

Abstract
Malaria, an infectious disease caused by eukaryotic parasites from the genus Plasmodium, afflicts hundreds of millions of people every year. Both the parasite and its host utilize protein kinases to regulate essential cellular processes. Bioinformatic analyses of parasite genomes predict at least 65 protein kinases, but their biological functions and therapeutic potential are largely unknown. We profiled 1,358 small molecule kinase inhibitors to evaluate the role of both the human and malaria kinomes in Plasmodium infection of liver cells, the parasites’ obligatory but transient developmental stage that precedes the symptomatic blood stage. The screen identified several small molecules that inhibit parasite load in liver cells, some with nanomolar efficacy, and each compound was subsequently assessed for activity against blood stage malaria. Most of the screening hits inhibited both liver and blood stage malaria parasites, which have dissimilar gene expression profiles and infect different host cells. Evaluation of existing kinase activity profiling data for the library members suggests several kinases are essential to malaria parasites, including cyclin-dependent kinases, glycogen synthase kinases, and phosphoinositide-3-kinases. CDK inhibitors were found to bind to Plasmodium protein kinase 5, but it is likely that these compounds target multiple parasite kinases. The dual stage inhibition of the identified kinase inhibitors makes them useful chemical probes and promising starting points for antimalarial development.
Keywords: Plasmodium, malaria, chemical probes, kinase, high-throughput screen
Introduction
Malaria continues to burden large parts of the world despite increased efforts to develop novel therapeutic agents.[1]Among the millions of cases each year there are hundreds of thousands of deaths, mostly in pregnant women and in children under the age of five years.[1] Protozoan parasites from the genus Plasmodium cause this life-threatening disease. Currently there are relatively few effective drugs that can inhibit Plasmodium parasites,[2] and the emergence of drug resistance threatens the efficacy of these therapeutics.[3] Recently drug resistance has emerged to artemisinin, which is the current first-line medication for malaria treatment.[4]To address these problems new antimalarial targets and chemotypes are needed, but our limited understanding of Plasmodium biology and lack of suitable tools to explore this biology hinder development.
The malaria parasite requires both an insect vector and a mammalian host to complete its life cycle.[5] Parasites in their sporozoite form are transmitted to humans by the bite of an infected mosquito. These sporozoites efficiently travel through the dermis and the blood stream with the goal of finding and infecting liver cells. In the host liver cells, the parasites proliferate and develop into merozoites, the form that infects red blood cells and causes malaria’s characteristic symptoms. Comparative proteomics and gene expression profiling have revealed significant differences between liver and blood stage parasites,[6] suggesting that both forms have diverse processes that may be targeted. In fact, antimalarial drug screening focused on either the blood[7] or liver stage[8] typically discovers compounds with stage selectivity. Treating malaria’s symptoms requires drugs with blood stage efficacy, but addressing malaria prevention – and conceivably eradication – requires drugs with liver stage efficacy.
Protein kinases in both the host and parasite represent a large protein family with therapeutic potential but their role in parasite infection remains poorly understood. Unlike human kinases for which a diverse assortment of commercially available biochemical assays are available, only a few assays exist for Plasmodium kinases. Signal transduction pathways in Plasmodium remain to be elucidated and only a few phospho-specific antibodies are available to allow for confirmation of intracellular kinase inhibition. Despite these limitations the importance of kinases in P. falciparum[9] and P. berghei[10] has been demonstrated with genetic approaches. P. falciparum is the deadliest human malaria strain and P. berghei is a more tractable rodent malaria strain that is widely used to investigate parasite biology. While genetic studies with these important malarial species have established several critical parasite kinases (reviewed in[11]), they have focused on the characterization of genetic knockouts, and no systematic study of the malarial host and parasite kinomes using small molecule probes has been reported.
Exploring the Plasmodium kinome with small molecule probes has many advantages. Elucidating the role of protein kinases in vivo in mammalian systems is often complicated by redundancies in protein function, like those observed in the cyclin dependent kinase (CDK) protein family. As a consequence, knocking out or knocking down just one gene may not reveal the role of the protein family. Almost all of the current kinase inhibitors in therapeutic use and under development are able to address this issue because they target multiple kinases with similar binding pockets.[12] Additionally, since many of these compounds hit multiple targets the promiscuity creates a polypharmacological approach[13] to reduce avenues for drug resistance. Finally, many kinase inhibitors are being advanced in clinical trials for a variety of disease indications, and compounds that also have antimalarial activity could be repurposed to reduce the cost and time of drug development. Therefore, we completed a malaria screen with a diverse array of kinase inhibitors, including several compounds in clinical trials, to identify chemical probes to evaluate Plasmodium parasite biology and molecules that may potentially be repurposed.
Results
Here, we report the screening results for a set of 1,358 small molecules that are biased for targeting an array of kinase families. The library was assembled from a collection of publicly disclosed kinase inhibitors developed by Roche and GlaxoSmithKline[14] (~50%) as well as compounds from other companies and in-house synthetic chemistry efforts (Figure S1), to probe the importance of different kinase families in liver stage malaria infection. Many library members are derived from novel inhibitor classes that were publicly disclosed in 2012[14]and these diverse molecules targeted over 30 different kinases. Importantly, while every member of the library had previously been screened against human kinases, to the best of our knowledge such a collection of kinase inhibitors has neither been compiled nor used to evaluate parasitic processes.
Liver stage malaria infection was evaluated in the presence of these compounds using a high-throughput liver stage malaria screen that utilizes parasites derived from live P. berghei-infected mosquitoes to infect human hepatoma HepG2 cells.[8] Actives were defined as molecules that decreased parasite load in hepatocytes by ≥95% at a low micromolar concentration as assessed using transgenic parasites containing a luciferase reporter.[15] Secondary assays included the evaluation of liver stage malaria inhibition with a different human hepatoma cell line (Huh-7 cells) and an alternative parasite detection method that utilized anti-Plasmodium antibodies with immunofluorescence detection.[16]
Active compounds were further characterized to determine dose-dependent activity against liver stage P. berghei. These does response curves were generated for parasite inhibition in both HepG2 and Huh-7 cells and only compounds that inhibited parasite load in both liver cell lines with an IC50 below 10 μM were considered hits. This screen identified 31 compounds as inhibitors of liver stage parasites in vitro with an IC50 less than 10 μM. The 31 screening hits were subsequently tested to determine dose-dependent activity against blood stage P. falciparum Dd2 parasites (discussed below).
Each screening hit was tested for liver cell toxicity with two different assays, one that measured protease activity using a fluorescence reporter (CellTiter-Fluor) and a second luminescence-based method that measured cellular ATP levels (CellTiter-Glo). Dose-response curves were generated to evaluate general toxicity to the host cell as a possible mode of action (Figure S2). All of the determined IC50 values were above 10μM with the exception of Torin2 inhibition of Huh-7 cells (IC50 ~ 3 μM). Torin2 inhibition of liver stage malaria is significantly more potent (0.039 μM) and thus cell toxicity cannot account for the compound’s antimalarial activity. As shown in Figure S2, none of the compounds inhibited liver cell viability (HepG2 or Huh-7 cells) at the concentrations used to study parasite inhibition.
Each member of the reported library has previously been profiled for kinase selectivity data or has been reported to inhibit a specific kinase target. While many kinase inhibitors have activity against multiple kinases, most were designed to target a single kinase family or one specific kinase. The kinase inhibition profiles of the screening hits were used to generate a hypothesis about potential targets (Figure 1). Based on this analysis, the CDK family was most frequently targeted, followed by the phosphatidylinositol 3-kinase-related kinase (PIKK) and glycogen synthase kinase 3 (GSK-3) families. Importantly the tested library was not biased for molecules targeting these kinase families. The Aurora, vascular endothelial growth factor (VEGF), and Raf kinase families were also the primary targets of multiple chemotypes. The discovery of diverse chemical scaffolds that bind to these kinase families and inhibit malaria supports the prediction that their targets are essential to the parasite.
Figure 1.

Target diversity of screening hits. Kinase families that are targeted by more than one library member are shown in the pie chart with the structures of representative inhibitors. Every kinase family shown except the PIM kinases was targeted by diverse chemotypes. Among the identified CDK inhibitors, the relative percent abundance of compounds with previously reported activity against specific human isoforms (CDK 1-7) is shown (white bars).
Dual stage inhibition
Transcriptional and proteomic data suggest significant differences in the biological processes of the various Plasmodium life stages.[6]Screening hits were selected based on their activity against liver stage Plasmodium parasites, and we subsequently generated blood stage P. falciparum Dd2 dose-response curves as described above. These experiments probe the efficacy of the compounds on more than one stage in the Plasmodium lifecycle. Additionally, P. falciparum is a human-infective malaria parasite whereas P. berghei is rodent-infective. Therefore testing for inhibition of P. falciparum not only examines dual stage activity (blood versus liver), but also interrogates activity against different species (human-infective parasites versus mouse-infective parasites).
Only a few of the screening hits were selective for liver stage malaria. Figure 2A shows dose-response curves for the Aurora kinase inhibitor CYC-116, which inhibited liver stage P. berghei significantly more potently than blood stage P. falciparum. However, most of the kinase inhibitors (~75%) reported inhibit blood stage malaria at the same level or more potently than liver stage parasites (Table 1). The representative dose-response curves for the CDK inhibitor SNS-032 are shown in Figure 2B, which was found to be a nonselective inhibitor. To quantitatively evaluate stage selectivity, blood stage IC50 values were divided by liver stage IC50 values. With this analysis ratios > 10 are deemed selective for liver stage malaria and values below 0.1 are selective for blood stage malaria. Figure 2C demonstrates that compounds known to target the PIKK and CDK kinase families do not exhibit stage selectivity.
Figure 2.

Characterization of screening hits. (A) Dose-response curves for P. berghei ANKA liver stage (dashed red curve) and P. falciparum 3D7 blood stage (solid red curve) are shown for CYC-116. (B) Dose-response curves for P. berghei ANKA liver stage (dashed blue curve) and P. falciparum 3D7 blood stage (solid blue curve) are shown for the CDK inhibitor SNS-032. Data in A and B were fit to a nonlinear regression equation (curves shown) to obtain IC50 values. (C) Stage selectivity of known human PIKK and CDK inhibitors for P. falciparum blood stage and P. berghei liver stage parasites determined by dividing the blood stage IC50 by the liver stage IC50. Values above 10 are selective for liver stage malaria and values below 0.1 are selective for blood stage malaria. All compounds that target these families inhibit both parasite stages. (D) Time dependence of CDK 1/2 inhibition. CDK 1/2 at 10 μM (black bars) or DMSO control (white bars) was added to P. berghei-infected liver cells 0 and 24 hours post infection. Parasite signal was read 36 hours post infection.
Table 1.
Anti-Plasmodium activity of kinase inhibitors.[a]
| Compound | Liver stage P. berghei IC50 (μM) |
Blood stage P. falciparum IC50 (μM) |
Primary human kinase target(s) |
|---|---|---|---|
| Torin2 | 0.039 | 0.00086 | mTOR |
| WZ12-073 | 0.37 | 6.7 | EGFR T790M |
| Torin1 | 0.41 | 0.024 | mTOR |
| CYC-116 | 0.46 | 4.5 | Aurora/VEGFR2 |
| CDK 1/2 inhibitor | 0.47 | 0.258 | CDK |
| CYT387 | 0.55 | 1.1 | JAK2 |
| RO0317886 | 0.81 | ND[b] | GSK-3[19] |
| GW805758X | 1.1[c] | ND | CDK/GSK33[20] |
| GW780056X | 1.1[c] | ND | CDK[20] |
| PIK-75 | 1.2[c] | 2.0 | PI3K |
| AS-601245 | 1.2 | 3.9 | JNK |
| GSK650394 | 1.2 | 4.8 | SGK |
| BAY439006 | 1.4 | 8.1 | Raf-1/VEGFR |
| RO0282155 | 1.4 | ND | CDK[21] |
| RO1153853 | 1.5 | ND | MAP[22] |
| Flavopiridol | 1.9[c] | 2.2 | CDK |
| ZSTK474 | 2.6[c] | 0.76[d] | PI3K |
| SNS-032 | 2.7[c] | 3.3 | CDK |
| GW827106X | 3.5 | ND | GSK-3[23] |
| PKC-412 | 4.0 | 0.57 | FLT3 |
| SNS-314 | 4.2 | 3.7 | Aurora |
| CVM-04-70 | 4.4 | > 10 | PIM |
| GSK429286A | 4.7 | 0.83 | ROCK1 |
| GW833373X | 5.5 | ND | GSK-3[23] |
| AT-7519 | 5.6[c] | 0.42 | CDK |
| CVM-04-089 | 5.8 | > 10 | PIM |
| GW405841X | 6.7 | ND | cRaf1[24] |
| SB-610251-B | 6.9 | ND | B-Raf[25] |
| GSK237701A | 7.0 | ND | PLK1[26] |
| GSK619487A | 7.3 | ND | AKT[27] |
| PPY-A | 8.3 | > 10 | Abl |
IC50 determined for P. berghei ANKA (liver stage) and P. falciparum Dd2 (blood stage).
ND, not determined.
Determined with antibody staining.
PubChem AID 2305.
Probing the host cell cycle
As obligate intracellular organisms, malaria parasites depend on specific host processes for survival in addition to their own machinery. The determined Plasmodium IC50 values (0.5 – 6 μM) are significantly greater than the human CDK IC50 values previously determined in whole cell assays (low nanomolar). As a consequence it is predicted that the compounds are binding to human CDKs at effective concentrations, but it is unknown if this binding inhibits malaria parasites.
To probe if CDK inhibitors reduce parasite load via a host target, their activity was measured after incubation with hepatocytes. HepG2 cells will divide every two days under the described culture conditions. Since CDK2 is known to mediate cell division in humans, the inhibition of parasites on a similar time scale would implicate the host target. Liver stage parasite load was reduced with only a 12-hour treatment with the CDK inhibitors (Figure 2D), suggesting that inhibition of parasites does not correlate with the host cell cycle or inhibition of the host cell cycle by these compounds. The mTOR inhibitor Torin1 and the PI3K inhibitor ZSTK474 were also found to inhibit parasites with only a 12-hour treatment with the inhibitors (Figure S3). Both mTOR and PI3K have multiple roles in host cell cycle progression and therefore these results provide further support that inhibition of the host cell cycle is not linked to parasite inhibition.
Molecular docking to PfPK5
To provide direct evidence for a parasite target we sought to determine if the identified CDK inhibitors interact with a parasite protein. Inhibitors of the human CDK family were the most abundant class of compounds identified among the screening hits, and within this group all had activity against human CDK2. In malaria parasites Plasmodium protein kinase 5 (PK5) is most similar to human CDK2 based on sequence analysis.[17] Molecular docking studies were conducted to explore the potential binding of these compounds to P. falciparum PK5 (PfPK5). The crystallographic structure of PfPK5 bound to the CDK inhibitors indirubin-5-sulphonate (1V0O) and purvalanol B (1V0P) has been reported,[18] and this structure was used to dock CDK 1/2 (Figure 3A) and SNS-032 (Figure S4A and S4B) onto the inhibitor binding site. The docking model predicts that residues Gln84 and Asp85 form hydrogen bonds with the thioamide proton and the primary amine proton on aminotriazole of CDK 1/2, respectively. The docking studies also predict that two hydrogen bonds form with SNS-032 and PfPK5, involving residues Ile10 and Leu82. Molecular docking therefore predicts that these compounds bind to PK5, but this hypothesis needed verification with biochemical studies.
Figure 3.

Binding to PfPK5. (A)Structure of CDK 1/2 docked onto the PfPK5 crystal structure (pdb 1V0O). The protein kinase (pink) and CDK 1/2 carbon (grey), hydrogen (grey), nitrogen (blue), sulfur (yellow) and oxygen (red) atoms are shown. Hydrogen bonds are predicted to form (black dotted lines) between the thioamide proton of CDK 1/2 and Gln 84, and between the primary amine proton on aminotriazole and Asp 85 (bond lengths < 2.5 Å). (B) Percent binding of commercially available kinase inhibitors to PfPK5 at 10 μM. Significant binding (> 50%) is only observed with the known CDK inhibitors, flavopiridol, SNS-032 and CDK 1/2, and the PIK inhibitor PIK-75. (C) Competitive binding plot of CDK 1/2 (red curve), flavopiridol (black curve) and SNS-032 (blue curve) to PfPK5 where decrease in relative signal correlates with compound binding. Data in C were fit to a nonlinear regression equation (curves shown) to obtain Kd values.
Inhibitor binding to PfPK5
We next tested our hypothesis that known inhibitors of human CDK2 will have activity against PfPK5. To this end, we profiled all commercially available screening hits in a PfPK5 active site-directed competition binding assay (DiscoveRx). This assay utilizes DNA-tagged purified protein and a bait ligand to capture protein on to a solid support. Protein binding to this support is competed with 10 μM inhibitor, and then bound protein is quantified using PCR.
Only the identified CDK inhibitors and PIK-75 bound to PfPK5 with a Kd< 10 μM(Figure 3B). The CDK inhibitors CDK 1/2, SNS-032 and flavopiridol as well as PIK-75 were tested at additional concentrations to generate a binding curve. Analysis of the results indicates that CDK 1/2 has a Kd of 0.32 μM for PfPK5 (Figure 3C). Flavopiridol and SNS-032 bound to the enzyme with Kd values of 3.0 μM and 7.0 μM, respectively. For both compounds, binding to PfPK5 was on the same order of magnitude as liver stage malaria inhibition. Interestingly, there was significant variability in hit potency of binding to PfPK5 when compared to previously reported inhibition of human CDK2. This observation indicates that these compounds are currently selective for the human enzyme but suggests that agents can be designed to shift selectivity to inhibit the parasite enzyme.
In vivo activity of kinase inhibitors
Cell-based phenotypic assays serve important functions in disease models, but in vivo experiments remain essential to validate activity and address drug development needs. The current work screened for compounds that inhibited liver stage malaria in the hopes of identifying potential prophylactic agents. Therefore to test these compounds in an animal model the drugs were administered before malaria infection. Initial tests with the CDK inhibitors flavopiridol, CDK 1/2, AT-5791 and SNS-032 at a single concentration revealed that SNS-032 had relatively limited toxicity to mice and was more potent at inhibiting liver stage malaria when compared to the other compounds. SNS-032 was further tested at a single oral dose (75 mg/kgb.w.) administered before malaria infection, and was found to reduce parasite load in mouse livers when compared to vehicle controls (Figure 4A). This finding is significant as SNS-032 is currently in clinical trials for patients with either chronic lymphocytic leukemia or multiple myeloma.
Figure 4.
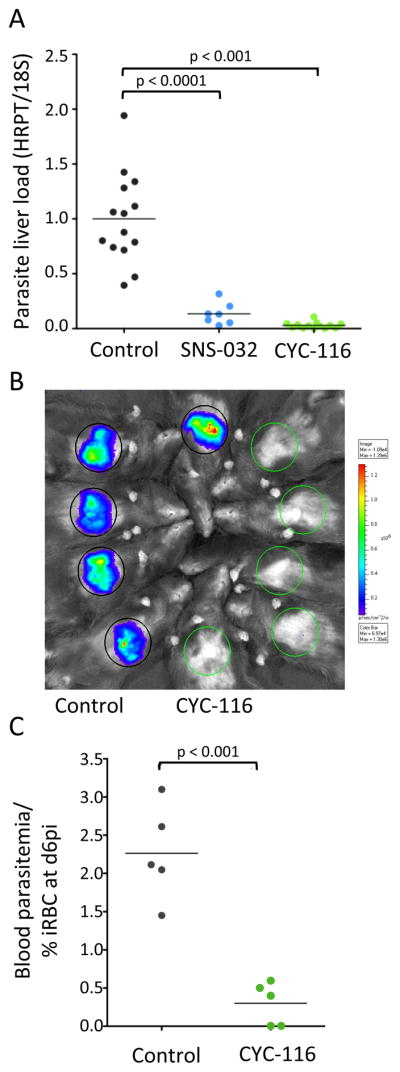
In vivo activity of SNS-032 and CYC-116. (A) Quantitation of relative parasite load in mouse livers after a single treatment by oral administration of 75 mg/kgb.w. SNS-032 (blue), 100 mg/kgb.w. CYC-116 (green), or equivalent amount of vehicle as a control (grey). (B) Whole-animal bioluminescence imaging of P. berghei infected mice livers with (green circles) or without (grey circles) treatment with 100 mg/kgb.w. CYC-116. (C) P.berghei parasitemia in mice after a single treatment by oral administration of 100 mg/kgb.w. CYC-116 (green) or equivalent amount of vehicle as a control (grey).
We decided to test a second screening hit for in vivo efficacy to determine if inhibitors without CDK activity are also effective in a mouse model system. Within the screening hit set, the Aurora kinase inhibitor CYC-116 had the highest selectivity for inhibiting liver stage over blood stage parasites (Figure 2A). CYC-116 is currently in Phase I clinical trials in patients with a range of advanced solid tumors. Due to these interesting properties CYC-116 was chosen for in vivo testing. A single oral treatment of CYC-116 (100 mg/kgb.w.) before malaria infection significantly reduced parasite load in mouse livers when compared to a vehicle control (Figure 4A and 4B). There was also a reduction in P. berghei parasitemia after treatment with CYC-116 when compared to the vehicle control (Figure 4C).
Bioinformatics of Plasmodium kinases
Most of the screening hits were designed to target a single protein or protein family. While many compounds target several kinases they often have one primary target. If the primary target was identified from only one hit chemotype it was not studied further as it was considered to be less robust. For primary targets that were identified with multiple chemotypes, the protein sequences were used in a BLAST search to identify the Plasmodium protein with the greatest similarity. Some primary targets yielded no reasonable BLAST results, such as the mammalian target of rapamycin (mTOR), but several searches to others human proteins identified a parasite protein with homology to the known human target. The parasite proteins that were identified are highlighted on a Plasmodium kinase tree (Figure 5), which was previously constructed.[10]
Figure 5.

Phylogenetic tree of P. berghei kinases. Kinases with homology to human kinases targeted by screening hits are highlighted. Several compounds target human proteins that have no identifiable homolog in Plasmodium parasites based on sequence analysis. Kinase tree replicated with permission from Dr. Oliver Billker.[10]
Discussion
The human kinome consists of more than 530 protein kinases, about 2.5% of the genome, while the malaria kinome has 65 predicted members which represent approximately 1.2% of the parasite genome.[28] Efforts to assign function to malaria proteins have been hampered by a lack of sequence similarity to known proteins, and as a consequence there may be additional Plasmodium kinases. None the less, the current estimate of both parasite and host protein kinases represents a target rich area for therapeutic intervention[11c] that remains to be elucidated. To probe the potential importance of protein kinases in malaria parasite infection, a diverse collection of kinase inhibitors was assembled and used as chemical probes. By screening this biased library of 1,358 kinase inhibitors, 31 compounds were identified as liver stage malaria inhibitors. The relatively high hit rate (2.3%) associated with screening this library underscores the importance of kinases in malaria parasite biology.
Survival of the malaria parasite depends both on its own intracellular pathways and those that occur within the host cell. Since every identified screening hit has a known affinity for host kinases, inhibition of malaria parasites can originate from an interaction with a parasite and/or host factor. None of the identified inhibitors influenced liver cell proliferation at the studied concentration range (Figure S2), but this observation does not exclude an interaction with host kinases. Indeed, many of the kinase inhibitors are known to interact with host kinases at the concentrations used to inhibit liver stage malaria. For example, CDK 1/2 inhibits purified human CDK2 with an IC50 of 0.5 nM and inhibits liver stage malaria with an IC50 of 470 nM. However there are several lines of evidence to suggest that inhibition of the host CDKs is not responsible for the inhibition of malaria parasites (discussed below).
A previously reported RNA interference (RNAi) study of host kinases showed that knockdown of the genes associated with the primary human kinase targets of the hits does not influence parasite infection.[29] The report identified MET, PKCζ, protein kinase lysine deficient 1 (PRKWNK1), serum and glucocorticoid inducible kinase 2 (SGK2), and STK35 as essential host factors for liver stage malaria (P. berghei) infection,[29] but among these MET was later shown not to be essential for all malaria species (P. falciparum and P. yoelii).[30] Thus inhibition of the predicted host targets is an unlikely mode of action for all of the reported screening hits except the SGK inhibitor GSK650394 (Table 1). While an off-target host interaction is possible for the other hits, most of the compounds inhibit the parasites’ blood and liver stages (Table 1) and with the inherent differences between erythrocytes and liver cells nonselective inhibition supports a parasite target that is essential to both stages. This dual stage activity also suggests the importance of the target(s) to both rodent and human parasite species. Based on this reasoning the majority of the compounds are likely to have parasite targets, but this proposal was further tested.
Many compounds that target the PIKK kinase family were identified as potent liver and blood stage malaria inhibitors (Figure 1, Table 1). Half of the inhibitors within this group have mTOR as a primary target, while the other half target phosphatidylinositol kinases (PIKs). Torin2, the most potent molecule identified from the screen, is known to inhibit mTOR with an IC50 value of ~ 2 nM, and at 10-100 fold higher concentrations it inhibits PI3K, ATM and ATR kinases.[31] Torin2 inhibits liver and blood stage malaria parasites with IC50 values of 40 nM and 1 nM (Table 1). Recently is was found that Torin2 also exhibits activity against liver and blood stage P. berghei in a mouse animal model.[32] Inhibition of blood stage parasites is on the same order of magnitude as mTOR inhibition, but liver stage parasite inhibition occurs at a 40-fold higher concentration. This higher IC50 value may be due to an interaction with PI3K, ATM or ATR kinases, or due to compound instability in the presence of metabolically active hepatocytes. There is no parasite protein with homology to mTOR, but a parasite phosphatidylinositol-3 kinase (PI3K) has been identified. The P. falciparum PI3K has been shown to be important for hemoglobin digestion,[33] a necessary function during the parasites’ blood stages. Hemoglobin digestion is a blood stage specific process, but the protein is expressed during the sporozoite form of P. falciparum based on a proteomic study.[34] In support of the role of PI3K during the liver stage, inhibitors of the PIKK family were not stage selective (Figure 2C). In fact Torin1, an mTOR inhibitor, and ZSTK474, a PI3K inhibitor, target a process that is essential for both early and late stages of parasite development in the liver (Figure S3). The expression of PI3K in sporozoites and the inhibition of liver stage parasites by known PI3K inhibitors suggest that the protein has another essential, yet currently undetermined role during the liver stage. Interestingly, the related protein PI4K was recently discovered to be the primary target of imidazopyrazines, a class of potent antimalarials, and this kinase was shown to be critical to blood and liver stage parasites.[35]Therefore the mode of action of these compounds could be through inhibition of PI3K and/or a parasite mTOR-like protein, but this awaits experimental confirmation.
Cyclin dependent kinases play essential roles in eukaryotes including cell division, cell differentiation, cell morphology and transcription. Humans have at least 20 CDKs, while the parasite has 5 predicted CDKs.[28] Based on analysis of the screening data CDK2, or a CDK2-like protein, is the most likely target of the CDK inhibitors because 100% of the hits classified as CDK inhibitors are known to inhibit this isoform (Figure 1). Many of the IC50 values reported for the CDK inhibitors (Table 1) were determined by antibody staining instead of the transgenic luciferase reporter due to complications with the luciferase assay that are specific to inhibitors of this kinase family. Overall several chemically diverse CDK inhibitors were identified as inhibitors of liver stage malaria with a range in potency (mid nM to low μM).
There are several indications that the CDK inhibitors are not functioning through a host target. First, there was no correlation between the previously reported IC50 values of human CDK2 inhibition in cell-based assays and the potency of liver stage malaria inhibition (Figure S4C) and second, knockdown of human CDKs did not significantly inhibit parasite infection of liver cells.[29] Another observation in support of a parasite target is the inhibition of both liver and blood parasite stages for all the identified CDK inhibitors (Figure 2B and 2C). A host-dependent inhibitor would likely have varying activity against parasites within erythrocytes versus liver cells. Additionally, there is a ~1,000-fold difference in potency between compound inhibition of malaria parasites and inhibition of human CDKs, suggesting that binding to host kinases is not responsible for parasite inhibition. Finally, there was no correlation between the host cell cycle and liver stage malaria viability (Figure 2D and Figure S3). Liver stage malaria parasites have a much faster proliferation rate (~10,000x parasites in 2 days) when compared to HepG2 cells. Their extraordinary proliferation rate likely makes processes involved in the parasite’s cell cycle especially susceptible to disruption by small molecules. Liver cells in vivo have a much slower proliferation rate than HepG2 cells which suggests there could be a large therapeutic window for compounds that target the cell cycle.
All of the commercially available screening hits were tested for their ability to bind to the P. falciparum PK5, a predicted cyclin dependent kinase that is homologous to human CDK1 and CDK2. Only four compounds significantly bound to the enzyme at 10 μM and three of these compounds were CDK inhibitors (CDK 1/2, flavopiridol, and SNS-032). This observed selectivity within the screening hits suggests that binding to human CDK1/CDK2 is a predictor for activity against PfPK5, and supports using affinity to human proteins to predict possible parasite targets. While these observations support the proposal that the parasite PK5 is a target for these compounds, there are many factors that suggest these inhibitors have more than one target.
An important feature for future drugs that target malaria kinases will be selectivity for parasite proteins over host homologs. While this work identified several molecules with antimalarial activity in vitro, engineering selectivity remains an important task. The identified CDK inhibitors are currently selective for host proteins, which highlights differences between the two proteins. This selectivity suggests that molecules with parasite selectivity could also be designed. Developing selectivity is challenging, but there have been notable successes: parasite GSK-3[36] and dihydroorotate dehydrogenase.[37]
Confirmation in an animal model is an important validation for the inhibitors identified by this phenotypic screen, as compounds identified with high-throughput screening often fail to exhibit in vivo activity. To test if these molecules exhibit in vivo efficacy two were chosen for experiments in a mouse malaria model system. These drugs were administered before malaria infection to evaluate their potential as prophylactic agents. CDK inhibitors were of particular interest because they represented the class of proteins most abundantly targeted by the screening hits. An initial test with four different CDK inhibitors revealed that SNS-032 had the greatest efficacy and lowest toxicity, which was not unexpected as SNS-032 is in clinical trials and was optimized for favorable pharmacokinetic and pharmacodynamic properties during its development. Here it was observed that a single treatment of 75 mg/kgb.w. before infection reduced parasite load in the liver when compared to the vehicle control (Figure 4A).
Genetic studies have shown that deletion of PK5 in P. berghei produces no obvious phenotype,[10] but the protein is thought to be essential for blood stage proliferation in P. falciparum.[9] We found that SNS-032 inhibits PK5 and exhibits activity against malaria in a mouse model. While these results may seem contradictory, they most likely suggest that the observed phenotype in mice and cell culture is from the interaction of CDK inhibitors with more than one kinase. This proposal may also be supported by the dual stage inhibition exhibited by the identified kinase inhibitors.
In addition to the CDK inhibitors, the drug CYC-116 was chosen for testing in the mouse model system. CYC-116 is an Aurora kinase inhibitor in clinical trials that is unique in this study for its selective inhibition of liver stage parasites (Figure 2A). With a single treatment of 100 mg/kgb.w. before infection, parasite load in both liver and blood parasitemia were reduced when compared to the vehicle control (Figure 4B and 4C). These results demonstrate that molecules identified with the high-throughput screen have in vivo liver stage malaria efficacy and suggest that SNS-032 and CYC-116 have potential as therapeutics to prevent malaria.
Current therapies largely function through inhibition of dihydrofolate reductase, the cytochrome bc1 complex, isoprenoid precursor biosynthesis, and hemozoin formation. Resistance to drugs targeting these systems has already been reported, requiring the development of new strategies to inhibit Plasmodium parasites. Small molecule inhibitors targeting parasite dihydroorotate dehydrogenase,[38] ATPase,[39] and lysyl-tRNA synthetase,[40] to name a few, are currently being explored. There are advantages to having a drug with multiple modes of action for malaria treatment, including the potential to limit the development of parasite drug resistance and to obtain multistage inhibition. Recent efforts to overcome malaria drug-resistance have focused on the development of hybrid molecules, coupling two inhibitors with distinct modes of action. However, polypharmacology presents another strategy using multiple modes of action to treat disease, as demonstrated by the success of several kinase inhibitors in cancer treatment. The multikinase inhibitor Nexavar is a popular example of this success. Nexavar was tested for inhibition of liver stage parasites to further explore the possibility that clinically used drugs that exhibit polypharmacology may yield new antimalarials. At a low micromolar concentration Nexavar completely inhibited parasite load in liver cells highlighting the potential to identify malaria inhibitors within multiple-action drugs.
Bioinformatic analysis identified Plasmodium kinases with homology to the primary human targets of the reported screening hits. Although Plasmodium proteins often have notable distinctions from other organisms, sequence similarity to proteins with known function was used to annotate the genome.[41] The use of eukaryotic model systems, such as yeast, has also proven to be useful for target identification.[40] Therefore despite differences between human and parasite kinases, sequence similarity remains an important route to evaluate the molecular target(s) of a compound. Using a BLAST search, ten different kinases were identified as potential targets based on their sequence similarity to known human targets. Not all BLAST queries yielded useful results, as some of the human proteins have no known parasite homolog. Using a previously reported phylogenetic tree of P. falciparum kinases,[10] we have highlighted proteins that may be targets of the screening hits based on their sequence similarity to the primary human targets of the compounds (Figure 5). Kinase inhibitors remain a rich area for malaria drug development and Figure 5 indicates as many as ten parasite proteins that may prove to have essential roles in parasite signaling pathways upon further investigation with chemical biology methods.
It is worth noting that several Plasmodium kinases exist with limited homology to human proteins. These atypical kinases also represent promising drug targets as they may be inhibited without affecting a host homolog. The library members used in this work, as well as many chemical tools for studying human kinases, cannot be used to study malaria’s atypical kinases and therefore our approach to predict kinase targets (Figure 5) excludes these proteins. Further biochemical investigations on the predicted targets of this study as well as on Plasmodium atypical kinases may shed light onto the biological roles of these proteins.
Conclusion
The successful inhibition of malaria in mice by kinase inhibitors supports the validity of phenotypic malarial screening and emphasizes the potential for further development of the identified compounds. Previous liver stage malaria screening of more diverse libraries revealed that many compounds selectively inhibit one parasite form, a result that was not reflected in Plasmodium kinome inhibitors. In fact, the majority of the kinase screening hits had blood and liver stage efficacy, indicating an essential role of the respective protein kinases for both parasite forms. Several challenges exist in further characterizing inhibitors that target Plasmodial kinases. It is likely that these compounds target several proteins, and unlike human kinases for which a diverse assortment of biochemical assays are commercially available to address inhibitor selectivity, only a few assays exist for Plasmodial kinases. Signal transduction pathways in parasites are poorly characterized and there are few phospho-specific antibodies that allow for confirmation of intracellular kinase inhibition. With the relatively limited resources for malaria research and the enormous effort of many researchers to develop kinase inhibitors, drug repurposing is an attractive route for drug development. Many kinase inhibitors in clinical trials target more than one protein. Targeting the parasite kinome through polypharmacology is an attractive option for malaria drug development. Not only does this strategy present a cheaper route to drug development, but also the inhibition of multiple targets could delay the emergence of drug resistance. Finally, these findings suggest that drug screening with a focus on kinase inhibitors may be an effective route to identifying probes to elucidate Plasmodium kinase function, which remains an interesting yet complex challenge.
Experimental Section
Materials
HepG2 cells (ATCC) were maintained in DMEM (Invitrogen), 10% FBS (Sigma) and 1% antibiotic-antimycotic (Invitrogen) in a standard tissue culture incubator (37°C, 5% CO2). Plasmodium-infected Anopheles stephensi mosquitoes were purchased from the New York University Langone Medical Center Insectary.
Library Screening
Compounds tested were the kinase inhibitors from the GSK Published Kinase Inhibitor Set (PKIS, 367 members tested at 11 μM), Roche (235 members tested at 11 μM), the Library of Integrated Network-based Cellular Signatures (LINCS, 88 members tested at 1.3 μM)[42] [42][38][37] and previously reported synthetic compounds (668 tested at 3.6 – 10 μM). The EMD Kinase Inhibitor 1 library (244 members) was tested previously.[8] Among the compounds tested only WZ12-051 (Figure S1A) has not been previously reported, but it is structurally similar to WZ4002.[43] Liquid chromatography–mass spectrometry and 1H nuclear magnetic resonance characterization of this compound is in the Supporting Information (Figure S1B and Figure S1C, respectively).
For assays, 15,000 HepG2 cells/well were added to 384-well microtiter plates in duplicate. After 18 – 24 hours at 37°C the media was exchanged and compounds (100 nL) were added to the plates with a pin array on a robot arm. The final concentration of DMSO was 0.3% (v/v) and compounds were between 1.3 – 11 μM. Atovaquone or halofuginone (1 μM) were used as positive controls for parasite inhibition. After 1 hour, luciferase-expressing P. berghei ANKA parasites[15] obtained from freshly dissected mosquitoes were added to the plates at 4,000 parasites/well, the plates were spun for 10 min at 1,000 rpm, and then they were incubated at 37°C. The final assay volume was 30 μL. After 42 – 47 hours at 37°C the parasite load and HepG2 viability in the presence of the compounds was assessed. To examine HepG2 viability CellTiter-Fluor (Promega) was added to the plates and fluorescence was measured. After the HepG2 viability reading Bright-Glo (Promega) was added to the plates and luminescence was measured, which correlates with parasite load. The relative signal intensity of each plate was evaluated with an EnVision (PerkinElmer) system. Data analysis was carried out using GraphPad Prism and all results are given as mean ± SD.
Secondary Assays
Blood stage assays were performed as previously described[44] and a hemolysis assay was also done as a control. Additionally, several secondary assays were performed to confirm screening actives. First, dose-response curves for inhibition of P.berghei parasite load in Huh-7 cells were generated for all compounds utilizing transgenic parasites with a luciferase reporter. Second, parasite load in HepG2 cells after P. berghei infection was assessed by immunofluorescence.[16] Briefly, 45 hours post-infection cells were washed 3 times with phosphate buffered saline (PBS) and then fixed with formaldyhyde. The primary antibody was 2E6 against P. berghei heat-shock protein 70[45] and the secondary antibody was conjugated to Alexa Fluor 488 (Invitrogen). Parasites and cells were visualized and quantitated with an IsoCyte Laser Scanning Cytometer (Molecular Devices). Compounds that did not inhibit parasite infection of Huh-7 cells or were not reproducible between luciferase and immunofluorescence detection systems were not considered hits. Lastly time course experiments were completed by adding hits (10 μM) 2 hours before or 3, 8, and 24 hours after sporozoite addition to HepG2 cells. All parasite levels were measured ~48 hours post-infection with an EnVision system.
PfPK5 Assays
Identified liver stage inhibitors were tested for activity against recombinant PfPK5 at 10 μM using an active-site directed competition binding assay by DiscoveRx Bioscience with KinomeScan™ Technology. The assay utilizes DNA-tagged protein and a bait ligand to capture protein on to a solid support. Kinase binding to this solid support in the presence and absence of test compounds is quantified using PCR. The assay is done in the absence of ATP and thus measures a dissociation constant. Compounds that showed binding at 10 μM were selected for Kd determination (DiscoveRx).
Molecular Modeling/Docking
Two ligand-bound structures of PfPK5, 1V0O and 1V0P,[18] are in The Protein Data Bank.[46] The ligand-binding site, as identified by the position of the ligand in the crystallographic structure, is isolated to a single monomer chain, so only a single chain was needed for further analysis. Comparison of the chains’ structures showed that they are effectively identical for each PDB structure, so chain A was selected. Computations were done using Schrodinger Suite 2012 with visualization performed in Maestro (Schrödinger, version 9.3). Each structure was prepared for docking independently using Schrodinger’s Protein Preparation Wizard[47] with the default parameters, which resulted in all water molecules being removed. Ligands were prepared using LigPrep (Schrödinger, version 2.5) and docking was done for crystallographic structure ligands, as well as the screening hits CDK 1/2 and SNS-032. For the crystallographic structure ligands, preparation was done with both the 3-dimensional structure of the ligand as is and generating input structures from SMILES strings. For CDK 1/2 and SNS-032, input structures were generated from SMILES strings. Docking was performed with Glide (Schrödinger, version 5.8)[48] using XP mode[49] with default parameters. Receptor grids were generated using the default parameters and images were generated in PYMOL.
Mouse Studies
Male C57BL/6 mice, aged 6–8 weeks and weighing 20–22 g were purchased from Charles River and housed in the pathogen-free facilities of the Instituto de Medicina Molecular, Lisbon. Compounds were dissolved in DMSO and further diluted to the final concentration in drinking water. Mice received 200 μL of compound solution per mouse (CYC-116: 100 mg/kg body weight; SNS-032: 75 mg/kg body weight), administered orally by gavage 1 hour before infection. Control groups received an equivalent amount of vehicle. Infection was performed through intravenous injection of 1x104 P. berghei-luciferase sporozoites. Parasite load in the livers was determined 42–44 hours after infection using an in vivo Imaging System (IVIS 100 and Spectrum; Caliper Life Sciences).[15] Statistically significant differences between groups were analyzed using the Mann Whitney test. All in vivo protocols were approved by the Animal Care Committee of the Instituto de Medicina Molecular and were performed according to the regulations of the European guidelines 86/609/EEG.
Supplementary Material
Acknowledgments
We thank David Drewry and William Zuercher (GSK, Raleigh-Durham) for helpful input concerning the GSK PKIS set and Paul Gillespie (Roche, Nutley) for kindly supplying RO1153853 and input with the Roche kinase inhibitor set. We thank Dr. Oliver Billker for sharing his phylogenetic analysis of malaria kinases and the ICCB-Longwood screening center where the screen was performed. We also thank Inês Albuquerque for preliminary mouse experiments with CDK 1/2. This work was supported by the Harvard Medical School – Portugal Program in Translational Research (M.M.M. and J.C.) and the NIH K99 Pathway to Independence Award (GM099796, to E.R.D.).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
Contributor Information
Dr. Emily R. Derbyshire, Email: emily_derbyshire@hms.harvard.edu.
Prof. Dr. Jon Clardy, Email: jon_clardy@hms.harvard.edu.
References
- 1.Wolrd Health Organization. World Malaria Report. 2013. [Google Scholar]
- 2.a) Kappe SH, Vaughan AM, Boddey JA, Cowman AF. Science. 2010;328:862–866. doi: 10.1126/science.1184785. [DOI] [PubMed] [Google Scholar]; b) Mazier D, Renia L, Snounou G. Nat Rev Drug Discov. 2009;8:854–864. doi: 10.1038/nrd2960. [DOI] [PubMed] [Google Scholar]
- 3.White NJ. J Clin Invest. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Derbyshire ER, Mota MM, Clardy J. PLoS Pathog. 2011;7:e1002178. doi: 10.1371/journal.ppat.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Flannery EL, Chatterjee AK, Winzeler EA. Nat Rev Microbiol. 2013;11:849–862. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL. Genome Biol. 2003;4:R9. doi: 10.1186/gb-2003-4-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA. Science. 2003;301:1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]; c) Silvestrini F, Bozdech Z, Lanfrancotti A, Di Giulio E, Bultrini E, Picci L, Derisi JL, Pizzi E, Alano P. Mol Biochem Parasitol. 2005;143:100–110. doi: 10.1016/j.molbiopara.2005.04.015. [DOI] [PubMed] [Google Scholar]; d) Westenberger SJ, McClean CM, Chattopadhyay R, Dharia NV, Carlton JM, Barnwell JW, Collins WE, Hoffman SL, Zhou Y, Vinetz JM, Winzeler EA. PLoS Negl Trop Dis. 2010;4:e653. doi: 10.1371/journal.pntd.0000653. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Williams CT, Azad AF. PLoS One. 2010;5:e10267. doi: 10.1371/journal.pone.0010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meister S, Plouffe DM, Kuhen KL, Bonamy GM, Wu T, Barnes SW, Bopp SE, Borboa R, Bright AT, Che J, Cohen S, Dharia NV, Gagaring K, Gettayacamin M, Gordon P, Groessl T, Kato N, Lee MC, McNamara CW, Fidock DA, Nagle A, Nam TG, Richmond W, Roland J, Rottmann M, Zhou B, Froissard P, Glynne RJ, Mazier D, Sattabongkot J, Schultz PG, Tuntland T, Walker JR, Zhou Y, Chatterjee A, Diagana TT, Winzeler EA. Science. 2011;334:1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derbyshire ER, Prudencio M, Mota MM, Clardy J. Proc Natl Acad Sci U S A. 2012;109:8511–8516. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solyakov L, Halbert J, Alam MM, Semblat JP, Dorin-Semblat D, Reininger L, Bottrill AR, Mistry S, Abdi A, Fennell C, Holland Z, Demarta C, Bouza Y, Sicard A, Nivez MP, Eschenlauer S, Lama T, Thomas DC, Sharma P, Agarwal S, Kern S, Pradel G, Graciotti M, Tobin AB, Doerig C. Nature communications. 2011;2:565. doi: 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- 10.Tewari R, Straschil U, Bateman A, Bohme U, Cherevach I, Gong P, Pain A, Billker O. Cell Host Microbe. 2010;8:377–387. doi: 10.1016/j.chom.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Doerig C, Abdi A, Bland N, Eschenlauer S, Dorin-Semblat D, Fennell C, Halbert J, Holland Z, Nivez MP, Semblat JP, Sicard A, Reininger L. Biochimica et biophysica acta. 2010;1804:604–612. doi: 10.1016/j.bbapap.2009.10.009. [DOI] [PubMed] [Google Scholar]; b) Doerig C, Billker O, Haystead T, Sharma P, Tobin AB, Waters NC. Trends Parasitol. 2008;24:570–577. doi: 10.1016/j.pt.2008.08.007. [DOI] [PubMed] [Google Scholar]; c) Lucet IS, Tobin A, Drewry D, Wilks AF, Doerig C. Future medicinal chemistry. 2012;4:2295–2310. doi: 10.4155/fmc.12.183. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Yang PL, Gray NS. Nature reviews Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight ZA, Lin H, Shokat KM. Nature reviews Cancer. 2010;10:130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knapp S, Arruda P, Blagg J, Burley S, Drewry DH, Edwards A, Fabbro D, Gillespie P, Gray NS, Kuster B, Lackey KE, Mazzafera P, Tomkinson NC, Willson TM, Workman P, Zuercher WJ. Nat Chem Biol. 2013;9:3–6. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- 15.Ploemen IH, Prudencio M, Douradinha BG, Ramesar J, Fonager J, van Gemert GJ, Luty AJ, Hermsen CC, Sauerwein RW, Baptista FG, Mota MM, Waters AP, Que I, Lowik CW, Khan SM, Janse CJ, Franke-Fayard BM. PLoS One. 2009;4:e7881. doi: 10.1371/journal.pone.0007881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derbyshire ER, Mazitschek R, Clardy J. ChemMedChem. 2012;7:844–849. doi: 10.1002/cmdc.201200045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halbert J, Ayong L, Equinet L, Le Roch K, Hardy M, Goldring D, Reininger L, Waters N, Chakrabarti D, Doerig C. Eukaryot Cell. 2010;9:952–959. doi: 10.1128/EC.00005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holton S, Merckx A, Burgess D, Doerig C, Noble M, Endicott J. Structure. 2003;11:1329–1337. doi: 10.1016/j.str.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 19.Gong L, Hirschfeld D, Tan YC, Heather Hogg J, Peltz G, Avnur Z, Dunten P. Bioorg Med Chem Lett. 2010;20:1693–1696. doi: 10.1016/j.bmcl.2010.01.038. [DOI] [PubMed] [Google Scholar]
- 20.Stevens KL, Reno MJ, Alberti JB, Price DJ, Kane-Carson LS, Knick VB, Shewchuk LM, Hassell AM, Veal JM, Davis ST, Griffin RJ, Peel MR. Bioorg Med Chem Lett. 2008;18:5758–5762. doi: 10.1016/j.bmcl.2008.09.069. [DOI] [PubMed] [Google Scholar]
- 21.Luk KC, Simcox ME, Schutt A, Rowan K, Thompson T, Chen Y, Kammlott U, DePinto W, Dunten P, Dermatakis A. Bioorg Med Chem Lett. 2004;14:913–917. doi: 10.1016/j.bmcl.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Trejo A, Arzeno H, Browner M, Chanda S, Cheng S, Comer DD, Dalrymple SA, Dunten P, Lafargue J, Lovejoy B, Freire-Moar J, Lim J, McIntosh J, Miller J, Papp E, Reuter D, Roberts R, Sanpablo F, Saunders J, Song K, Villasenor A, Warren SD, Welch M, Weller P, Whiteley PE, Zeng L, Goldstein DM. J Med Chem. 2003;46:4702–4713. doi: 10.1021/jm0301787. [DOI] [PubMed] [Google Scholar]
- 23.Tavares FX, Boucheron JA, Dickerson SH, Griffin RJ, Preugschat F, Thomson SA, Wang TY, Zhou HQ. J Med Chem. 2004;47:4716–4730. doi: 10.1021/jm040063i. [DOI] [PubMed] [Google Scholar]
- 24.Lackey K, Cory M, Davis R, Frye SV, Harris PA, Hunter RN, Jung DK, McDonald OB, McNutt RW, Peel MR, Rutkowske RD, Veal JM, Wood ER. Bioorg Med Chem Lett. 2000;10:223–226. doi: 10.1016/s0960-894x(99)00668-x. [DOI] [PubMed] [Google Scholar]
- 25.Takle AK, Bamford MJ, Davies S, Davis RP, Dean DK, Gaiba A, Irving EA, King FD, Naylor A, Parr CA, Ray AM, Reith AD, Smith BB, Staton PC, Steadman JG, Stean TO, Wilson DM. Bioorg Med Chem Lett. 2008;18:4373–4376. doi: 10.1016/j.bmcl.2008.06.070. [DOI] [PubMed] [Google Scholar]
- 26.Emmitte KA, Adjabeng GM, Andrews CW, Alberti JG, Bambal R, Chamberlain SD, Davis-Ward RG, Dickson HD, Hassler DF, Hornberger KR, Jackson JR, Kuntz KW, Lansing TJ, Mook RA, Jr, Nailor KE, Pobanz MA, Smith SC, Sung CM, Cheung M. Bioorg Med Chem Lett. 2009;19:1694–1697. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- 27.Heerding DA, Rhodes N, Leber JD, Clark TJ, Keenan RM, Lafrance LV, Li M, Safonov IG, Takata DT, Venslavsky JW, Yamashita DS, Choudhry AE, Copeland RA, Lai Z, Schaber MD, Tummino PJ, Strum SL, Wood ER, Duckett DR, Eberwein D, Knick VB, Lansing TJ, McConnell RT, Zhang S, Minthorn EA, Concha NO, Warren GL, Kumar R. J Med Chem. 2008;51:5663–5679. doi: 10.1021/jm8004527. [DOI] [PubMed] [Google Scholar]
- 28.Ward P, Equinet L, Packer J, Doerig C. BMC genomics. 2004;5:79. doi: 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prudencio M, Rodrigues CD, Hannus M, Martin C, Real E, Goncalves LA, Carret C, Dorkin R, Rohl I, Jahn-Hoffmann K, Luty AJ, Sauerwein R, Echeverri CJ, Mota MM. PLoS Pathog. 2008;4:e1000201. doi: 10.1371/journal.ppat.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaushansky A, Kappe SH. Nat Med. 2011;17:1180–1181. doi: 10.1038/nm.2456. author reply 1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Xu C, Kirubakaran S, Zhang X, Hur W, Liu Y, Kwiatkowski NP, Wang J, Westover KD, Gao P, Ercan D, Niepel M, Thoreen CC, Kang SA, Patricelli MP, Wang Y, Tupper T, Altabef A, Kawamura H, Held KD, Chou DM, Elledge SJ, Janne PA, Wong KK, Sabatini DM, Gray NS. Cancer research. 2013;73:2574–2586. doi: 10.1158/0008-5472.CAN-12-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanson KK, Ressurreicao AS, Buchholz K, Prudencio M, Herman-Ornelas JD, Rebelo M, Beatty WL, Wirth DF, Hanscheid T, Moreira R, Marti M, Mota MM. Proc Natl Acad Sci U S A. 2013;110:E2838–2847. doi: 10.1073/pnas.1306097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaid A, Ranjan R, Smythe WA, Hoppe HC, Sharma P. Blood. 2010;115:2500–2507. doi: 10.1182/blood-2009-08-238972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. Nature. 2002;419:520–526. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- 35.McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung BK, Chatterjee AK, McCormack SL, Manary MJ, Zeeman AM, Dechering KJ, Kumar TR, Henrich PP, Gagaring K, Ibanez M, Kato N, Kuhen KL, Fischli C, Rottmann M, Plouffe DM, Bursulaya B, Meister S, Rameh L, Trappe J, Haasen D, Timmerman M, Sauerwein RW, Suwanarusk R, Russell B, Renia L, Nosten F, Tully DC, Kocken CH, Glynne RJ, Bodenreider C, Fidock DA, Diagana TT, Winzeler EA. Nature. 2013;504:248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fugel W, Oberholzer AE, Gschloessl B, Dzikowski R, Pressburger N, Preu L, Pearl LH, Baratte B, Ratin M, Okun I, Doerig C, Kruggel S, Lemcke T, Meijer L, Kunick C. J Med Chem. 2013;56:264–275. doi: 10.1021/jm301575n. [DOI] [PubMed] [Google Scholar]
- 37.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. J Biol Chem. 2005;280:21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 38.a) Booker ML, Bastos CM, Kramer ML, Barker RH, Jr, Skerlj R, Sidhu AB, Deng X, Celatka C, Cortese JF, Guerrero Bravo JE, Crespo Llado KN, Serrano AE, Angulo-Barturen I, Jimenez-Diaz MB, Viera S, Garuti H, Wittlin S, Papastogiannidis P, Lin JW, Janse CJ, Khan SM, Duraisingh M, Coleman B, Goldsmith EJ, Phillips MA, Munoz B, Wirth DF, Klinger JD, Wiegand R, Sybertz E. J Biol Chem. 2010;285:33054–33064. doi: 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. J Med Chem. 2011;54:3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Phillips MA, Rathod PK. Infectious disorders drug targets. 2010;10:226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, Aust T, McCormack SL, Plouffe DM, Meister S, Schuierer S, Plikat U, Hartmann N, Staedtler F, Cotesta S, Schmitt EK, Petersen F, Supek F, Glynne RJ, Tallarico JA, Porter JA, Fishman MC, Bodenreider C, Diagana TT, Movva NR, Winzeler EA. Cell Host Microbe. 2012;11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.http://www.lincsproject.org/.
- 43.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. Proc Natl Acad Sci U S A. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuji M, Mattei D, Nussenzweig RS, Eichinger D, Zavala F. Parasitol Res. 1994;80:16–21. doi: 10.1007/BF00932618. [DOI] [PubMed] [Google Scholar]
- 46.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic acids research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Journal of computer-aided molecular design. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 48.a) Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]; b) Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 49.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.