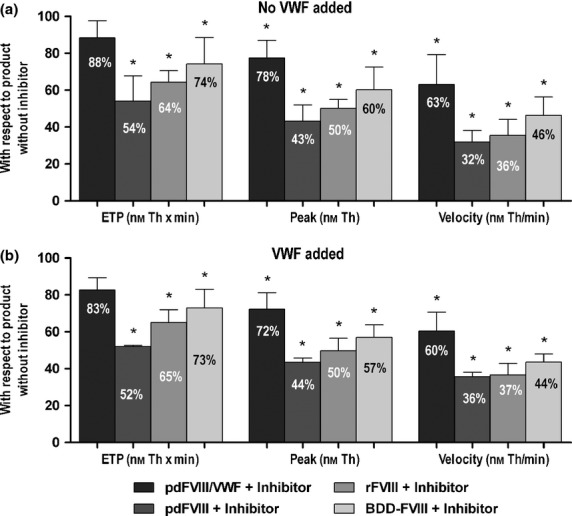
Thrombin Generation Assay (Endogenous Thrombin Potential [ETP], peak thrombin and velocity) performed on FVIII concentrate samples in the presence of antibody anti-C2 (ESH-8), with or without VWF added (pdFVIII/VWF: plasma-derived FVIII/VWF complex; pdFVIII: plasma-derived FVIII; rFVIII: full-length recombinant FVIII; BDD-rFVIII: B-domain deleted recombinant FVIII) (n = 4–6); *P < 0.01 with respect to the value of the product without inhibitor (100%).
Discussion
Detailed knowledge of the mechanisms of the FVIII and VWF interaction with regard to the role of VWF in interfering with FVIII antibody recognition is still incomplete 22. In the clinical setting this uncertainty is reflected in the controversy around which type of therapeutic concentrate should be used to minimize FVIII immunogenicity and maximize success in immune tolerance strategies 9,18,23,24. In this experimental study we used different combinations of VWF with isolated plasma-derived and recombinant FVIII concentrates to kinetically analyse inhibitor titres and thrombin generation vs. normal plasma and vs. pdFVIII/VWF complex purified from normal plasma. The reduced inhibitor reactivity to plasma and pdFVIII/VWF complex concentrates compared to concentrates of isolated FVIII, even after binding to VWF, is highlighted.
Kinetic results from the haemophilia-mimic case (i.e. addition of the FVIII concentrate to previously mixed VWF + inhibitor) showed that the BU titres of the inhibitor pool against pdFVIII/VWF were null at t = 0 and increased over time in a pattern almost identical to that of normal plasma. In contrast, for isolated FVIII products (pdFVIII, rFVIII and BDD-rFVIII) BU titres were immediately detectable at t = 0, and the BU values at the subsequent time points were significantly higher than those found in plasma (or also to pdFVIII/VWF). Whenever the results of this study are considered, it is important to denote that the differential inhibitor reactivity results observed between the FVIII concentrates were obtained against a pool of inhibitors from different patients instead of an inhibitor from a single patient. Since the Bethesda assay is normally performed in plasma, we believe testing of isolated FVIII concentrates should be performed, as previously suggested, if these concentrates are going to be used in a patient with inhibitors 12,17,18,25.
Interestingly, the kinetics of inhibitor reactivity was independent of FVIII and VWF incubation sequence. Inhibitor curves were similar for the haemophilia-mimic case and the factors-mixture case in which FVIII was allowed to bind to VWF prior to contact with the inhibitor. Results of the controls performing the Bethesda assay at t = 0 and 2 h, showed no differences with or without preincubation of BDD-rFVIII with VWF. In accordance with previous studies 13, this indicates that the FVIII-VWF binding is fast, not dependent on incubation time.
These results strongly indicate that the protective action of VWF on FVIII is more efficient when both molecules are bound constitutively as in plasma or in the pdFVIII/VWF complex concentrates, than in the compound resulting from the combination of isolated FVIII (plasma-derived or recombinant) and VWF proteins. Moreover, in the context of haemophilia treatment, these results would suggest that isolated FVIII infused into a haemophilic patient with inhibitors would be less protected by VWF than the FVIII found in pdFVIII/VWF complex concentrates. However, caution is warranted when extrapolating an in vitro approach to an in vivo situation.
Previous studies have suggested that post-translational sulphation of tyrosine residues in FVIII, which is critical for VWF binding 26,27, may be incomplete in recombinant FVIII 28,29. This could affect the ability to form a rFVIII+VWF complex that is completely identical to the native human pdFVIII/VWF. However, in our results, isolated pdFVIII plus VWF reacted to inhibitors in a manner similar to the rFVIII products. This strongly suggests that the native pdFVIII/VWF complex is not structurally or functionally identical to the combination of isolated FVIII and VWF proteins, independent of the possible differential characteristics of plasma and recombinant FVIII.
Current knowledge of VWF secretion and remodelling from vascular endothelial cells provides possible explanations. VWF is synthesized as ultra-large multimers (ULVWF) by endothelial cells and megakaryocytes. In endothelial cells, ULVWF is stored in the Weibel-Palade bodies, which can be secreted either constitutively or upon endothelial stimulation 30. Endothelial cells have also been reported to synthesize FVIII 31–33. During release from endothelial cells, ULVWF multimers in an unfolded conformation remain transiently bound to the cell surface to be cleaved by ADAMTS13 into smaller VWF multimers 34. After secretion into plasma, the VWF propeptide dissociates from VWF due to a change in pH 35 and functional circulating VWF adopts a globular conformation 36. Since constitutive binding of FVIII to the D'–D3 domain of VWF is enabled by the release of the VWF propeptide 37, it is plausible that this event takes place before VWF adopts the globular conformation. Given the much larger size of VWF with respect to FVIII (up to 20 000 vs. 330 kDa, respectively), there is a high probability that the constitutively bound FVIII is protected inside the globular VWF molecule. By contrast, exogenously infused FVIII would have more probabilities of binding to the surface of the circulating globular VWF molecule, hence being more exposed (with antigenicity implications) and also more accessible to inhibitors.
It is known that VWF-containing FVIII concentrates generate more thrombin than isolated FVIII concentrates when added to FVIII-deficient plasma with the presence of inhibitor 12. In this current report, TGA studies confirmed that FVIII in the pdFVIII/VWF complex concentrates is protected to a higher degree against an inhibitor (in this case anti-C2 antibody) compared to the other FVIII products. Moreover, no parameter (ETP, peak thrombin and velocity) was modified by the prior addition of VWF to isolated FVIII concentrates, which indicates that the FVIII+VWF mixture did not increase the protection of FVIII from anti-C2 antibody.
In summary, these data indicate a difference in sensitivity to FVIII inhibitors for different types of FVIII concentrates. Plasma and native pdFVIII/VWF complex showed higher residual FVIII activity after incubation with inhibitor compared to isolated FVIII concentrates, even after VWF incubation. This suggests that there may be structural and/or functional differences between the native pdFVIII/VWF complex and the FVIII+VWF compound formed from the isolated proteins. These differences are likely to be responsible for the higher protection conferred against inhibitory activity for the native pdFVIII/VWF complex. In addition, our results strongly support the recommendation to perform Bethesda Assay titration on different FVIII concentrates prior to treatment of inhibitor patients.
Acknowledgments
The authors wish to thank Jesica Castillo, Carlota Gelabert, Francisca Doncel and Meritxell Mira for their expert technical assistance. Writing support for the preparation of this manuscript was provided under the direction of the authors by Jordi Bozzo Ph.D. and Latoya Mitchell Ph.D. at Grifols. Michael K Woodward, June Davis Ph.D. and Eva Bastida Ph.D. at Grifols are acknowledged for their careful critical review of the manuscript.
Authors contribution
MIB and BDR were involved in study design, analysis and interpretation of data and manuscript preparation; JIJ and SG were involved in study design, the interpretation of data and revision of the manuscript. All the authors read and approved the final version of the paper.
Disclosures
The authors are employees of Grifols.
References
- Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9:418–35. doi: 10.1046/j.1365-2516.2003.00780.x. [DOI] [PubMed] [Google Scholar]
- Astermark J, Altisent C, Batorova A, et al. Non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16:747–66. doi: 10.1111/j.1365-2516.2010.02231.x. [DOI] [PubMed] [Google Scholar]
- Gouw SC, van der Bom JG, Marijke van den Berg H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109:4648–54. doi: 10.1182/blood-2006-11-056291. [DOI] [PubMed] [Google Scholar]
- Delignat S, Repesse Y, Navarrete AM, et al. Immunoprotective effect of von Willebrand factor towards therapeutic factor VIII in experimental haemophilia A. Haemophilia. 2012;18:248–54. doi: 10.1111/j.1365-2516.2011.02679.x. [DOI] [PubMed] [Google Scholar]
- Gringeri A, Ofosu FA, Grancha S, Oldenburg J, Ewing NP, Federici AB. Understanding FVIII/VWF complex–report from a symposium of XXIX WFH meeting 2010. Haemophilia. 2012;18:469–75. doi: 10.1111/j.1365-2516.2011.02655.x. [DOI] [PubMed] [Google Scholar]
- Dmoszynska A, Kuliczkowski K, Hellmann A, et al. Clinical assessment of Optivate(R), a high-purity concentrate of factor VIII with von Willebrand factor, in the management of patients with haemophilia A. Haemophilia. 2011;17:456–62. doi: 10.1111/j.1365-2516.2010.02446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klukowska A, Komrska V, Jansen M, Laguna P. Low incidence of factor VIII inhibitors in previously untreated patients during prophylaxis, on-demand treatment and surgical procedures, with Octanate(R): interim report from an ongoing prospective clinical study. Haemophilia. 2011;17:399–406. doi: 10.1111/j.1365-2516.2010.02428.x. [DOI] [PubMed] [Google Scholar]
- Klukowska A, Windyga J, Batorova A. Clinical efficacy of a novel VWF-containing FVIII concentrate, Wilate((R)), in the prophylaxis and treatment of bleeding episodes in previously treated haemophilia A patients. Thromb Res. 2011;127:247–53. doi: 10.1016/j.thromres.2010.11.030. [DOI] [PubMed] [Google Scholar]
- Iorio A, Halimeh S, Holzhauer S, et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: a systematic review. J Thromb Haemost. 2010;8:1256–65. doi: 10.1111/j.1538-7836.2010.03823.x. [DOI] [PubMed] [Google Scholar]
- Mannucci PM. The role of natural VWF/FVIII complex concentrates in contemporary haemophilia care: a guideline for the next decade. Haemophilia. 2012;18(Suppl 2):2–7. doi: 10.1111/j.1365-2516.2012.02794.x. [DOI] [PubMed] [Google Scholar]
- Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368:231–9. doi: 10.1056/NEJMoa1208024. [DOI] [PubMed] [Google Scholar]
- Salvagno GL, Astermark J, Ekman M, et al. Impact of different inhibitor reactivities with commercial factor VIII concentrates on thrombin generation. Haemophilia. 2007;13:51–6. doi: 10.1111/j.1365-2516.2006.01400.x. [DOI] [PubMed] [Google Scholar]
- Shi Q, Kuether EL, Schroeder JA, et al. Factor VIII inhibitors: von willebrand factor makes a difference in vitro and in vivo. J Thromb Haemost. 2012;10:2328–37. doi: 10.1111/j.1538-7836.2012.04902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragni MV. VWF: factor VIII protector and friend. J Thromb Haemost. 2012;10:2324–7. doi: 10.1111/j.1538-7836.2012.04922.x. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Arai M, Amano K, Kagawa K, Fukutake K. Factor VIII inhibitor antibodies with C2 domain specificity are less inhibitory to factor VIII complexed with von Willebrand factor. Thromb Haemost. 1996;76:749–54. [PubMed] [Google Scholar]
- Vlot AJ, Koppelman SJ, van den Berg MH, Bouma BN, Sixma JJ. The affinity and stoichiometry of binding of human factor VIII to von Willebrand factor. Blood. 1995;85:3150–7. [PubMed] [Google Scholar]
- Astermark J, Voorberg J, Lenk H, et al. Impact of inhibitor epitope profile on the neutralizing effect against plasma-derived and recombinant factor VIII concentrates in vitro. Haemophilia. 2003;9:567–72. doi: 10.1046/j.1365-2516.2003.00802.x. [DOI] [PubMed] [Google Scholar]
- Berntorp E, Ekman M, Gunnarsson M, Nilsson IM. Variation in factor VIII inhibitor reactivity with different commercial factor VIII preparations. Haemophilia. 1996;2:95–9. doi: 10.1111/j.1365-2516.1996.tb00022.x. [DOI] [PubMed] [Google Scholar]
- Amano K, Arai M, Koshihara K, et al. Autoantibody to factor VIII that has less reactivity to factor VIII/von Willebrand factor complex. Am J Hematol. 1995;49:310–7. doi: 10.1002/ajh.2830490409. [DOI] [PubMed] [Google Scholar]
- Chanarin I. Investigation of a prolonged activated partial thromboplastin time. In: Chanarin I, editor. Laboratory Haematology: An Account of Laboratory Technique. London: Churchill Livingstone; 1989. pp. 293–306. [Google Scholar]
- Varadi K, Turecek PL, Schwarz HP. Thrombin generation assay and other universal tests for monitoring haemophilia therapy. Haemophilia. 2004;10(Suppl 2):17–21. doi: 10.1111/j.1365-2516.2004.00936.x. [DOI] [PubMed] [Google Scholar]
- Terraube V, O'Donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010;16:3–13. doi: 10.1111/j.1365-2516.2009.02005.x. [DOI] [PubMed] [Google Scholar]
- Kallas A, Talpsep T. von Willebrand factor in factor VIII concentrates protects against neutralization by factor VIII antibodies of haemophilia A patients. Haemophilia. 2001;7:375–80. doi: 10.1046/j.1365-2516.2001.00530.x. [DOI] [PubMed] [Google Scholar]
- Kruse-Jarres R. Current controversies in the formation and treatment of alloantibodies to factor VIII in congenital hemophilia A. Hematology Am Soc Hematol Educ Program. 2011;2011:407–12. doi: 10.1182/asheducation-2011.1.407. [DOI] [PubMed] [Google Scholar]
- Sukhu K, Keeling DM, Giangrande PL. Variation in inhibitor reactivity in acquired haemophilia A with different concentrates. Clin Lab Haematol. 2000;22:287–90. doi: 10.1046/j.1365-2257.2000.00328.x. [DOI] [PubMed] [Google Scholar]
- Leyte A, van Schijndel HB, Niehrs C, et al. Sulfation of Tyr1680 of human blood coagulation factor VIII is essential for the interaction of factor VIII with von Willebrand factor. J Biol Chem. 1991;266:740–6. [PubMed] [Google Scholar]
- Michnick DA, Pittman DD, Wise RJ, Kaufman RJ. Identification of individual tyrosine sulfation sites within factor VIII required for optimal activity and efficient thrombin cleavage. J Biol Chem. 1994;269:20095–102. [PubMed] [Google Scholar]
- Grancha S, Navajas R, Maranon C, Paradela A, Albar JP, Jorquera JI. Incomplete tyrosine 1680 sulphation in recombinant FVIII concentrates. Haemophilia. 2011;17:709–10. doi: 10.1111/j.1365-2516.2010.02454.x. [DOI] [PubMed] [Google Scholar]
- Mikkelsen J, Thomsen J, Ezban M. Heterogeneity in the tyrosine sulfation of Chinese hamster ovary cell produced recombinant FVIII. Biochemistry. 1991;30:1533–7. doi: 10.1021/bi00220a013. [DOI] [PubMed] [Google Scholar]
- Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117:5033–43. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shovlin CL, Angus G, Manning RA, et al. Endothelial cell processing and alternatively spliced transcripts of factor VIII: potential implications for coagulation cascades and pulmonary hypertension. PLoS ONE. 2010;5:e9154. doi: 10.1371/journal.pone.0009154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the predominant and possibly exclusive source of plasma factor VIII. Blood. 2014;123:3706–13. doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697–705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–9. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- Huang RH, Wang Y, Roth R, et al. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc Natl Acad Sci U S A. 2008;105:482–7. doi: 10.1073/pnas.0710079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourik MJ, Valentijn JA, Voorberg J, Koster AJ, Valentijn KM, Eikenboom J. von Willebrand factor remodeling during exocytosis from vascular endothelial cells. J Thromb Haemost. 2013;11:2009–19. doi: 10.1111/jth.12401. [DOI] [PubMed] [Google Scholar]
- Bendetowicz AV, Morris JA, Wise RJ, Gilbert GE, Kaufman RJ. Binding of factor VIII to von willebrand factor is enabled by cleavage of the von Willebrand factor propeptide and enhanced by formation of disulfide-linked multimers. Blood. 1998;92:529–38. [PubMed] [Google Scholar]