

Abstract
Traumatic brain injury (TBI) induces severe harm and disability in many accident victims and combat-related activities. The heat shock proteins Hsp70/Hsp110 protect cells against death and ischemic damage. In this study, we used mice deficient in Hsp110 or Hsp70 to examine their potential requirement following TBI. Data indicate that loss of Hsp110 or Hsp70 increases brain injury and death of neurons. One of the mechanisms underlying the increased cell death observed in the absence of Hsp110 and Hsp70 following TBI is the increased expression of ROS-induced p53 target genes Pig1, Pig8 and Pig12. To examine whether drugs that increase the levels of Hsp70/Hsp110 can protect cells against TBI, we subjected mice to TBI and administered Celastrol or BGP-15. In contrast to Hsp110 or Hsp70i-deficient mice that were not protected following TBI and Celastrol treatment, there was a significant improvement of wild-type mice following administration of these drugs during the first week following TBI. In addition, assessment of neurological injury shows significant improvement of Contextual and Cued Fear Conditioning tests and beam balance in wild-type mice that were treated with Celastrol or BGP-15 following TBI compared to TBI-treated mice. These studies indicate a significant role of Hsp70/Hsp110 in neuronal survival following TBI and the beneficial effects of Hsp70/Hsp110 inducers toward reducing the pathological consequences of TBI.
Keywords: Hsp70, Hsp110, knockout mice, closed cortical impact, Celastrol, BGP-15
Introduction
Traumatic brain injury (TBI) can affect everyone, leading to long-term disabilities such as reduced cognitive functions (Chen and D’Esposito 2010, Humphreys, Wood et al. 2013). In addition, patients who have suffered moderate to severe TBI have an increased risk of developing neurodegenerative disorders such as Alzheimer’s disease (Johnson, Stewart et al. 2010, Sivanandam and Thakur 2012). TBI has short- and long-term consequences on the organism. The short-term consequences of TBI result from the initial mechanical damage leading to death of neurons and astrocytes and breakage of the blood vessels (Kumar and Loane 2012). The long-term consequences of TBI lead to many cellular and molecular events such as impairment of ion homeostasis, production of ROS and inflammatory responses leading to cerebral edema (Kumar and Loane 2012). A number of neuroprotective treatments have been employed, including a few clinical trials that have not shown substantial beneficial effects (Maas, Roozenbeek et al. 2010, Kumar and Loane 2012). In order to improve the injury outcome following TBI, it is important to understand the cellular and molecular alterations that occur during the hours and days following TBI. Events such as neurogenesis, neuroinflammation, and the process of proteostasis in general are important parameters that require further investigation to fully understand the pathological aspects of TBI.
The Hsp70 machinery consists of constitutively expressed (Hsc70) and the stress-inducible Hsp70 (Hsp70i). Hsp70i as well as other Hsps including Hsp110 are induced following exposure of the cells to stress and they are known to protect cells against cell death. Hsp110 is a nucleotide exchange factor for Hsp70i and binds to Hsp70 after ATP hydrolysis and catalyzes ADP-ATP exchange facilitating folding of denatured proteins (Hartl, Bracher et al. 2011). Hsp110 homologs are capable of binding and stabilizing the unfolded proteins, but cannot fold the denatured proteins without the help of Hsp70i (Polier, Dragovic et al. 2008). In terms of the cellular protective mechanisms, Hsp110 function is less well understood; however, Hsp70 has been shown to bind to Apaf1, inhibiting the formation of apoptosome and preventing caspase 3 activation (Beere, Wolf et al. 2000, Saleh, Srinivasula et al. 2000). Hsp70 is also a glucocorticoid receptor chaperone and modulates inflammatory response. As such, Hsp70 together with glucocorticoid receptor ligand has been shown to repress TNFα-stimulated IkBα degradation and NFkB p65 nuclear import (Beck, Drebert et al. 2013). More recently, Hsp70 has also been shown to bind RIP3, which converts TNF-induced cell death from apoptosis to necrosis (necroptosis) (Narayan, Lee et al. 2012). During necroptosis, RIP3 interacts with RIP1, forming complex IIb, followed by cell death (Narayan, Lee et al. 2012). These studies indicate a critical role of the Hsp110/Hsp70 in multiple processes that are required for cellular survival. As such, there are a number of examples of increased expression of the Hsp70i following exposure of cells to hypoxia and ischemic injury (Xu, Xiong et al. 2011, Qi, Liu et al. 2012) and transgenic mice expressing elevated levels of Hsp70i are protected against TBI (Kim, Kim et al. 2013). Data from one study suggest that Hsp70 protects against ischemia/reperfusion-induced kidney injury through T-regulatory cells (Kim, Jung Cho et al. 2013) and inhibition of Hsp70 by quercetin reduces this protective function. In addition, treatment of mice with geranylgranylacetate (GGA) that increases the Hsp70i levels in cells is neuroprotective when it was administered 48 hours prior to brain trauma (Zhao, Faden et al. 2013).
In this study, we used mice deficient in Hsp110 or Hsp70 to examine their response to TBI. We found that hsp110- or hsp70i-deficient mice are significantly more sensitive and exhibit increased neuronal death following TBI. The potential mechanism underlying the increased response to injury in hsp70i or hsp110-deficient mice compared to wild-type mice is the enhanced expression of p53-induced genes (Pigs). Our results also show a significant improvement of neuronal injury when mice were exposed to inducers of Hsp70/Hsp110 post-TBI. These studies suggest a potential benefit of using drugs to induce specific Hsps to reduce the pathological effects of TBI.
Methods and Materials
Mouse lines and genotyping
Generation of hsp110-deficient mice has previously been described (Eroglu, Moskophidis et al. 2010). All mouse lines were in C57BL/6 genetic background. Generation of hsp70i-/- mice that harbor deletion of both the Hsp70.1 and Hsp70.3 (Huang, Mivechi et al. 2001) genes will be reported elsewhere (Cho, W. K., et.al., Manuscript under consideration). Briefly, generation of hsp70.1 and hsp70.3 (named hsp70i)-deficient mouse line was as follows: As the hsp70.1 and hsp70.3 genes are located only 10kb apart on the same chromosome, mice carrying both targeted genes cannot be realistically obtained by intercrossing of single deficient mice generated in the lab (Huang, Mivechi et al. 2001). Therefore, a gene-deletion strategy involving replacement of the entire hsp70.1 and hsp70.3 coding sequences, including the intergenic region was pursued. This approach resulted in disruption of both functional hsp70i genes. The mice with the germ-line transmission of the deleted hsp70i genes were intercrossed and homozygous mutant mice were born with the expected Mendelian frequency, indicating that Hsp70i is not essential for embryo survival and confirming published observations (Hunt, Dix et al. 2004). The targeting vector was constructed by replacing 15 kb of genomic DNA, including the start codon of hsp70.3 and stop codon of hsp70.1 alleles (Huang, Mivechi et al. 2001) with a neomycin resistance gene flanked by two loxP sequences and LacZ reporter gene. The vector was designed so that the promoter of the hsp70.3 gene drives a LacZ reporter expression. Southern blotting analyses of genomic DNA digested with EcoRI and hybridized with a probe external to the targeted vector yielded a 12 kb fragment for the wild-type (WT) and 9 kb fragment for targeted allele, correspondingly.
For routine genotyping of hsp110-/- mice, DNA was used in multiplex PCR analysis to verify a WT 242bp, and a mutant 405bp fragments, using primer 1:5’-ACATAAG-GCTGAGCGATTGG-3’; primer 2: 5’-ATGTAGCAGCTCTGTGAGCCTAC-3’; primer 3: 5’-CAGGAAGATCGCACTCCAG-3’. For genotyping of hsp70i-/- mice the following primers were used; primer 1: 5’-GACAGTAATCGGTGCCCAAG-3’; primer 2: 5’-AGATCACC-ATCACCAACGACAAG-3’; primer 3: 5’-GTCGCTACCATTACCAGTTG-3’. PCR analysis generated a WT fragment of 510 bp, and a mutant fragment of 680 bp. Hsp70i-/- mouse line is normal and fertile and they were maintained as heterozygote intercrosses.
Stress model and drug treatment
All animal procedures were approved by the Institutional Animal Care and Use Committee. Controlled Cortical Impact (CCI) was performed on 8-12-week-old male mice (Laird, Sukumari-Ramesh et al. 2010). Briefly, mice were anesthetized and a 3.5mm craniotomy was made in the right parietal bone between the bregma and lambda with the medial edge one mm lateral to the midline, leaving the dura intact. Mice were impacted at 4.5m/s with a 20ms dwell time and one mm depression using a three mm diameter convex tip and were allowed to recover. Sham-treated mice were also treated comparably and received vehicle injections, but were not impacted. The TBI was observed on coronal sections that corresponded to 1.1 mm bregma to -2.92mm bregma. For the in vivo drug treatments, mice were treated with 1mg/kg of Celastrol (Li, He et al. 2012) at 30 minutes and 6 hours, and then again once daily for 5 days post-TBI given intraperitoneally. BGP-15 was administered orally at 15mg/kg (Gehrig, van der Poel et al. 2012) immediately before, and then at 6 hours, and again once daily for 5 days post-TBI. In all experiments the mortality rate due to TBI that occurred within the first few hours was 5%. There was no mortality due to TBI and drug treatment.
To determine the brain water content, we used the wet-dry method, which is a sensitive way of determining brain edema. At 24 hours post-injury, brain water content was quantified in a 3mm coronal tissue section of the ipsilateral cortex (or corresponding contralateral cortex), centered on the impact site. Brain tissues were weighed (wet-weight) immediately, then dehydrated at 65°C for 48 hours and reweighed to quantify the dry weight. The percentage of water content in the tissue samples was calculated using the formula: wet-weight (−) dry weight/wet weight×100 (Laird, Sukumari-Ramesh et al. 2010).
Magnetic resonance imaging
Images of mouse brain were acquired using a Bruker (Billerica, MA) 7T PharmaScan 20 cm horizontal bore MRI spectrometer with a micro imaging gradient insert (950 mT/m) and a 35 mm volume coil to transmit and receive at 1H frequency (Homma, Jin et al. 2007). For all sessions, anesthetized mice were placed in the MR scanner. Breathing and body temperature were maintained at 35 respirations/minute and at 37°C, respectively. The images generated were T2- weighted with a RARE pulse sequence; matrix = 256 × 256; TE/TR = 56.0/2800 ms; slice thickness = 0.5 mm; FOV = 2.56 × 2.56 cm; and NA = 5. Two sets of high-resolution, T2-weighted axial images were acquired for use in injury segmentation. The position of the first slice for the second set was offset from the first set by 0.5 mm, allowing both sets to be incorporated into a single, interleaved three-dimensional image volume. This composite image volume covered a 17.5 mm length of brain tissue along the axial axis, encompassing most or all of the olfactory lobe to the anterior cerebellum. Image intensity for each image set was slightly different, resulting mainly from receiver gain settings determined during an automatic prescan stage in the scanning sequence. The process of combining the two image sets into one was as follows. Histograms of pixel image intensity were calculated for each image set using identical intensity bins, after multiplying one of the image sets by a gain factor (g). If the two histograms were identical after multiplication with a gain factor, the number of pixels in a given bin should be equal between both histograms. The logarithm of the number of pixels belonging to each bin (lnbin) was taken, and the difference in lnbin between both histograms was computed for each bin. The absolute values of these differences were summed over all bins, giving a measure of difference between the histograms (hdiff) that should be minimized for the appropriate gain factor. Finally, g was adjusted in steps between 0.6 and 1.2 mm; the value of g yielding the minimum of hdiff was accepted and the intensity of even-numbered slices was multiplied by g. After adjustment of the slice-to-slice intensities, the injury was segmented out using ImageJ (NIH) image processing software. The edema sections were segmented from the rest of the brain by choosing a section of the image with clear edema, measuring the brightness of the injured area in pixel intensity over the area with averages and standard deviations (SDs); then the standard value of injury was set as average +2 SDs. The image set was filtered with the standard value set to the lower threshold; the resulting image set was used as a palette to manually segment out the injury slice by slice throughout the brain using the original image set as a reference. The resulting segmented 3-dimensional area was measured by pixel count and the sum count was converted to volume equivalent of the injured area (Homma, Jin et al. 2007, Kimbler, Shields et al. 2012).
Immunoblotting
Brain cortical tissues (3mm coronal of the ipsilateral cortex centered around the impact site) were homogenized in RIPA buffer and processed for immunoblotting as previously described (Eroglu, Moskophidis et al. 2010). Antibodies and dilutions used were as follows; βActin (sc-4778, 1:1000), Hsp110 (sc-1804, 1:1000, sc-6241 (1:300) for IHC); PUMAa/b (sc-377015, 1:1000); p53 (sc-6243, 1:1000) (Santa Cruz); Hsp70i (SMC-100, 1:1000, StressMarq); Bcl2 (2870S, 1:1000, Stressgen); Tau and p-Tau (1:2000 for western blotting and 1:400 for IHC, Generous gift from Dr. P. Davies, Albert Einstein College of Medicine, NY); Bim (1:1000, 2933), BclXL (1:1000, 2762); Hsp25 (1:1000, H2289,) (Cell Signaling); For quantitation of Western blots, the intensity of the target bands was quantified using NIH Image J 1.47v software, normalized to β-Actin and presented as fold-change relative to sham-treated group.
Histology and immunohistochemistry (IHC)
Brains were fixed in 10% formalin and embedded in paraffin. For immunostaining, 7 μm tissue sections were deparaffinized in xylene and rehydrated in a series of alcohol/water mixtures. For H&E staining, tissue sections were stained with Hematoxylin and rinsed with tap water. Slides were dipped in Scott’s tap water, washed, dipped in 95% alcohol and then stained with Eosin. After staining, sections were dehydrated in alcohol, placed in xylene and covered. For IHC staining, following antigen retrieval (boiling for 30 minutes in 10 mM sodium citrate plus 0.05% Tween 20, pH6.0), tissue sections were treated with PBS plus 3% BSA for 1 hour at 25°C to reduce nonspecific staining. Tissue sections were incubated in primary antibody for 16 hours at 4°C. Antibody/antigen was detected with Cy3- or FITC-conjugated secondary antibody. Nuclei were stained with DAPI (Eroglu, Moskophidis et al. 2010). The immunostained tissue sections were analyzed by a Zeiss Axio Image M1 fluorescent microscope. The antibodies used were: GFAP (Z0334, 1:400, Dako); CD11b (hybridoma, 1:400, gift of Dr. D. Moskophidis); βTubulin III (2276-1, 1:500, Epitomics); Ki67 (RM-9106-S0, 1:200, Thermoscientific); Iba1 (019-19741, 1:400, Wako). For apoptosis, tissue sections were processed and used for TUNEL assay according to manufacturer’s instruction (Millipore). For quantification of TUNEL and IHC staining, the positively stained cells were counted around the injured site (-0.1mm to -1.58mm bregma) under a 40x objective lens using at least 10 sections per mouse. Total cell number was then calculated for a total of 3-5 mice and then the mean was calculated. Further statistical analysis was performed as described below. For most IHC staining the results were expressed as the number of positive cells per section. For IbaI and GFAP IHC staining due to large number of positively stained cells, we counted 5 randomly defined views per tissue section and the results were expressed as the number of the positive cells per view. To determine the extent of axonal injury, serial coronal sections corresponding to -0.58 mm to -0.94 mm posterior to bregma were immunostained using β-tubulin III antibody. Under 100x magnification, β-tubulin III-positive neurons were identified in the ipsilateral cortex between the dorsal peak of the corpus callosum and up to 4mm from the midline. The length of each axon was determined using the Zeiss Axio imager M1 microscope on the Axiovision software. To determine the extent of tissue loss following TBI, brain tissue sections from -0.34 mm and -1.58 mm posterior to bregma were stained with H&E and captured using Zeiss Axio imager M1 microscope. The areas of the ipsilateral and contralateral cortices were calculated using NIH ImageJ 1.47v software. The amount of tissue loss was expressed as the ratio between two cortices: 100 x (contralateral–ipsilateral) divided by contralateral. In all cases, individual quantifying the data was blinded with respect to the groups’ identity.
Microarray analyses and qRT-PCR
For microarray analyses, brain tissue section (3mm coronal of the ipsilateral cortex centered around the impact site) of sham or TBI-treated WT or hsp110-/- 8-10 week-old male mice (n=5 mice each) were subjected to total RNA isolation using Triazol reagent (Invitrogen) followed by purification using RNeasy (Qiagen). Total RNA samples were then subjected to Affymetrix Mouse gene 1.0 ST Array (GRU Integrative Genomic Core Facility). Quality assessment of each CEL file was performed using Affymetrix Expression Console Software according to Affymetrix standard protocol (Quality Assessment of Exon and Gene 1.0 ST Arrays, Affymetrix). Relative log expression (RLE) was used to identify outlier samples. In order to monitor labeling and hybridization quality, we used polyA-control RNAs (Lys, Phe, Thr and Dap) and bacterial spike-in controls (BioB, BioC, BioD and Cre), respectively. The CEL files were imported into Partek Genomics Suite using GC-RMA normalization. The probesets were annotated using the MoGene-1_0-st-v1 Probeset Annotations and Transcript Cluster Annotations. The differential expressions were calculated using ANOVA of the Partek Express Software (Januchowski, Zawierucha et al. 2013). Gene expression was measured by averaging the signal intensities of probes. For each pair comparison, fold change was computed and genes above 1.5 fold change were retained as differentially expressed genes (DEGs). DEGs for both pairs were generated and sorted based on the fold change for hsp110-/- sample. The 50 up and 50 down-regulated gene from the sorted DEGs were used to generate a heatmap using R Bioconductor with the average linkage and Euclidean distant (Gentleman, Carey et al. 2004). Biological Pathways Associated with significantly regulated genes (fold change >1.0 and < 1.0) were generated using the ‘Biological Pathways’ subset of Gene Ontology included in the DAVID System (DAVID, http://david.abcc.ncifcrf.gov/) (Huang da, Sherman et al. 2009). In the Figure presented, blue bars indicate number of genes and red bars indicate the significance values (negative log10 of p-values).
For qRT-PCR, total RNA was isolated from mouse brain tissue (n=3-5 mice) using Triazol reagent (Invitrogen). The cDNAs were generated using iScript cDNA synthesis Kit (BioRad). The primer sequence to detect the cDNA were as follows: Hsp110, F: 5’-CAGGTA-CAAACTGATGGTCAACA-3’, R: 5’-TGAGGTAAGTTCAGGTGAAGGG-3’; Hsp70i, F: 5’-AGGTGCTGGACAAGTGCCAG-3’, R: 5’-AACTCCTCCTTGTCGGCCA-3’, growth hormone (GH), F:5’-CAAAGAGTTCGAGCGTGCCTA-3’, R: 5’-AGAAGCGAAGCAA-TTCCATGTC-3’; PrL, F:5’-CAGGGGTCAGCCCAGAAAG-3’, R:5’-TCACCAGCGGAAC-AGATTGG-3’ ;CCL2, F:5’- TTAAAAACCTGGATCGGAACCAA-3’, R:5’- GCATTAGCTTCAGATT-TACGGGT-3’; CCL3, F: 5’-TGTACCATGACACTCTGCAAC-3’, R:5’- CAAC-GATGAATTGGCGTGGAA-3’; Pig1, F: 5’-TGGACACTTCCGAGGT-TGTCT-3’, R:5’-CCTCACGGCCCCATTTG-3’; Pig8, F: 5’-GGCATCTGTACCATCTCAAAGCT-3’, R: 5’-TCGACGCTGTTCCTCTCTCTTC-3’; Pig12, F: 5’-GGCTTGGG-ATCCAGAGATGTC-3’, R: 5’-GTGGCAGAGCTGCAGAAAGAA-3’; Lif1, F: 5’-AGCTA-TGTGCGCCTAACATGA-3’, R: 5’-CGACCATCCGATACAGCTCC-3’. qRT-PCR System (Applied Biosystems) and SYBR Green PCR supermix were used for the reactions. Samples were normalized to β-actin. Results were presented as mean ± SD relative to control.
Neurological injury determination
Behavioral studies were performed and analyzed in the Institutional Small Animal Behavioral Core (Wilson and Terry 2013). The tests performed were Fear Conditioning (Context-dependent Freezing and Cued-dependent Freezing), Rotarod, Beam Balance, and Flexion Reflex. The contextual task was run 24 hours after the completion of the training task. The appearance of the chamber was comparable to the one used in the training task (i.e. square walls all around, barred floors, and white light). The animals were allowed to explore the chamber for 5 minutes and their freezing was recorded. The cued task was run 48 hours after the completion of the training task. The appearance of the chamber was changed. Then animals were allowed to explore the “new” chamber for 2 minutes before being re-exposed to the tone for 3 minutes. Freezing was scored for 5 minutes total. The testing apparatus was the Med Associates using the program Video Freeze. It records the animals movements and then motion thresholds (generally 18) and a minimum freezing duration (generally 1 sec) so that what qualifies as a freeze can be identified when video is monitored. For Rotarod tests, mice were placed on a rotating cylindrical rod that accelerates from 4 to 45 revolutions/minute over a period of 5 minutes. Mice were provided four trials each day over 2 days, and the time to either drop from the rod or turn one complete rotation was determined (Eroglu, Moskophidis et al. 2010, Wilson and Terry 2013). We also measured the ability of mice to balance on a 1.2 cm diameter bar fixed at a height of 40 cm. The ability of untreated or drug-treated mice to remain on the bar was assessed. The flexion reflex was performed as described previously (Tupper and Wallace 1980). Briefly, the hind legs of mice were pinched, and the presence or absence of withdrawal reflex was recorded and calculated as percentages of mice responding per group. Neurological injury assessment was performed using 8-10 mice for each group. The results are expressed as the mean± SD.
Statistical consideration
The number of mice used for each time point and/or genotype was n=3-10. Multiple sections of brain tissues (n=10 or more) from each mouse were analyzed for each histological or immunohistological study. In all the experiments, quantification of the data was performed in a random manner to exclude counting bias. For statistical analyses, data were expressed as mean +/- SD. The p value was calculated by two-tailed Student’s t-test. Differences between groups were considered significant at p<0.05.
Results
Hsp110 and hsp70i -deficient mice are susceptible to CCI
To explore whether deletion of specific chaperones increases pathology observed during CCI and examine the pathways that may be affected, we used mice deficient in hsp110 or hsp70i and exposed them to CCI. Figure 1A shows the brain water content, a sensitive measure of edema that is associated with severity of the damage 24 hours after hsp110-deficient mice were exposed to TBI. Results indicate that hsp110-/- mice show significantly more brain edema compared to wild-type (WT) mice following exposure to TBI. Before mice were euthanized, we determined the extent of brain damage and edema volume using MR imaging of the mice exposed to CCI (Figure 1B-C). Consecutive images of the brain sections (Figure 1B, sections 1-8) show that hsp110-/- mice exhibited areas of damage that appeared more severe than those observed in WT mice. From the MR images, we also determined the edema volume and we found that hsp110-/- mice exhibit increased brain edema compared to WT mice.
Figure 1. Hsp110-/- mice sustain increased brain injury post-TBI.
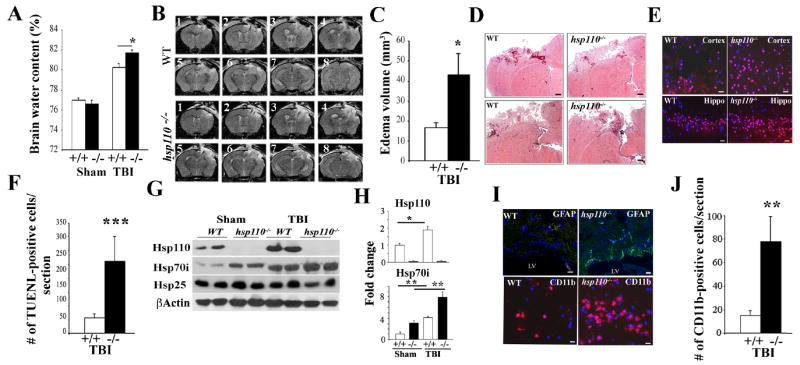
WT (+/+) and hsp110-/- (-/-) mice were subjected to CCI or were sham-treated and analyzed 24 hours post-TBI.
(A) Brain water content was quantified from the ipsilateral cortex post-TBI. Data presented as mean+/-SD (n=5 mice). *p<0.05.
(B-C) The T2-weighted MR images were analyzed to determine the overall brain volume and corresponding edema volume for each imaged mouse. Consecutive images are presented as 1-8. Quantification of cerebral edema from the MRI data is presented in (C). Data presented as mean+/- SD. *p<0.05.
(D) Representative coronal brain sections stained with H&E at 24 hours post-TBI. Scale bars:200 μm.
(E-F) Apoptotic cells were stained in the cortex and hippocampal (Hippo) regions close to the injured site (-0.1mm to -1.58mm bregma). Scale bar:20μm. Quantification of the TUNEL-positive cells is presented in (F). Data are presented as mean+/-SD.**p<0.01.
(G-H) Immunoblotting analyses showing brain cell lysates of sham or TBI-treated mice using the indicated antibodies. β-Actin is loading control. Quantification of the immunoblots is presented in (H). Data are presented as mean+/-SD.*p<0.05,**p<0.01.
(I-J) IHC analyses showing increased GFAP (upper panel) and infiltrating CD11b (lower panel) immune cells post-TBI in the cortical area (-0.1mm to -0.94mm bregma). Scale Bars:20μm (upper); 10μm (lower panel). Quantification of the CD11b-positive cells post-TBI is presented. Data are presented as mean +/- SD. ** p<0.01.
In all panels, DAPI represents nuclei staining. In panels B to J, n=3-5 mice per group.
The histopathological examination of the WT and hsp110-/- mice using H&E staining showed significant damage in the ipsilateral cortex 24 hours post-TBI (Figure 1D). No histopathology was observed in the contralatetal side or the sham-treated mice that were treated with the same surgical procedures, but without the trauma (not presented). Trauma induces apoptotic cell death and measurement of apoptosis using TUNEL assays in the cortex and hippocampus area showed significantly more apoptotic cells in the ipsilateral region in the hsp110-/- mice compared to WT (Figure 1E and F). To examine the extent of Hsps expression following TBI we performed immunoblotting analyses of sham and TBI-treated brain tissue. Figure 1G shows the increased expression of Hsp110 and Hsp70i following TBI. Expression of Hsp25 was not significantly affected in the brain cell lysates of TBI-treated WT and hsp110-/- mice. Quantification of the data is presented in Figure 1H. In untreated brain, Hsp110 is normally expressed at higher levels constitutively than the Hsp70i (Huang, Mivechi et al. 2001, Eroglu, Moskophidis et al. 2010) and there appears to be compensatory increase in the expression of Hsp70i in hsp110-/- sham-treated brain tissue. Brain trauma causes astrogliosis that can be detected using GFAP immunostaining and IHC staining of brain tissue sections indicate increased GFAP-positive cells in hsp110-/- compared to WT mice (Figure 1I). In addition, CD11b immunostaining suggests increased infiltration of the inflammatory cells in the injured site (Figure 1I). Quantification of CD11b-positive cells in TBI-treated brain tissue sections are presented in Figure 1J.
We then examined the critical role of Hsp110 partner protein, Hsp70 following TBI using mice deficient in the two inducible hsp70 genes. The results of hsp70i-/- mice exposed to TBI were comparable to that observed in hsp110-deficient mice (Figure 2). The increased damage observed following analyses of H&E stained brain tissue sections post-TBI and the amount of brain water content was significantly higher in the absence of Hsp70i compared to WT mice (Figure 2A-B). The impacted areas of the brain of hsp70i-/- mice exhibited increased apoptosis and CD11b infiltrating cells (Figure 2C-E). Figure 2F shows immunoblotting of brain tissue lysates showing the absence of Hsp70i in hsp70-/- mice. Immunoblotting experiments showed that the expression of Hsp110 was reduced in mice deficient in hsp70i that were exposed to TBI, perhaps due to increased cellular apoptosis observed in brain tissues of these mice. There was an increase in the level of Hsp70i and, to a lesser extent, Hsp110 in the brain tissue of WT mice exposed to TBI (Figure 2F and G).
Figure 2. Hsp70i-/- mice sustain increased brain injury post-TBI.
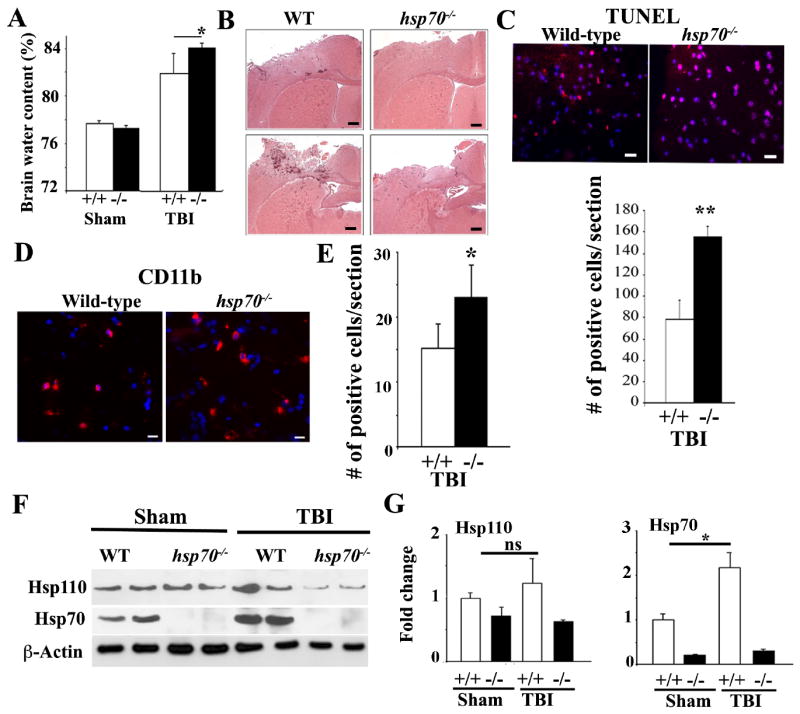
WT (+/+) and hsp70i-/- (-/-) mice were subjected to CCI or were sham-treated and analyzed 24 hours post-TBI.
(A) Brain water content was quantified from the ipsilateral cortex post-TBI. Data are presented as mean+/-SD (n=5 mice).* p<0.05.
(B) Representative photographs of coronal brain sections stained with H&E post-TBI. Scale Bars: 200 μm.
(C) Apoptotic cells in the cortical region close to the injured site (-0.1mm to -1.58mm bregma) are presented. Scale bar: 20 μm. Quantification of the TUNEL-positive cells in (lower panel) is presented in the lower panel. Data are presented as mean+/-SD.** p<0.01.
(D-E) IHC analyses of CD11b-positive cells post-TBI (-0.1mm to -1.58mm bregma). Scale bar:10μm. Quantification of the CD11b-positive cells is presented in panel E. Data is presented as mean+/-SD.* p<0.05.
(F-G) Immunoblotting analyses showing sham or TBI-treated brain cell lysates of cortical regions using the indicated antibodies. β-actin is loading control. Quantification of the immunoblots is presented in (G). Data are presented as mean+/-SD.*p<0.05, ns=not significant.
In all panels, DAPI (blue) represents nuclei staining. In panels B to G, n=3-5 mice per group.
Taken together, the data show that both hsp110-/- and hsp70-/- mice exhibit increased response to damaging effects of TBI compared to WT mice.
Hsp110 is highly expressed in the adult brain (Eroglu, Moskophidis et al. 2010), however, there is no information available regarding the role of Hsp110 in various biological processes or following brain injury. To learn more regarding the expression of genes that could be altered following exposure of mice to TBI in the absence of hsp110 gene we performed Affymetrix Mouse gene 1.0 ST Array analyses of injured brain tissue 24 hours following exposure of WT or hsp110-/- mice to sham or TBI treatment. From this, we identified 270 genes whose expression were significantly up or down-regulated (p<0.05) when we compared injured brain tissues of WT and hsp110-/- sham- or TBI- treated mice. Table S1 presents the total number of genes affected, and Table S2 shows the list of 270 genes whose expression changed significantly between the groups (p<0.05). Figure S1 and Table S3 show the results of the relative gene expression for the 50 up and 50 down-regulated genes, a heatmap and the functional significance of these 100 genes. We then tested the expression of number of genes that were significantly altered in WT and hsp110-/- brain tissue following TBI by qRT-PCR (Figure S1B). In these experiments, we also tested the expression of the same genes in hsp70i-/- sham or TBI-treated mice for comparison. We found that the expression of Hsp110 was absent in hsp110-/- brain tissues, and Hsp110 expression level was higher in WT and hsp70i-/- TBI-treated brain tissues relative to sham. qRT-PCR of Hsp70i cDNA showed that there was no expression for this gene in hsp70i-/- brain tissue and expression of hsp70i gene was higher in WT and hsp110-/- TBI-treated brain tissues relevant to sham. We also tested the expression level of growth hormone (GH) and prolactin (Prl) that we found to be significantly higher in WT compared to hsp110-/- brain tissue following TBI (3.5 and 2.7 -fold, respectively). Data confirmed that there is a reduction of GH expression in brain tissue of hsp110-/- sham relative to WT sham (Figure S1B). In addition, the level of GH in WT was significantly higher following TBI compared to hsp110-/- and hsp70i-/- mice. In the case of GH, we also tested the level of GH in the brain tissue and serum of WT, hsp10-/- and hsp70i-/- mice using ELISA and we found the level of GH to be lower in hsp10-/- brain tissue and serum following TBI (Figure S1B). The level of GH in the serum of hsp70i-/- mice was also lower following TBI, but did not reach significance. qRT-PCR data for prolactin (Prl) was comparable to GH.
We also tested the expression levels of chemokines CCl2 and CCL3 and leukemia inhibitory factor (Lif) in sham and TBI-treated brain tissues of WT, hsp110-/- and hsp70i-/- mice. As the data presented in Table S2 shows, we found the expression of CCL2 and CCL3 to be lower in WT sham versus WT TBI (2.6 and 3.3-fold, respectively) and lower in hsp110-/- sham versus hsp110-/- TBI (3.5 and 3.6-fold, respectively), and qRT-PCR confirmed these findings (Figure S1B). The level of CCL2 was also higher in TBI-treated hsp70i-/- brain tissue compared to TBI-treated WT mice. CCL2 and CCL3 are chemokines ligand 2 and 3 and are known to induce inflammatory responses. We also found leukemia inhibitory factor to be 1.8-fold lower in WT mice exposed to TBI versus TBI-treated hsp110-/- mice by microarray analyses, and qRT-PCR confirmed the result. Lif was also significantly higher in the hsp70i-/- brain tissue exposed to TBI compared to TBI-treated WT mice.
Using the microarray data, we then analyzed the biological pathways that were associated with significantly altered genes (Table S4) and selected pathways relevant to tissue injury post CCI have been presented in Figure S1C. In general, numbers of biological pathways were affected between the groups and they included those involved in the inflammatory response, chemotaxis, response to wounding and stress when we compared WT sham versus WT TBI -treated groups and hsp110-/- sham versus hsp110-/- TBI-treated groups. Expression of 3 genes associated with oxidation/reduction pathway (cytochrome P450s such as Cyp2a5, Cyp2g1 and Cyp2a4) was significantly altered when tissues of WT sham versus hsp110-/- sham-treated mice were compared. The expression of larger number of biological pathways and genes were altered when tissues of WT and hsp110-/- TBI-treated groups were compared, and these included response to external stimulus and wounding (Figure S1C and Table S4).
Taken together, using gene expression profiling we found an increase in the expression level of genes involved in the proinflammatory response, oxidation/reduction and response to stress following exposure of hsp110-/- and hsp70i-/- mice to TBI compared to WT.
Mice treated with Celastrol post-TBI exhibit lower levels of brain injury
To examine whether an increase in the level of expression of Hsp70/Hsp10 is beneficial following exposure of WT mice to TBI, we subjected mice to TBI with, or without, treating them with Celastrol (Li, He et al. 2012). At 24 hours, and at 1 and 4 weeks following exposure of WT mice to brain trauma and drug treatment, we examined the consequences of drug treatment on TBI. As the results in Figure 3A (upper panels) indicate, exposure of mice to two treatments of Celastrol (30 minutes and 6 hours post-TBI) leads to a significant improvement in histopathology as evidenced following examination of H&E staining of brain tissue (Figure 3A), and brain water content following Celastrol treatment of sham and TBI-treated WT mice (Figure 3B). MR images at 24 hours post-TBI show significant improvement of edema volume when WT mice were treated with Celastrol post-TBI (Figure 3C and D).
Figure 3. Celastrol treatment of mice post-TBI improves response to injury.

WT mice were subjected to CCI or were sham-treated and received vehicle or Celastrol at 30 minutes and 6 hours (named 24-hour), or treated with TBI plus Celastrol at 30 minutes, 6, 24, 48, 72, 96, and 120 hours post-TBI (named one-week).
(A-B) Representative photographs of coronal brain sections stained with H&E post-TBI (A). Scale bars:200 μm. Arrows point to the injured site. Brain water content was quantified from the contra- and ipsi-lateral cortex at 24 hours post-TBI (B). Data presented as mean+/-SD (n=5 mice). **p<0.01.
(C-D) MR images of WT mice treated with vehicle or Celastrol were taken at 22 hours post-TBI. The T2-weighted MR images were analyzed to determine the overall brain and corresponding edema volume. Consecutive images are presented as 1-6. Quantification of brain water content from the MRI data is presented in panel (D). Data are presented as mean+/-SD.*p<0.05.
(E-F) Apoptotic cells in the cortical region close to the injured site (-1.34mm to -1.58mm bregma) are presented. Scale bar: 20μm. Quantification of the apoptotic cells is presented in (F). Data is presented as mean+/-SD; *** p<0.001.
(G-H) Representative IHC analyses of CD11b-positive cells in the cortical region close to the injured site (-0.1mm to -1.58mm bregma) in untreated (vehicle), or Celastrol-treated mice post-TBI. Scale bar:10μm. Quantification of CD11b-positive cells is presented in (H). Data is presented as mean + /-SD.*p<0.05.
(I-K) Representative IHC staining of Iba1-positive cells in the cortical region close to the injured site (-0.1mm to -1.58mm bregma) post-TBI (I-J). Scale bars: 200μm(I); 10μm(J). Quantification of Iba1-positive cells in the striatum close to the injured site post-TBI (K). Data are presented as mean+/-SD.*p<0.05.
(L) Representative IHC analyses showing Hsp110-immunostaining at 24 hours, and one-week post-TBI using -0.1mm to −0.94mm bregma region.
(M-N) Immunoblotting analyses showing sham or TBI-treated brain cell lysates of cortical regions close to the injured site using the indicated antibodies. β-actin is loading control. Quantification of the immunoblots is presented in (N). Data are presented as mean +/-SD.* p<0.05,**p<0.01,***p<0.001.
In all panels DAPI represents nuclei staining. In panels C to N, n=3-5 mice per group.
To examine the effects of increasing the levels of the Hsp70/Hsp110 expression further using Celastrol treatment for 5 days post-TBI, we exposed mice to TBI and treated them with Celastrol at 30 minutes and 6, 24, 48, 72, 96 and 120 hours post-TBI and determined the tissue injury and recovery using several criteria. As the data in Figure 3A (lower panels) indicate, H&E staining of brain tissue show reduced injury at the impact site in mice that were treated with TBI and Celastrol compared to mice that were treated with TBI alone. The number of apoptotic cells was also significantly lower in the brain tissue of mice that were exposed to TBI and treated with Celastrol compared to mice that were exposed to TBI alone at both 24 hours, or one-week post-TBI (Figure 3E-F). The pathological improvement of TBI-treated mice following Celastrol treatment also correlated with decreased number of CD11b inflammatory cell infiltration into the injured site (Figure 3G-H). Quantification of the number of glia around the injured site (known to increase following injury (Woodcock and Morganti-Kossmann 2013) also show a reduced number in mice that received Celastrol for 24 hours or 5 days post-TBI (Figure 3I, J and K). The results in Figure 3L shows that there is an increase in the number of cells expressing Hsp110 close to the injury site in mice that were treated with Celastrol compared to untreated mice following TBI. Immunoblotting of brain cell lysates show significant increase in Hsp70i following TBI, and TBI plus Celastrol-treated brain tissue 24 hours following treatment (Figure 2M-N). The levels of Hsp70i remained elevated in both TBI, and TBI plus Celastrol one week-post injury but it did not reach significance (Figure 3M-N).
Astrogliosis is a hallmark of brain injury that can be detected using GFAP expression. We noted improvement in the pathology observed post-TBI plus Celastrol using GFAP immunostaining (Figure 4A, upper panel). Interestingly, mice treated with TBI also exhibited an increased number of Ki67-positive cells in the subventricular zone close to the injured site (Figure 4A, lower panel). Ki67-positive neurons can usually be detected in the subventricular zone post-TBI (Rola, Mizumatsu et al. 2006). Quantification of the number of GFAP and Ki67-positive cells post-treatment are presented in Figure 4B.
Figure 4. Celastrol treatment of mice post-TBI improves neurological response.
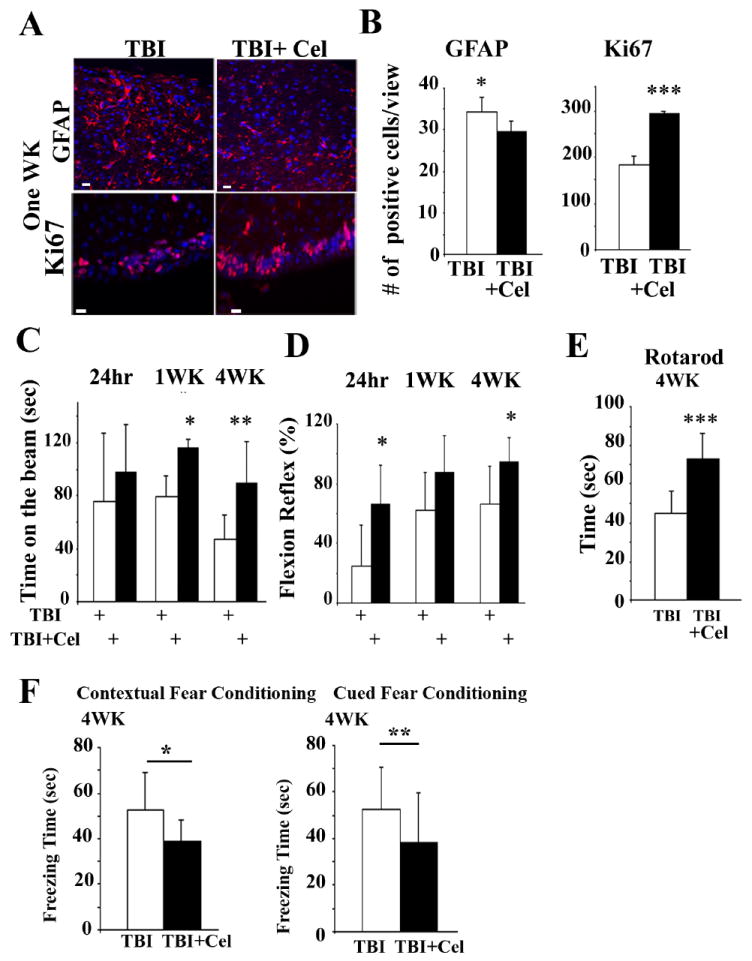
(A-B) Representative IHC staining showing GFAP (upper panels) and Ki67-positive cells (lower panels). Scale bar: 10μm. Quantification of the positive cells is presented in panel B. For GFAP, sections analyzed corresponded to -0.10 mm to -0.7 mm bregma, and for Ki67 corresponded to -0.58mm to -0.94mm to bregma. In all panels, DAPI represents nuclei staining. Data is presented as mean+/-SD. *p<0.05, *** p<0.001. n=3 mice.
(C-D) Time spent on the beam and flexion reflex were determined in mice at the indicated times. Celastrol treatment post-TBI for the 4-week group was the same as for the 1-week group indicated above. Data are presented as mean +/- SD (n=8 mice). *p<0.05,**p<0.01.
(E) Motor activity using Rotarod test measured 4 weeks post-TBI or TBI plus Celastrol-treated groups (n=9 mice). Data is presented as mean +/- SD. ***p<0.001.
(F) Mice were subjected to TBI or TBI plus Celastrol for 5 days. At 4 weeks post-injury mice were subjected to Centextual and Cued Fear Conditioning tests (n=8 mice). Data is presented as mean +/- SD. *p<0.05, **p<0.01.
We then performed behavioral analyses following treatment of mice with Celastrol for a period of 5 days post-TBI using number of neurological parameters. We found that there was significant improvement in the time that mice spent on the beam (Figure 4C), and the flexion reflex (Figure 4D) at 24 hours, and at 1, and 4 weeks post-TBI in mice that were treated with Celastrol. Cohorts of mice were also left for 4 weeks following 5 days of Celastrol treatment post-TBI, for determination of motor activity and cognitive function. We examined motor activity using the Rotarod test and Contextual and Cued fear conditioning tests to measure hippocampus-dependent associative learning. We found significant improvement of motor activity in TBI-treated mice that were treated with Celastrol (Figure 4E). We also found significant improvement in Contextual and Cued Fear Conditioning tests in Celastrol-treated group (Figure 4F).
Wild-type mice treated with BGP-15 post-TBI exhibit lower levels of brain injury
To examine further the benefits of increasing Hsp levels following TBI on brain injury, we subjected WT mice to TBI with, or without treatment of mice with BGP-15, a drug that is known to increase the levels of Hsp70/Hsp110 and protect mice against muscular dystrophy (Gehrig, van der Poel et al. 2012). We examined the consequences of drug treatment on TBI at 24 hours, and at 1 week after TBI and BGP-15 administration. As the results in Figure 5A indicate, exposure of mice to two treatments of BGP-15 (0 and 6 hours post-TBI (24-hour time-point), or additional 5 daily doses (one-week-time point) lead to a significant improvement in the observed histopathology using H&E staining (Figure 5A). The data in Figure 5B-E show that the number of apoptotic cells, and infiltrating CD11b inflammatory cells were significantly lower in TBI and BGP-15-treated mice. We also observed reduced numbers of IbaI-positive glia cells, that normally increases following brain injury, in mice that were treated with TBI and BGP-15 (Figure 5F-G). Interestingly, we observed elevated expression of Hsp110 using IHC staining of brain tissue in TBI-treated mice that were treated with BGP-15 for the first 24 hours or 5 days post-TBI (Figure 5H). Disruption of axons following TBI is a common occurrence (Woodcock and Morganti-Kossmann 2013). We therefore, used IHC staining to detect the expression of β-tubulin III, which detects neurons and axons following TBI or TBI plus BGP-15 treatment. Data indicate that there was significant improvement in the axonal length in mice treated with BGP-15 post-TBI compared to WT (Figure 5I-J). GFAP expression also increases following brain injury. IHC staining of brain tissue of mice treated with TBI, or TBI plus BGP-15 indicate significantly lower GFAP-positive cells in mice treated with BGP-15 (Figure 5K-L).
Figure 5. BGP-15 treatment of mice leads to improved post-TBI response.

WT mice were treated with TBI and received vehicle or BGP-15 immediately before (0 hour) and at 6, 24, 48, 72, 96 and 120 hours post-TBI. 24 hours group only received the first two treatments.
(A) Representative photographs of coronal brain sections stained with H&E. Scale Bars: 200 μm.
(B-C) Apoptotic cells in the cortical region close to the injured site (-0.1mm to −1.58mm bregma) are presented. Scale bar:10μm. Quantification of apoptotic cells post-TBI (C). Data is presented as mean +/- SD. ***p<0.001.
(D-E) Representative IHC staining of CD11b-positive cells in the cortical region (-0.1mm to −1.58mm bregma) are presented. Scale bar: 10 μm. Quantification of CD11b-positive cells is presented in (E). Data presented as mean +/- SD.** p<0.01.
(F-G) Representative IHC staining of Iba1-positive cells in the cortical region (-0.1mm to −0.94mm bregma) are presented. Scale bar=10μm. Quantification of IbaI-positive cells is presented in (G). Data presented as mean+/- SD. *p<0.05, ***p<0.001.
(H) Representative IHC staining of Hsp110-positive cells in the cortical region (-0.1mm to −0.94mm bregma) are presented. Scale bar=10μm.
(I-J) Representative IHC staining of β-Tubulin III in the cortical region (-0.58mm to −0.94mm bregma) are presented. Scale bar=10 μm. Quantification of axon-length is presented in (J). Data presented as mean +/- SD. **p<0.01, *** p<0.001.
(K-L) Representative IHC staining of GFAP-positive cells in the cortical region (-0.1mm to -0.94mm bregma) are presented. Scale bar=10 μm. Quantification of GFAP-positive cells is presented in (L). Data presented as mean +/- SD. ** p<0.01.
(M-N) Representative IHC staining of Ki67-positive cells is presented. Scale bar=10μm. Quantification of Ki67-positive cells is presented in (N). Data presented as mean +/- SD. ***p<0.001.
(O) Immunoblotting analyses of cortical region close to the injured site (3mm coronal of the ipsilateral cortex centered around the impact site) using the indicated antibodies. Brain cell lysates from two treated mice are presented. (-) untreated brain cell lysates. Quantification of the immunoblots is presented in the lower panels. Data presented as mean +/- SD.*p<0.05,**p<0.01.
(P) Flexion reflex and time spent on the beam were measured in mice post-TBI. Data is presented as mean +/- SD (n=8 mice). *p<0.05.
In all panels, DAPI (blue) represents nuclei staining. In panels A to O, n=3-5 mice per group.
Neurogenesis is a critical event that occurs following TBI. Therefore, to examine if there are changes in Ki67-positive cells when mice were treated with BGP-15, IHC staining was performed. Quantification of the number of Ki67-positive cells in the subventricular zone indicate significantly higher numbers of Ki67-positive cells in mice treated with BGP-15 post-TBI compared to those that received TBI alone (Figure 5M-N). Immunoblotting experiments of brain tissue lysates indicated elevated levels of Hsp110 and Hsp70 following TBI and Celastrol (Figure 5O). We also performed neurological assessment of TBI-treated mice that were treated with vehicle or BGP-15. Flexion reflex and the time mice spend on the beam of TBI and BGP-15-treated mice was significantly improved both at 24 hours, and at one-week post-TBI (Figure 5P).
TBI-treated brain tissue exhibit hyperphosphorylated Tau (P-Tau), which is significantly reduced following treatment of mice with Hsp70 inducers
It has been reported that TBI-treated brain tissue extracts exhibit P-Tau. This is important since this could lead to increased neuronal death post-TBI and contribute to persistence of pathological effects that are observed post-TBI. To examine if we observe P-Tau in brain tissue following exposure of mice to CCI model, we performed immunoblotting experiments of brain tissue lysates in mice that were treated with vehicle, Celastrol or BGP-15 24 hours or one-week post-TBI. We found a background level of P-Tau (pS202) in sham-treated brain tissue of WT mice while P-Tau levels increased significantly both after 24 hours and at one-week post TBI (Figure S2A). Interestingly, mice treated with Celastrol or BGP-15 exhibits P-Tau only at 24 hours post-TBI and P-Tau appeared to recover to the background levels at one-week post-TBI. We found that the level of Hsp70 is also increased following TBI, and following treatment of mice with TBI and Hsp70 inducers (Celastrol and BGP-15) (Figure S2A-C). In the case of hsp110-/- brain tissue extracts both sham- or TBI-treated brain cell lysates exhibited the presence of P-Tau (Figure S2A-C) (Eroglu, Moskophidis et al. 2010). IHC analyses also show increased levels of P-Tau (pS202) at 24 hours and one-week post-TBI when mice were treated with vehicle. However, there was significant improvement on clearance of p-Tau when mice were treated with Celastrol or BGP-15 at 24 hours, but more significantly at one-week post-TBI (Figure S2D- E). There was no change in the level of total Tau.
Hsp110 and Hsp70-deficient mice exhibit increased expression of p53 and p53-induced genes in brain tissue following TBI
As noted before, Celastrol and BGP-15 are known to affect multiple biological pathways. However, these drugs also increase the level of Hsp70i through activation of Hsf1which can also increase the level of other inducible Hsps, such as Hsp110. To probe if hsp70i-/- or hsp110-/- mice exposed to TBI plus Celastrol exhibit protection of brain tissue as in WT mice, we exposed WT, hsp110-/- and hsp70i-/- mice to TBI or TBI plus Celastrol and determined the area of tissue injury and number of apoptotic cells present after 24 hours. Our data show that the extent of injury following TBI at -1.54 to -1.58 mm bregma were 8.1+/-0.2 for WT, 10.7+/-1.1 for hsp110-/- and 10.5+/-0.5 for hsp70i-/- mice, and following TBI plus Celastrol the extent of injury in same area was 3.7+/- 1.3 for WT, 8.8+/-1.2 for hsp110-/- and 9.7+/-1 for hsp70i-/- mice. In brief, our data indicate no significant changes in the damage areas following TBI and Celastrol treatment in hsp110-/- or hsp70i-/- mice compared to TBI alone, while Celastrol treatment of TBI-treated WT mice was protective compared to TBI-treated WT mice (p<0.001). Comparable data was observed following TUNEL assays (data not shown).
We then attempted to gain insight into the potential mechanism underlying the increased sensitivity of hsp110 or hsp70 -deficient brain tissues following exposure of mice to TBI compared to WT mice. Thus, we performed immunoblot analyses to detect the expression of the pro-apoptotic gene p53 that has been shown to play a role in neuronal cell death following brain trauma (Plesnila, von Baumgarten et al. 2007). In these experiments, we also tested if Celastrol treatment of TBI-treated WT, hsp110 or hsp70-deficient mice protected the brain tissue of WT mice, but not those of the knockout mice. Data presented in Figure 6A indicate that sham-treated mice of all genotypes express background levels of p53 as expected, and p53 expression is enhanced upon exposure of mice to TBI. Interestingly, brain tissue lysates of only the WT mice exposed to TBI plus Celastrol (Celastrol was administered at 30 minutes and 6 hours post-TBI), exhibit reduced level of p53 expression (Figure 6A, compare samples in lanes 4 and 7, and Figure S3 for quantitation of the data). However, comparably treated hsp110 and hsp70-deficient mice did not show this reduction in p53 expression levels upon Celastrol treatment (Figure 6A, compare samples in lanes 5 and 6 to 8 and 9, and Figure S3 for quantitation of the data). Expression of the pro-apoptotic genes that are p53 targets, such as Puma and Bim also confirmed p53 activation following TBI, and the reduction in their expression was only observed in Celastrol-treated WT brain tissue (Figure 6A, compare samples in lanes 4 and 7, and Figure S3 for quantitation of the data). Additionally, the expression of anti-apoptotic genes, Bcl2 and Bcl-XL were only up-regulated in WT brain tissue that were exposed to TBI plus Celastrol, and not in the knockout brain tissues comparably treated (Figure 6A, compare samples in lanes 4 and 7, and Figure S3 for quantitation of the data).
Figure 6. p53 target genes are up-regulated following TBI.
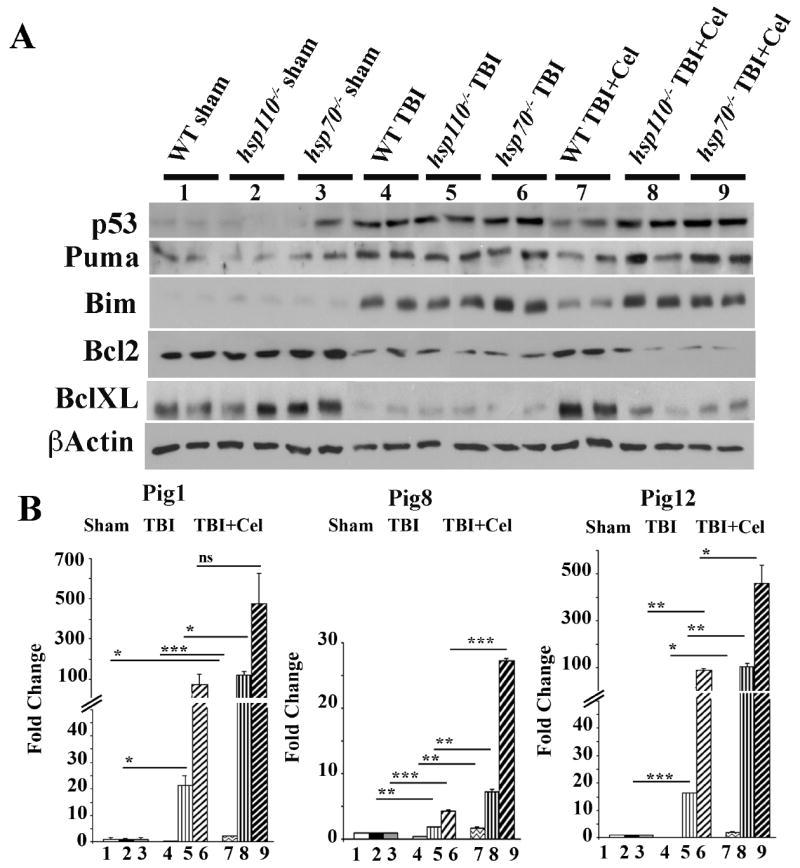
WT, hsp110-/- or hsp70i-/- mice were treated with TBI and received vehicle or Celastrol at 30 minutes and 6 hours post-TBI. 24 hours later brain tissues (3mm coronal of the ipsilateral cortex centered around the impact site) were processed for immunoblotting or cDNA preparation. (A) Brain tissues of treated mice were analyzed by immunoblotting using the indicated antibodies. β-actin is loading control. Quantification of the immunoblots is presented in Figure S3. (B) qRT-PCR for the indicated genes are presented. Samples were normalized to β-actin and sham for each group. Data presented as mean +/- SD. *p<0.05, **p<0.01, ***p<0.001. ns=not significant. In panels A and B, samples 1-9 are as follows: 1. WT sham, 2. hsp110-/- sham, 3. hsp70-/- sham, 4. WT TBI, 5. hsp110-/- TBI, 6. hsp70i-/- TBI, 7. WT TBI + Celastrol, 8. hsp110-/- TBI+ Celastrol, 9. hsp70i-/- TBI + Celastrol. In panels A-B, n=3 mice per group.
These data indicate that hsp110 and hsp70i-deficient brain cells are more sensitive to TBI and express elevated levels of p53 target genes, and these mice are not protected following TBI and Celastrol treatment.
To further investigate the mechanisms underlying the increased sensitivity of hsp110 and hsp70i knockout mice to TBI relative to WT, we performed qRT-PCR of a set of p53-induced genes (Pigs) that are known to be activated by reactive oxygen species (ROS) (Abbas, Maccio et al. 2010). Examining the mRNA expression levels of Pig1, Pig8 and Pig12 whose expression are enhanced by ROS, we found that the expression level of these genes were mainly induced in hsp110 and hsp70-deficient brain tissues, and not in WT brain tissue following TBI. Furthermore, Celastrol treatment was ineffective in reducing the levels of Pigs in hsp110 or hsp70-deficient brain tissue. Indeed, with the exception of Pig8 in the case of Hsp110, there was an enhancement of Pig1, Pig8 and Pig12 mRNA expression levels in hsp110 and hsp70i-deficient mice treated with TBI and Celastrol.
These data indicate that the increased brain tissue damage following TBI in hsp110 and hsp70-deficient mice may be due to an enhanced ROS production and expression of p53-driven ROS-induced genes.
Discussion
TBI imposes a significant burden of harm and mortality. TBI has short-term effects, such as mechanical injury, that lead to edema and death of neurons, glia and astrocytes and long-term consequences that are associated with a variety of chemical and metabolic alterations, such as production of chemokines, pro-inflammatory cytokines, as well as hypothalamic-pituitary dysfunction that leads to neurological problems (Kumar and Loane 2012, Woodcock and Morganti-Kossmann 2013). Clinical trials for the treatment of TBI thus far have proven unsuccessful (Maas, Roozenbeek et al. 2010), and better treatment strategies are needed for this devastating condition.
Hsps are induced following exposure of the cells to a variety of stress conditions. Enhanced expression of Hsps lead to inhibition of protein aggregation in a number of neurodegenerative model systems as well as following exposure of mice to ischemic reperfusion and TBI (Hartl, Bracher et al. 2011, Kim, Kim et al. 2013). Hsp70/Hsp110 and Hsp40 are involved in protein folding and linking the protein folding to Hsp90 and its co-chaperones for further folding or degradation through the UPS (Hartl, Bracher et al. 2011). In terms of apoptosis, Hsp70 has been shown to reduce the Fas-induced apoptotic response. Hsp70 also binds to JNK, and inhibition of its kinase activity is protective to apoptotic cells (Gabai, Mabuchi et al. 2002). Absence of Hsp70i also leads to increased brain hemorrhage and increased expression of matrix metalloproteinases that are involved in break down of extracellular matrix worsening the brain damage (Kim, Kim et al. 2013). In addition, Hsp70 can inhibit apoptosis by directly binding to Apaf1, preventing the formation of the apoptosome (Beere, Wolf et al. 2000). Formation of an apoptosome complex leads to the activation of caspase 3 and apoptosis. The expression of pro-apoptotic p53 gene has been shown to be up-regulated following brain trauma (Napieralski, Raghupathi et al. 1999). p53 target genes such as proapoptotic genes, Puma and Bax induce neuronal cell death. Analyses of post-seizure injury in mice indicate that the induction of Puma correlates best with p53-induced cell death (Engel, Gomez-Villafuertes et al. 2012). Using Hsp70 inducers to increase the Hsp70 levels was shown to reduce p53 activation, and this was shown to be protective to CA1 neurons (Bonner, Concannon et al. 2010). Following stress stimuli, Hsp70 has been found in the nuclei, and its protective effects have been associated with reduction in ROS-induced DNA damage. We therefore, analyzed p53 target genes that are implicated in ROS production and ROS action 24 hours following TBI in WT, hsp70 and hsp110-deficient brain tissues (Figure 6). We found elevated levels of p53-induced genes (Pigs) in the injured brain in the absence of Hsp70i and less significantly, in the injured hsp110-deficient brain tissue. Interestingly, WT mice did not show an increase in the expression of Pigs at 24 hours post-TBI. In our studies, we tested the mRNA expression levels of Pig1, Pig 8 and Pig 12. Pig1 is a member of galectin family and enhances superoxide generation (Liu, Hsu et al. 1995). Pig 8 also generates ROS following treatment of cells with etoposide (Lehar, Nacht et al. 1996). Pig 12 is a homolog of microsomeal glutathione transferase and participates in redox reactions (Lee and DeJong 1999, Abbas, Maccio et al. 2010). Our data indicate that ROS production may be one reason for the increased sensitivity of hsp70i and hsp110 -deficient mice to TBI. Interestingly, Celastrol was in most cases ineffective to protect the hsp70i- and hsp110-deficient brain tissue with respect to lowering p53 levels, or lowering the expression of p53 target genes, suggesting that the protective effect of Celastrol could be at least in part through pathways associated with Hsp70 expression. Although we did not find upregulation of ROS-induced p53 targets (Pigs) in WT mice at 24 hours post TBI, the ROS-induced cell death in WT mice following TBI cannot be excluded. Further experiments are needed to determine the kinetics of ROS-induced p53- target genes in WT and knockout mouse lines which may show enhanced expression of Pigs also in the WT mice at later time points following TBI.
Since there are limited data available regarding the role of Hsp110 in the CNS protection against injury, we performed gene expression profiling of WT and hsp110-/- brain tissue following exposure of mice to TBI. Data suggest altered gene expression in the critical pathways of inflammatory response, response to stress and wounding as well as other critical pathways (Figure S1 and Table S4). We found reduction in GH in hsp110-/- brain tissue and serum and reduction in GH has been noted in patients following TBI, and administration of GH to TBI patients improves cognition (Garcia-Aragon, Lobie et al. 1992, Devesa, Reimunde et al. 2012). We also found alterations in the expression of chemokines, CCL2 and CCL3 in the brain tissue following TBI. CCL2 has been shown to stimulate proliferation and differentiation of neurons from bone marrow-derived mesenchymal stem cells that are candidate for neurogenesis (Lee, Kang et al. 2013), and CCL3 (and CCL2) are generated by microglia during cuprizone-induced demyelination (Buschmann, Berger et al. 2012). Lif1 is an essential factor for embryonic and adult neurogenesis as well, and it can stimulate neuronal stem/progenitor cells proliferation following ischemia (Covey and Levison 2007).
Furthermore, to examine the usefulness of increasing the levels of Hsps following CCI to reduce brain injury, in this study, we selected two drugs that are known Hsp70/Hsp110 inducers to determine if they can protect early events occurring in the brain following CCI. We found that Celastrol treatment of mice post-TBI significantly protects brain tissue following TBI. Celastrol is a Hsp70 inducer and a natural product from Celastrace family of plants and a proteasome inhibitor and has been shown to reduce the inflammatory response through inhibition of NFkB and inflammatory cytokines such as TNFα and IL6 (Li, He et al. 2012). Celastrol has been shown to be protective against cerebral ischemia, Alzheimer’s disease and SLE as well as rheumatoid arthritis (Li, He et al. 2012). The triterpenoid Celastrol and other specific structural analogues were originally discovered through screen for drugs to treat ALS and Huntington’s disease (Westerheide, Bosman et al. 2004). Celastrol inhibits the neurotoxicity associated with MPTP and 3-nitropropionic acid–induced neurodegeneration in mice (Trott, West et al. 2008). BGP-15 is a pharmacological inducer of Hsp70 and has been shown to confer protection against obesity-induced insulin resistance and it is in the phase II clinical trials for treatment of diabetes. Treatment of mice with BGP-15 results in elevation of Hsp70i in the diaphragm muscle of mdx mouse model (dystrophin-null muscular dystrophy) leading to reduction in the creatine kinase and an increase in the muscle strength and extended the life span (Gehrig, van der Poel et al. 2012). BGP-15 also prevents metabolic side effects of the atypical antipsychotics. BGP-15 induction of Hsp expression is presumably via Rac1 signaling and inhibition of Hsf1 acetylation lengthening the activity of Hsf1 thereby increasing the expression of all stress-inducible Hsp levels (e.g., Hsp110, Hsp70i) (Literati-Nagy, Kulcsar et al. 2009, Gombos, Crul et al. 2011). BGP-15 also affects ischemic reperfusion injury in the heart through PARP activity (Szabados, Literati-Nagy et al. 2000). Hydroxylamines (HA) such as BGP-15 more specifically impact stressed cells rather than the non-stressed cells. Hydroxylamine derivatives such as Bimoclomol, Arimoclomol and BRX-220 are useful in treating wound healing a complication induced by diabetes in rats and they delay progression of ALS. The NG094 which is another HA derivative reduces poly Q-dependent paralysis in C. elegans and other models (Gombos, Crul et al. 2011, Haldimann, Muriset et al. 2011).
In conclusion, our results show that deletion of Hsp70 and Hsp110 in mice increases brain injury following CCI. We show that p53-induced genes (Pigs) that are elevated following increase in oxidative stress and ROS production may potentially be the reason for the increased sensitivity of hsp70i and hsp110 -deficient mice to CCI compared to WT. In addition, we show that both Celastrol and BGP-15 given to mice following TBI protects the brain tissue against cellular apoptosis, reduce inflammatory cell infiltration and gliosis, and increases Ki-67 positive cells and improves behavior. It is anticipated that a transient increase in the level of Hsps soon after TBI may be beneficial to reduce TBI pathology.
Supplementary Material
Acknowledgments
This work was supported by VA Merit Award 1I01BX000161 and in part by NIH grants CA062130, CA132640 (NFM), CA121951 (DM) and NS065172 (KMD). The funding agencies had no role in the design, data collection or analysis of the data, decision to publish, or preparation of the manuscript. The authors wish to thank Christopher Middleton for performing the MRI at the GRU Core Imaging Facility for Small Animals (CIFSA) and Integrative Genomic Core at GRU.
Footnotes
Authors have no conflicts of interest. The data presented do not represent the view of the Veteran Administration.
Contributor Information
Binnur Eroglu, Email: Beroglu@gru.edu.
Donald E. Kimbler, Email: d.kimbler@neu.edu.
Junfeng Pang, Email: Jpang@gru.edu.
Justin Choi, Email: JEChoi@gru.edu.
Demetrius Moskophidis, Email: Dmoskofidis@gru.edu.
Krishnan M. Dhandapani, Email: KDahandapani@gru.edu.
Nahid F. Mivechi, Email: nmivechi@gru.edu.
References
- Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, You MJ, Hirschi KK, Lozano G. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7(5):606–617. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck IM, Drebert ZJ, Hoya-Arias R, Bahar AA, Devos M, Clarisse D, Desmet S, Bougarne N, Ruttens B, Gossye V, Denecker G, Lievens S, Bracke M, Tavernier J, Declercq W, Gevaert K, Vanden Berghe W, Haegeman G, De Bosscher K. Compound A, a selective glucocorticoid receptor modulator, enhances heat shock protein Hsp70 gene promoter activation. PLoS One. 2013;8(7):e69115. doi: 10.1371/journal.pone.0069115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2(8):469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Bonner HP, Concannon CG, Bonner C, Woods I, Ward MW, Prehn JH. Differential expression patterns of Puma and Hsp70 following proteasomal stress in the hippocampus are key determinants of neuronal vulnerability. J Neurochem. 2010;114(2):606–616. doi: 10.1111/j.1471-4159.2010.06790.x. [DOI] [PubMed] [Google Scholar]
- Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J Mol Neurosci. 2012;48(1):66–76. doi: 10.1007/s12031-012-9773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AJ, D’Esposito M. Traumatic brain injury: from bench to bedside [corrected] to society. Neuron. 2010;66(1):11–14. doi: 10.1016/j.neuron.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Covey MV, Levison SW. Leukemia inhibitory factor participates in the expansion of neural stem/progenitors after perinatal hypoxia/ischemia. Neuroscience. 2007;148(2):501–509. doi: 10.1016/J.NEUROSCIENCE.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devesa J, Reimunde P, Devesa P, Barbera M, Arce V. Growth hormone (GH) and brain trauma. Horm Behav. 2013;63:331–344. doi: 10.1016/j.yhbeh.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, Miras-Portugal MT, Henshall DC, Diaz-Hernandez M. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2012;26(4):1616–1628. doi: 10.1096/fj.11-196089. [DOI] [PubMed] [Google Scholar]
- Eroglu B, Moskophidis D, Mivechi NF. Loss of Hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid beta. Mol Cell Biol. 2010;30(19):4626–4643. doi: 10.1128/MCB.01493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabai VL, Mabuchi K, Mosser DD, Sherman MY. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol Cell Biol. 2002;22(10):3415–3424. doi: 10.1128/MCB.22.10.3415-3424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Aragon J, Lobie PE, Muscat GE, Gobius KS, Norstedt G, Waters MJ. Prenatal expression of the growth hormone (GH) receptor/binding protein in the rat: a role for GH in embryonic and fetal development? Development. 1992;114(4):869–876. doi: 10.1242/dev.114.4.869. [DOI] [PubMed] [Google Scholar]
- Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, Lynch GS. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature. 2012;484(7394):394–398. doi: 10.1038/nature10980. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombos I, Crul T, Piotto S, Gungor B, Torok Z, Balogh G, Peter M, Slotte JP, Campana F, Pilbat AM, Hunya A, Toth N, Literati-Nagy Z, Vigh L, Jr, Glatz A, Brameshuber M, Schutz GJ, Hevener A, Febbraio MA, Horvath I, Vigh L. Membrane-lipid therapy in operation: the HSP co-inducer BGP-15 activates stress signal transduction pathways by remodeling plasma membrane rafts. PLoS One. 2011;6(12):e28818. doi: 10.1371/journal.pone.0028818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann P, Muriset M, Vigh L, Goloubinoff P. The novel hydroxylamine derivative NG-094 suppresses polyglutamine protein toxicity in Caenorhabditis elegans. J Biol Chem. 2011;286(21):18784–18794. doi: 10.1074/jbc.M111.234773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Homma S, Jin X, Wang G, Tu N, Min J, Yanasak N, Mivechi NF. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27(30):7974–7986. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang L, Mivechi NF, Moskophidis D. Insights into regulation and function of the major stress-induced hsp70 molecular chaperone in vivo: analysis of mice with targeted gene disruption of the hsp70.1 or hsp70.3 gene. Mol Cell Biol. 2001;21(24):8575–8591. doi: 10.1128/MCB.21.24.8575-8591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys I, Wood RL, Phillips CJ, Macey S. The costs of traumatic brain injury: a literature review. Clinicoecon Outcomes Res. 2013;5:281–287. doi: 10.2147/CEOR.S44625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, Pandita TK. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol. 2004;24(2):899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januchowski R, Zawierucha P, Andrzejewska M, Rucinski M, Zabel M. Microarray-based detection and expression analysis of ABC and SLC transporters in drug-resistant ovarian cancer cell lines. Biomed Pharmacother. 2013;67(3):240–245. doi: 10.1016/j.biopha.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11(5):361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim N, Zheng Z, Lee JE, Yenari MA. The 70 kDa heat shock protein protects against experimental traumatic brain injury. Neurobiol Dis. 2013;58C:289–295. doi: 10.1016/j.nbd.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MG, Jung Cho E, Won Lee J, Sook Ko Y, Young Lee H, Jo SK, Cho WY, Kim HK. The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4CD25Foxp3 regulatory T cells in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2014;85:62–71. doi: 10.1038/ki.2013.277. [DOI] [PubMed] [Google Scholar]
- Kimbler DE, Shields J, Yanasak N, Vender JR, Dhandapani KM. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PLoS One. 2012;7(7):e41229. doi: 10.1371/journal.pone.0041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Laird MD, Sukumari-Ramesh S, Swift AE, Meiler SE, Vender JR, Dhandapani KM. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? J Neurochem. 2010;113(3):637–648. doi: 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Kang JE, Lee JK, Bae JS, Jin HK. Bone-marrow-derived mesenchymal stem cells promote proliferation and neuronal differentiation of Niemann-Pick type C mouse neural stem cells by upregulation and secretion of CCL2. Hum Gene Ther. 2013;24(7):655–669. doi: 10.1089/hum.2013.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, DeJong J. Microsomal GST-I: genomic organization, expression, and alternative splicing of the human gene. Biochim Biophys Acta. 1999;1446(3):389–396. doi: 10.1016/s0167-4781(99)00112-8. [DOI] [PubMed] [Google Scholar]
- Lehar SM, Nacht M, Jacks T, Vater CA, Chittenden T, Guild BC. Identification and cloning of EI24, a gene induced by p53 in etoposide-treated cells. Oncogene. 1996;12(6):1181–1187. [PubMed] [Google Scholar]
- Li Y, He D, Zhang X, Liu Z, Dong L, Xing Y, Wang C, Qiao H, Zhu C, Chen Y. Protective effect of celastrol in rat cerebral ischemia model: Down-regulating p-JNK, p-c-Jun and NF-kappaB. Brain Res. 2012;1464:8–13. doi: 10.1016/j.brainres.2012.04.054. [DOI] [PubMed] [Google Scholar]
- Literati-Nagy B, Kulcsar E, Literati-Nagy Z, Buday B, Peterfai E, Horvath T, Tory K, Kolonics A, Fleming A, Mandl J, Koranyi L. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: a proof of concept randomized double-blind clinical trial. Horm Metab Res. 2009;41(5):374–380. doi: 10.1055/s-0028-1128142. [DOI] [PubMed] [Google Scholar]
- Liu FT, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson WR., Jr Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am J Pathol. 1995;147(4):1016–1028. [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7(1):115–126. doi: 10.1016/j.nurt.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napieralski JA, Raghupathi R, McIntosh TK. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Brain Res Mol Brain Res. 1999;71(1):78–86. doi: 10.1016/s0169-328x(99)00155-2. [DOI] [PubMed] [Google Scholar]
- Narayan N, Lee IH, Borenstein R, Sun J, Wong R, Tong G, Fergusson MM, Liu J, Rovira, Cheng HL, Wang G, Gucek M, Lombard D, Alt FW, Sack MN, Murphy E, Cao L, Finkel T. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature. 2012;492(7428):199–204. doi: 10.1038/nature11700. [DOI] [PubMed] [Google Scholar]
- Plesnila N, von Baumgarten L, Retiounskaia M, Engel D, Ardeshiri A, Zimmermann R, Hoffmann F, Landshamer S, Wagner E, Culmsee C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007;14(8):1529–1541. doi: 10.1038/sj.cdd.4402159. [DOI] [PubMed] [Google Scholar]
- Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133(6):1068–1079. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Qi D, Liu H, Niu J, Fan X, Wen X, Du Y, Mou J, Pei D, Liu Z, Zong Z, Wei X, Song Y. Heat shock protein 72 inhibits c-Jun N-terminal kinase 3 signaling pathway via Akt1 during cerebral ischemia. J Neurol Sci. 2012;317(1-2):123–129. doi: 10.1016/j.jns.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, Potts MB, Fike JR. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp Neurol. 2006;202(1):189–199. doi: 10.1016/j.expneurol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2(8):476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci Biobehav Rev. 2012;36(5):1376–1381. doi: 10.1016/j.neubiorev.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Szabados E, Literati-Nagy P, Farkas B, Sumegi B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochem Pharmacol. 2000;59(8):937–945. doi: 10.1016/s0006-2952(99)00418-9. [DOI] [PubMed] [Google Scholar]
- Trott A, West JD, Klaic L, Westerheide SD, Silverman RB, Morimoto RI, Morano KA. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol Biol Cell. 2008;19(3):1104–1112. doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupper DE, Wallace RB. Utility of the neurological examination in rats. Acta Neurobiol Exp (Wars) 1980;40(6):999–1003. [PubMed] [Google Scholar]
- Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279(53):56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Terry AV., Jr Variable maternal stress in rats alters locomotor activity, social behavior, and recognition memory in the adult offspring. Pharmacol Biochem Behav. 2013;104:47–61. doi: 10.1016/j.pbb.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Xiong X, Ouyang Y, Barreto G, Giffard R. Heat shock protein 72 (Hsp72) improves long term recovery after focal cerebral ischemia in mice. Neurosci Lett. 2011;488(3):279–282. doi: 10.1016/j.neulet.2010.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Faden AI, Loane DJ, Lipinski MM, Sabirzhanov B, Stoica BA. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1897–1908. doi: 10.1038/jcbfm.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.