

Abstract
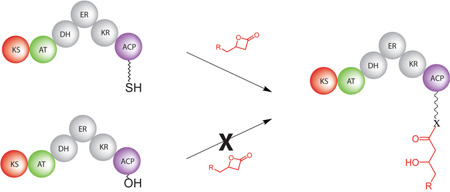
As the key component of many biosynthetic assemblies, acyl-carrier proteins offer a robust entry point for introduction of small molecule probes and pathway intermediates. Current labeling strategies primarily rely on modifications to the phosphopantetheine cofactor or its biosynthetic precursors followed by attachment to the apo form of a given carrier protein. As a greatly simplified alternative, direct and selective acylation of holo-acyl-carrier proteins using readily accessible β-lactones as electrophilic partners for the phosphopantetheine-thiol has been demonstrated.
Site-specific labeling of intact proteins remains a formidable challenge largely due to the multitude of potential nucleophilic sites in most biomacromolecules. β-lactones have received significant attention as electrophilic tagging agents capable of reacting with typical amino acid functionalities.1, 2 Our interest in polyketide biosynthesis has spurred questions of whether these structures might be used to directly label acyl carrier proteins (ACPs) via the phosphopantetheine (Ppant)-thiol. β-hydroxythioesters, formed by addition of the thiol to a β-lactone, are common components of polyketide intermediates rendering this strategy particularly appealing.
Acyl carrier proteins, the primary point of attachment for the growing polyketide chain, are inarguably the work-horse of modular polyketide synthases (PKSs).3 They must recognize and properly associate with ketosynthase (KS), acyl transferase (AT), ketoreductase (KR), dehydratase (DH), enoyl reductase (ER), and in some cases, thioesterase (TE) domains during polyketide maturation. To function, each apo-ACP is posttranslationally modified with a coenzyme A-derived PPant group to produce holo-ACP. The specific transferases responsible for this transformation have continually displayed broad tolerance for acyl groups on the terminal thiol.4 As a result, several groups have taken advantage of this for loading ACPs with a variety of substrates. However, we reasoned that the Ppant group may be uniquely reactive, given an appropriate electrophilic partner, and therefore, targetable for direct acylation. As we ideally wished to maintain polyketide-like functionality in the product, β-lactones seemed most suitable. The work described herein provides a greatly simplified alternative to traditional means of selectively loading ACPs.
The simplest test of selective Ppant acylation is to expose our reagents to isolated holo- and apo-ACP domains. Successful agents will exhibit selectivity for the holo structures, without non-specific acylation of the apo ones. To begin assessing our direct acylation approach, an alkyne-bearing β-lactone, compound 1 (Figure 1), was prepared according to the method of Nelson and coworkers.5 Isolated ACP domains from modules 2 (ACP2) and 3 (ACP3) of the 6-deoxyerythronolide B synthase (DEBS) were overexpressed and purified in both apo and holo forms using BL21 and BAP1 cells, repectively.
Figure 1.

Structures of β-Lactones prepared for direct acylation of acyl-carrier proteins
Of course, to be truly useful, our probes must avoid reaction with nucleophiles present on other common PKS domains. Since KS active site cysteines have been previously shown to be competent nucleophiles for β-lactone ring opening6, a KS-AT didomain from DEBS module 6 (KS-AT6) was overexpressed and purified as previously reported.7 We were encouraged by the previous report of Sieber and coworkers where minimal reactivity between compound 1 and the KS domain of bacterial fatty acid synthase was observed.6 It remained to be seen, however, whether the purified DEBS KS domain would behave in a similar fashion. The stage was now set to examine both efficiency and selectivity of ACP-acylation with β-lactones.
Tandem proteolysis-mass spectrometry has been shown previously to be a powerful means of detecting PKS-bound small molecules in vitro.8 Thorough trypsinolysis of apo- and holo-ACPs produced a series of peptide fragments including one containing the conserved DSL motif, to which the Ppant is attached in samples derived from BAP1 as determined by LC-MS (see supporting information). Incubation of holo-ACP2 and 3 with varied equivalents of 1 at pH 7 for 1 hour followed by exhaustive trypsinolysis and LC-MS, to determine the fraction acylated, produced saturation curves which indicated that a roughly 50-fold excess of lactone is required for complete acylation (Figure 2). Gratifyingly, both apo-ACPs showed no acylation with 50 equivalents of lactone, providing further evidence that the reaction is occuring on the Ppant arm as this is the only difference between the apo and holo forms of ACP (see supporting information). The analogous saturation curve for KS-AT6 showed 25–50% less acylation than ACP for the range of lactone equivalents tested. Most importantly, at 10 lactone equivalents, the ACPs were approximately 35–50% loaded while the KS was mostly unreactive after 1 hour incubation. Standard deviations for these experiments were generally within 1–4% suggesting that the differential reactivity observed was genuine.
Figure 2.

Saturation curves as determined by tandem proteolysis-mass spectrometry for ACP3 (Yellow), ACP2 (Blue) and KS-AT6 (red) with compound 1. Equivalents of lactone are per protein molecule.. Each data point is the average of three experiments. Standard deviations are shown as black error bars. Lines are added for clarity.
While tandem proteolysis/LC-MS confirms acylation of the PPant group, not all proteolysis products readily ionize under the conditions used. In addition, we could not rule out the potential for acylation of the peptide fragments over the intact protein. Therefore, an alternative, gel-based assay was designed according to previous work by Sieber and coworkers.6 Each ACP (apo and holo) and KS-AT6 was incubated for 1 hour with 10 equivalents of 1 followed by 1,3-dipolar cycloaddition (“click”) reaction with rhodamine azide.9 SDS-PAGE analysis of the resulting mixtures showed bright fluorescent bands corresponding to holo-ACP2 and 3 while the apo-ACPs and KS-AT6 displayed only background fluorescence (Figure 3). Not only does this provide initial indication that β-lactones can be selective for ACP over KS but other potential competing residues such as surface cysteines, serines, and lysines can also be avoided.
Figure 3.

PAGE analysis of DEBS ACP3 (apo and holo), ACP2 (apo and holo), KS-AT3 and KS-AT6 reaction with compound 1 and subsequent click reaction with rhodamine-azide. Lanes are marked above with the corresponding protein component. Markers to the left indicate expected bands for the indicated species. The right two lanes depict transfer of the acylation product from preloaded ACP to KSAT indicating that the intermediates are functional. β-lactone was applied at 10× relative to protein unless otherwise noted. ACP2 contains a C-terminal linker region accounting for its larger mass.
To examine the functionality of the acyl-ACP intermediates and provide evidence of thioester over thioether formation, preloaded ACP2 and ACP3(10× compound 1) were mixed with KS-AT6. After one hour, the click reaction was performed as before and the proteins separated by gel electrophoresis. Bright bands corresponding to the KS-AT6 didomain indicate that the β-hydroxythioester is efficiently transferred from the ACPs (Figure 3, right). It should be noted that direct KS-acylation is not observed under the conditions used in these experiments (Figure 3, left)
To examine β-lactone behavior with the next level of complexity, we overexpressed and purified apo and holo forms of the intact module 2 from spinosyn synthase (SpnB). This particular module was chosen because it contains KS, AT, DH, ER, KR, and ACP domains which represent the vast majority of synthase components present in a given assembly. Both apo and holo forms of spinosyn module 2 were incubated for 1 hour with 10, 50, and 75 equivalents of 1 followed by “click” reaction with rhodamine azide and PAGE analysis as before (Figure 4). As before, the only difference between apo and holo forms was the absence or presence of the Ppant group, respectively. To our satisfaction, only the holo-protein showed significant fluorescence indicating that Ppantthiol is the only competent partner for acylation amongst a multitude of alternative nucleophiles. Interestingly, minimal acylation of the apo-form was observed even at very high equivalents of lactone.
Figure 4.

SDS-PAGE analysis of SpnB (apo and holo) reaction with 10, 50, and 75 equivalents of compound 1 and subsequent click reaction with rhodamine-azide. Lanes are marked above with the corresponding protein component and equivalents of lactone.
With increased confidence that tandem proteolysis-mass spectrometry provides an accurate means of evaluating ACP and KS acylation, we began examination of a diverse panel of β-lactone structures. Compounds 2–5 were added to holo-ACP2 and 3 as well as KS-AT6 at 10 and 50 fold excess. Proteolysis and LC-MS anaylsis were performed after 1 hour incubation as above. Acylation efficiencies, calculated as percent loading, were obtained from peak heights of acylated peptides divided by the total peptide (acylated plus unreacted) peak heights for a given LC-MS run (Table 1).
Table 1.
Calculated % loading for ACP2, ACP3, and KS-AT6 with compounds 1–5 at 10 and 50 lactone equivalents.
| Compound | Lactone Equiv. |
ACP2 | ACP3 | KSAT6 |
|---|---|---|---|---|
| 1 | 10 | 33% | 55% | 7% |
| 50 | 93% | 97% | 64% | |
| 2 | 10 | 78% | 64% | 26% |
| 50 | 97% | 97% | 65% | |
| 3 | 10 | 53% | 62% | 41% |
| 50 | 96% | 94% | 62% | |
| 4 | 10 | 8% | 13% | 6% |
| 50 | 32% | 36% | 14% | |
| 5 | 10 | 7% | 15% | 27% |
| 50 | 28% | 35% | 58% |
While all of the compounds tested acylate both ACP and KS to some degree, an interesting trend emerges from this study. The monosubstituted, alkyl-lactones, compounds 1–3, are quite reactive toward both ACPs tested. In contrast, the disubstituted lactone, compound 5, shows little ACP-acylation activity even at 50 equivalents. KS-AT6, on the other hand, is more reactive than ACP2 and 3 toward compound 5 but significantly less reactive with compounds 1–3 than the ACPs. Finally, the monosubstitued, aromatic-lactone, compound 4, reacts slugishly with both ACP and KS. These data offer promising preliminary evidence that β-lactones can provide an efficient and selective means of acylating carrier proteins. The differential reactivities observed for ACP versus KS suggests that future optimization of ACP-selectivity should focus on monosubstituted lactones. Efforts to further explore this structure-selectivity relationship are currently underway in our laboratory.
In summary, we have successfully demonstrated direct acylation of isolated holo-ACP domains with functionalized β-lactones. The apo form of this ACP is unreactive suggesting that β-hydroxythioester formation takes place on the Ppant arm of mature carrier proteins. In addition, ACPs can be selectively acylated in the presence of competing PKS active sites. This method offers a synthetically straightforward means of loading key PKS components with choice substrates.
Supplementary Material
Acknowledgment
This work was supported by start-up funding from the University of Massachusetts, Amherst (to N.A.S). The authors would like to thank Professor Chaitan Khosla (Stanford University) for the generous gifts of BAP1, pVYA05, pNW06, and pAYC11. We also thank Professors Alan Kennan (Colorado State University) and James Chambers (University of Massachusetts) for helpful discussions.
Footnotes
Supporting Information Available. Detailed descriptions of synthetic, molecular biology and analytical procedures. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Drahl C, Cravatt BF, Sorensen EJ. Angew. Chem. Int. Ed. 2005;44:5788. doi: 10.1002/anie.200500900. 1. [DOI] [PubMed] [Google Scholar]; (b) Kim DH, Park JI, Chung SJ, Park JD, Park NK, Han JH. Bioorg. Med. Chem. 2002;10:2553. doi: 10.1016/s0968-0896(02)00108-6. [DOI] [PubMed] [Google Scholar]
- 2.Bottcher T, Sieber SA. J. Am. Chem. Soc. 2008;130:14400. doi: 10.1021/ja8051365. [DOI] [PubMed] [Google Scholar]
- 3.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu. Rev. Biochem. 2007;76:195. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 4.(a) Mercer AC, Meier JL, Torpey JW, Burkart MD. ChemBioChem. 2009;10:1091. doi: 10.1002/cbic.200800838. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kapur S, Worthington A, Tang Y, Cane DE, Burkart MD, Khosla C. Bioorg. Med. Chem. Lett. 2008;18:3034. doi: 10.1016/j.bmcl.2008.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Worthington AS, Rivera H, Torpey JW, Alexander MD, Burkart MD. ACS Chem. Biol. 2006;1:687. doi: 10.1021/cb6003965. [DOI] [PubMed] [Google Scholar]; (d) Meier JL, Mercer AC, Rivera H, Burkart MD. J. Am. Chem. Soc. 2006;128:12174. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]; (e) Clarke KM, Mercer AC, La Clair JJ, Burkart MD. J. Am. Chem. Soc. 2005;127:11234. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]; (f) Sieber SA, Walsh CT, Marahiel MA. J. Am. Chem. Soc. 2003;125:10862. doi: 10.1021/ja0361852. [DOI] [PubMed] [Google Scholar]
- 5.Nelson SG, Wan Z, Peelen TJ, Spencer KL. Tetrahedron Lett. 1999;40:6535–6539. [Google Scholar]
- 6.Bottcher T, Sieber SA. Angew. Chem. Int. Ed. 2008;47:4600. doi: 10.1002/anie.200705768. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kim C-Y, Alekeyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]; (b) Chen AY, Schnarr NA, Kim C-Y, Cane DE, Khosla C. J. Am Chem. Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Schnarr NA, Chen AY, Cane DE, Khosla C. Biochemistry. 2005;44:11836. doi: 10.1021/bi0510781. [DOI] [PubMed] [Google Scholar]; (b) Hicks LM, O'Connor SE, Mazur MT, Walsh CT, Kelleher NL. Chem. Biol. 2004;11:327. doi: 10.1016/j.chembiol.2004.02.021. [DOI] [PubMed] [Google Scholar]; (c) Hong H, Appleyard AN, Siskos AP, Garcia-Bernardo J, Staunton J, Leadlay PF. FEBS J. 2005;272:2373. doi: 10.1111/j.1742-4658.2005.04615.x. [DOI] [PubMed] [Google Scholar]
- 8.(a) Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (b) Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (c) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.