

Abstract
Clinical application of potent anthracycline anticancer drugs, especially doxorubicin (DOX), is limited by a toxic cardiac side effect that is not fully understood and preventive strategies are yet to be established. Studies in genetically modified mice have demonstrated that focal adhesion kinase (FAK) plays a key role in regulating adaptive responses of the adult myocardium to pathological stimuli through activation of intracellular signaling cascades that facilitate cardiomyocyte growth and survival. The objective of this study was to determine if targeted myocardial FAK activation could protect the heart from DOX-induced de-compensation and to characterize the underlying mechanisms. To this end, mice with myocyte-restricted FAK knock-out (MFKO) or myocyte-specific expression of an active FAK variant (termed SuperFAK) were subjected to DOX treatment. FAK depletion enhanced susceptibility to DOX-induced myocyte apoptosis and cardiac dysfunction, while elevated FAK activity provided remarkable cardioprotection. Our mechanistic studies reveal a heretofore unappreciated role for the protective cyclin-dependent kinase inhibitor p21 in the repression of the pro-apoptotic BH3-only protein Bim and the maintenance of mitochondrial integrity and myocyte survival. DOX treatment induced proteasomal degradation of p21, which exacerbated mitochondrial dysfunction and cardiomyocyte apoptosis. FAK was both necessary and sufficient for maintaining p21 levels following DOX treatment and depletion of p21 compromised FAK-dependent protection from DOX. These findings identify p21 as a key determinant of DOX resistance downstream of FAK in cardiomyocytes and indicate that cardiac-restricted enhancement of the FAK/p21 signaling axis might be an effective strategy to preserve myocardial function in patients receiving anthracycline chemotherapy.
Keywords: Anthracycline cardiomyopathy, apoptosis, mitochondria, cardiomyocyte, cell cycle
1. Introduction
The anthracycline doxorubicin (DOX) is one of the most frequently used chemotherapeutic agents as it is highly effective in the treatment of both solid and hematological malignancies. However, cumulative dose-dependent cardiac toxicity limits the therapeutic efficacy of this drug [1, 2]. Even with strict limitations of lifetime dosage a significant fraction of DOX-treated patients display signs of cardiac injury. The most common and devastating cardiovascular risk is chronic or delayed cardiomyopathy that, once initiated, rapidly progresses to congestive heart failure with 1-year mortality rates approaching 50% [1, 2]. Acute toxicities that develop immediately after treatment (mainly due to electrical disturbances) or sub-acute toxicities that occur within one week following treatment are also quite prevalent [1, 2]. Clearly a detailed understanding of the mechanisms underlying these various clinical presentations will provide an opportunity to develop effective treatment strategies to reduce or prevent detrimental off-target effects on the cardiovascular system.
DOX exposure can trigger both cancer cell and myocyte apoptosis, and this form of myocyte injury has been linked to the development of both sub-acute and chronic cardiomyopathies. DOX forms an inactive adduct between DNA and topoisomerase II (Top2) which inhibits Top2-dependent strand ligation during DNA synthesis[3] and very recent studies indicate that Top2β is required for the adverse cardiac remodeling that accompanies chronic DOX treatment [4]. In rapidly cycling cancer cells inhibition of Top2 leads to accumulation of single- and double-strand DNA breaks and initiates a DNA damage repair pathway resulting in activation of the tumor suppressor, p53 and initiation of p53-dependent apoptosis[5]. However, it is presently unclear to what extent the DNA damage response is causal for initiating myocyte apoptosis following DOX treatment[6-8]. DOX also induces oxidative stress via the generation of reactive oxygen species (ROS) and alters iron homeostasis and there is evidence to support a role for each of these pathways in DOX-induced cardiac toxicity[1, 2]. Notably, mitochondrial dysfunction is a common consequence of DOX-mediated genotoxic and oxidative myocyte stress and the resultant loss of mitochondrial membrane potential leads to cytochrome c release and activation of effector caspase-mediated apoptosis.
Importantly, the so called intrinsic mitochondrial-mediated programmed cell death cascade can be reversed by activation of pro-survival signals such as those mediated by integrin-dependent adhesive complexes. Integrins are heterodimeric cell-surface receptors composed of α and β subunits, which physically link extracellular matrices to the intracellular actin cytoskeleton and these adhesion complexes are necessary for maintaining tissue integrity, conveying tensile strength, and for the transduction of growth and survival signals [9]. Indeed, aberrant expression of integrins is associated with tumorigenesis and resistance to cytotoxic therapies. For example, activation of the α2β1 integrin rendered malignant T cells resistant to DOX-dependent apoptosis [10], while inhibition of β1 integrin signaling sensitized various types of cancer cells to radiotherapy and chemotherapy[11-13]. Since integrins lack intrinsic kinase activity, transduction of integrin-mediated survival signals requires cytoplasmic signaling molecules. Focal adhesion kinase (FAK), a nonreceptor protein tyrosine kinase, associates with the cytoplasmic tails of all β1-containing integrins and its activity is critical for integrin signaling, including the signals that mediate cancer cell resistance to cytotoxic agents [11, 14-19].
Interestingly, we and others have shown that FAK provides critical survival signaling throughout cardiac development and in adult hearts when subjected to pathological stress [20-24]. Herein we show that mice with myocyte-restricted depletion of FAK exhibited exacerbated DOX-induced cardiomyopathy, while those engineered to confer enhanced FAK activation in cardiomyocytes exhibited remarkable cardioprotection, indicating that cardiac-restricted enhancement of FAK activity might be an effective strategy to preserve myocardial function in this setting. Our mechanistic studies reveal a heretofore unappreciated consequence of DOX treatment on depletion of p21Cip1 (p21). We show that cytoplasmic p21 is cytoprotective in cardiomyocytes, and that activation of FAK reduced DOX cardiotoxicity, at least in part, by up-regulation of p21.
2. Materials and Methods
An expanded Methods section is available in the online-only Data Supplement.
2.1. Animals
Myocyte-restricted FAK knockout (MFKO) mice were generated using the Cre/LoxP technology, and cardiac-specific SuperFAK (SF) transgenic mice (SF2) were generated using a myosin heavy chain promoter as described previously [20, 24, 25]. Cardiomyopathy was induced by a single injection of DOX (20mg/kg, i.p.) with 0.9% NaCl as a control. All procedures were approved by the IACUC at the University of North Carolina, Chapel Hill.
2.2. Statistics
Results are expressed as mean ± SEM. Student’s unpaired t test was used to compare values between 2 groups. One-way analysis of variance with the Bonferroni/Dunnett post-hoc analysis was used to determine the difference among multiple groups. Differences were considered significant at P < 0.05.
3. Results
3.1. FAK antagonizes DOX-induced cardiomyopathy
We previously showed that conditional deletion of FAK from the myocardium of adult mice did not affect basal cardiac performance or myocyte viability [20] but exacerbated ischemia/reperfusion-induced cardiac stress [23]. In the present study, we sought to determine if endogenous FAK also plays an important cardioprotective role in chemotherapy-induced cardiotoxicity. To this end, we injected MFKO and wild-type (WT) littermate control mice with DOX (20mg/kg, i.p. bolus that was previously shown to induce sub-acute cardiomyopathy [26]) and monitored heart function by echocardiography for up to 5 days following treatment. As shown in Figure 1A,B, DOX-induced cardiac dysfunction was exacerbated in MFKO mice as evidenced by a significant decrease in ejection fraction (EF) and fractional shortening (FS) at days 3 and 5 following treatment.
Figure 1.

FAK antagonizes doxorubicin (DOX)-induced cardiomyopathy and myocyte apoptosis in vivo. (A-C) Myocyte-restricted FAK knockout mice (MFKO, n=5) and wild-type littermate control mice (WT, n=9) received a single injection of DOX (20mg/kg, i.p.) and heart function was monitored by echocardiography before DOX injection, and at day 3 and day 5 post DOX injection. DOX treatment led to a more pronounced decrease in ejection fraction (EF, A) and fractional shortening (FS, B) in MFKO compared to WT mice. (C) Myocyte apoptosis (measured by dual staining with TUNEL and anti-cardiac troponin T) was exacerbated in MFKO hearts at day 5 post DOX injection. * P<0.05; ** P<0.01 vs. WT. (D-F) Cardiomyocyte-specific transgenic mice expressing a superactivatable variant of FAK (SF2, n=8) and non-transgenic littermate control mice (NTG, n=8) were treated with DOX as described above and heart function was monitored at baseline, and at day 5 and day 14 post DOX injection. Both EF (D) and FS (E) were better preserved in DOX treated SF2 mice. (F) Apoptosis was significantly reduced in SF2 hearts compared to NTG hearts at day 1 and 14 post DOX injection. * P<0.05; ** P<0.01 vs. NTG. n=3-6 mice per group at each time point. Data are mean ± SEM.
Based on these data, we predicted that enhanced activity of this intrinsic pro-survival signal might impart resistance to cardiac toxicity induced by DOX. FAK activation proceeds by a 2-step process that involves integrin- or growth factor-induced dimerization and autophosphorylation of Tyr397 which directs Src to bind to and phosphorylate FAK on 2 additional sites within the activation loop (Tyr576 and Tyr577) that lead to further augmentation of FAK activity [25]. We recently generated mice with cardiac-restricted expression of a super-activatable FAK variant (SuperFAK, SF) that contained mutations within the FAK activation loop (Lys578Glu/Lys581Glu) that mimic the charge transfer (and enhanced catalytic activity) induced by Src phosphorylation [24, 27]. We reported that these so-named SuperFAK mice (SF2) exhibited elevated allosteric activation of FAK in the myocardium and that these mice were protected from ischemia/reperfusion-induced cardiac de-compensation[24]. To determine the extent to which SuperFAK might confer resistance to cardiac stress induced by genotoxic agents, we treated SF2 and non-transgenic littermate control mice (NTG) with DOX as described above. Echocardiography revealed that DOX treatment impaired heart function in NTG mice as evidenced by a significant decrease in EF and FS at 5 and 14 days following treatment, while both parameters were well preserved in SF2 mice (Figure 1D,E). Hearts from NTG mice also exhibited significantly decreased end-systolic posterior wall and intraventricular septal thickness and increased LV end-systolic dimension than those from similarly treated SF2 mice (Supplemental Figure 1). Since anthracylines commonly induce appetite suppression we also monitored body weight in these mice. As shown in Supplemental Figure 2A, DOX injection led to a similar extent of weight loss in NTG and SF2 mice, indicating that these mice experienced an equivalent level of stress. Collectively, these results demonstrate that FAK antagonizes DOX-induced cardiotoxicity in vivo.
3.2. FAK conferred resistance to DOX-induced cardiomyocyte apoptosis
To explore whether cardioprotection by FAK might result from mitigating DOX-dependent myocyte apoptosis, we evaluated TUNEL staining in hearts from DOX-treated mice (Supplemental Figure 2B). Compared with WT mice, MFKO hearts exhibited a significant increase in TUNEL labeling at end-stage (i.e. day 5; Figure 1C). However, a significant reduction in TUNEL-positive myocytes was observed in SF2 hearts when compared to treated littermate controls at both 1 and 14 days post-injection (Figure 1F). Moreover, DOX injection led to myocyte vacuolization and myofibrillar loss, which was exacerbated in MFKO mice, but attenuated in SF2 mice (Supplemental Figure 2C).
To uncover the signaling mechanism(s) by which FAK confers protection from anthracyline cardiotoxicity, we infected primary neonatal rat cardiomyocytes (NRCMs) with adenoviruses expressing green fluorescent protein (GFP) or SF. NRCMs are a widely used model system to interrogate downstream pathways in differentiated cardiomyocytes and importantly, previous studies determined that treatment with DOX (1 to 2μM) promotes apoptotic death of these cells within a 24 to 48h window [28]. As shown in Figure 2, DOX induced significantly higher numbers of apoptotic GFP-expressing myocytes than SF-expressing cells as assessed by TUNEL staining, caspase 3 cleavage, and cleavage of the caspase 3 substrate PARP (Figure 2A,C). As well, the metabolic capacity of SF infected cells was better preserved following DOX treatment as assessed by the ability of the cells to reduce the tetrazolium dye, MTT (Figure 2B).
Figure 2.

FAK is necessary and sufficient for cardiomyocyte survival in response to DOX treatment. (A-C) Neonatal rat cardiomyocytes (NRCM) were non-infected (NI), or infected with GFP or SF adenoviruses before treatment with DOX (1μM) for 24h. (A) Overexpression of SF attenuated cardiomyocyte apoptosis as assessed by TUNEL staining. Results are mean±SEM of 4 independent experiments. * P<0.05. (B) MTT reduction assay revealed that SF preserved metabolic activity of cardiomyocytes following DOX treatment. ** P<0.01; *** P<0.001. (C) Activation of FAK, as revealed by higher levels of phospho-FAK (Y397) in SF-expressing cells, inhibited DOX-induced cleavage of caspase 3 and its downstream target PARP. (D) NRCMs were transfected with control (si-Control) or FAK siRNA (si-FAK) prior to treatment with DOX (2 μM) for 24h. TUNEL staining revealed that knockdown of FAK increased DOX-dependent apoptosis. Data represent mean±SEM of 4 independent experiments. * P<0.05 vs. si-Control. (E) An MTT reduction assay revealed that knockdown of FAK reduced cardiomyocyte survival following DOX treatment. ** P<0.01. (F) NRCMs were transfected with si-Control or si-FAK before treatment with DOX (2 μM) for 16h. Western blotting revealed that depletion of FAK enhanced DOX-induced PARP cleavage.
We next used FAK-specific small interfering RNAs to confirm a role for endogenous FAK in mediating myocyte survival in this setting. As shown in Figure 2F, treatment of cultured cardiomyocytes with FAK siRNAs led to an approximate 90% reduction of FAK levels as assessed by Western analysis. While FAK depletion alone did not alter basal cell survival, it sensitized cardiomyocytes to DOX-dependent apoptosis as assessed by TUNEL staining, MTT reduction, and PARP cleavage (Figure 2D-F). Taken together, these data indicate that FAK activity is both necessary and sufficient to protect cardiomyocytes from DOX-induced programmed cell death.
3.3. FAK-dependent DOX resistance correlated with p21 levels
While several reports have shown that FAK depletion sensitizes various cancer cells to cytotoxic chemotherapy, the underlying mechanisms are not yet clear [29]. In some cell types, FAK promotes cell survival by trans-locating to the nucleus, binding to, and inducing the degradation of the tumor suppressor p53 [30, 31]. To determine whether a similar pathway is operative in cardiomyocytes, we first explored whether SF expression led to reduced protein levels of p53 and its transcriptional target, the cyclin-dependent kinase inhibitor (CDKI) p21. Contrary to reported findings in fibroblasts and tumor cells [30, 31], we found that p53 levels were not altered by SF expression in hearts from DOX-challenged mice (Supplemental Figure 3A). This discrepancy may be related to the sub-cellular locale of FAK in these various cell types. Indeed, the aforementioned studies indicated that nuclear localized FAK bound to p53 and targeted it for MDM2-dependent ubiquitination and degradation [30] and we have found very little nuclear FAK in cardiomyocytes (Supplemental Figure 3B). A somewhat surprising finding was that p21, which has been recently shown to confer chemoresistance to cisplatin[32], was elevated in these hearts and SF induced a significant and dose-dependent increase in basal levels of p21 in cardiomyocytes (Figure 3A and 3B). Interestingly, treatment with pifithrin-α (30 μM) fully attenuated p53-mediated induction of p21 but did not alter SF-mediated up-regulation of p21 (Supplemental Figure 3C,D). In support of a critical role for endogenous FAK in the regulation of p21, protein levels of p21 were dramatically reduced in FAK-depleted cardiomyocytes in vitro and in vivo (Figure 3C,D). The half-life of p21 was similar in GFP- and SF-infected cardiomyocytes, suggesting that FAK does not affect p21 protein stability (Figure 3E, Supplemental Figure 4). Instead, we found that SF expression markedly increased p21 mRNA levels (Figure 3F), and subsequent experiments in which SF- or GFP-expressing cardiomyocytes were treated with actinomycin D (5 μg/ml) revealed that elevated FAK activity reduced the rate of p21 mRNA degradation, suggesting that FAK signaling regulates p21 message stability (Figure 3G).
Figure 3.

FAK-dependent DOX resistance correlates with p21 levels. (A) NRCMs were infected with vehicle, GFP, or SF adenoviruses at multiplicity of infection (MOI) of 10, 50, and 250. Overexpression of SF increased p21 protein levels in a dose-dependent manner. (B) SF2 hearts maintained higher protein levels of p21 than controls at day 14 post DOX injection. Blots are representative of 3-5 hearts per group. (C) NRCMs were transfected with control (si-Control) or FAK siRNA (si-FAK). Depletion of FAK reduced p21 protein levels in cardiomyocytes. (D) Protein levels of p21 were lower in MFKO than in WT heart extracts at day 5 post DOX injection. (E) NRCMs were infected with GFP or SF adenoviruses prior to incubation with the protein synthesis inhibitor cycloheximide (CHX, 10μg/ml) for various periods of time. Western blotting revealed that activation of FAK did not alter p21 protein stability. (F) Semi-quantitative RT-PCR of p21 in NRCMs infected with vehicle, GFP, or SF adenoviruses. Overexpression of SF elevated mRNA levels of p21. Results are representative of 3 independent experiments. (G) Semi-quantitative RT-PCR of p21 in NRCMs infected with GFP, or SF adenoviruses prior to treatment with the RNA synthesis inhibitor actinomycin D (Act-D, 5μg/ml) for various periods of time. Overexpression of SF prevented degradation of p21 mRNA.
3.4. p21 localized primarily to the cytoplasm of cardiomyocytes and was necessary for resistance to DOX-induced apoptosis
Although nuclear p21 is well known for mediating cell cycle arrest, recent reports indicate that cytoplasmic p21 promotes cell survival and limits the effectiveness of anticancer agents[32-34]. Notably, we found that p21 is predominantly cytoplasmic in NRCMs and in adult hearts (as assessed by sub-cellular fractionation and immunofluorescent staining; Figure 4A-D). To determine whether p21 plays a critical cyto-protective role in cardiomyocytes, we depleted p21 in NRCMs using siRNA and evaluated the induction of apoptosis by DOX. Similar to our findings in FAK-depleted cells (Figure 2D-F), siRNA-mediated reduction of p21 alone did not influence cell survival (Supplemental Figure 5), but rendered cardiomyocytes hypersensitive to DOX-induced apoptosis as assessed by PARP cleavage (Figure 4E), TUNEL labeling (Figure 4F,G) and MTT reduction (Figure 4H). We recognize that neonatal cardiomyocytes exhibit some cell cycle activity while adult cardiomyocytes are post-mitotic; a difference that could be particularly important with respect to the function of p21. For this reason, we repeated some of our major findings in cardiomyocytes isolated from 10-day-old rats (which we confirmed were bi-nucleated and had withdrawn from the cell cycle as has been previously reported [35]). Importantly, as shown in Supplemental Figure 6, silencing p21 in these non-cycling cardiomyocytes also rendered these cells more susceptible to DOX-induced apoptosis. Together these data indicate that p21 functions as a pro-survival factor in stressed cardiac myocytes.
Figure 4.

p21 localizes primarily to the cytoplasm of cardiomyocytes and is necessary for DOX resistance. (A) Western blotting of nuclear (Nuc) and cytosolic (Cyto) fractions from NRCMs. Histone H3 served as a nuclear loading control and GAPDH as a cytosolic loading control. (B) Immunofluorescent staining of NRCMs for p21 (green), nuclei (DAPI, blue), and cardiomyocyte marker α-actinin (red). Scale bar = 10μm. (C) Subcellular fractionation of adult mouse hearts revealed that p21 localizes predominantly to the cytosol. (D) Immunohistochemical staining of adult heart sections for p21 (green), cardiac troponin T (cTnT, red) and nuclei (DAPI, blue). Scale bar = 10μm. (E) NRCMs were transfected with control (si-Control) or p21 siRNA (si-p21) before treatment with DOX (2μM) for 24h. Western blotting revealed that depletion of p21 enhanced DOX-induced PARP cleavage. (F,G) NRCMs were transfected with si-Control or si-p21 before treatment with vehicle or DOX (1μM) for 36h. Apoptosis was evaluated by TUNEL staining. (F) Representative images of cells stained for TUNEL (green), nuclei (DAPI, blue), and cardiomyocyte marker α-actinin (red). Scale bar = 50μm. (G) Quantification of TUNEL-positive myocytes following DOX treatment. Data are mean±SEM of 4 independent experiments * P<0.05 vs. si-Control. (H) An MTT reduction assay revealed that knockdown of p21 reduced cardiomyocyte survival following DOX treatment. ** P<0.01.
3.5. Long-term treatment with DOX induced proteasomal degradation of p21
While DOX has been reported to induce p21 transcription [36], a kinetic analysis using apoptotic-inducing concentrations of DOX in NRCMs showed that DOX induced an initial up-regulation of p21 (apparent at 4 hr post-treatment) followed by a dramatic decline below basal levels that was noticeable between 16 and 24 hrs post-treatment (Figure 5A). Both the temporal nature and dose-dependence of DOX-mediated p21 down-regulation correlated with the extent of cardiomyocyte apoptosis as assessed by PARP cleavage (Figure 5B,C). Importantly, SF expression was capable of maintaining p21 levels following DOX treatment in vitro and in vivo (see Figure 3B, Supplemental Figure 7). As shown above, p21 is a short-lived protein and previous studies revealed that its turnover is tightly controlled by proteolytic pathways such as the proteasome pathway (Supplemental Figure 8) [37, 38]. Since DOX is known to activate the myocardial ubiquitin proteasome system [39], we reasoned that this pathway might account for the low levels of p21 apparent in DOX-treated cardiomyocytes. Indeed, we found that treatment of NRCMs with the 26S proteasome inhibitor, MG132, prevented DOX-induced down-regulation of p21, indicating that DOX triggered p21 degradation by the proteasome pathway (Figure 5D).
Figure 5.

Long-term treatment with DOX induced proteasomal degradation of p21. (A) NRCMs were treated with DOX (1μM or 2μM) for various periods of time. Protein levels of p21 were transiently up-regulated at 4h and dramatically decreased at later time points. (B) Western blotting revealed that DOX-induced reduction of p21 protein levels was associated with apoptosis as assessed by PARP cleavage. Results are representative of 3 independent experiments. (C) NRCMs were treated with various doses of DOX for 24h. Again, DOX-induced down-regulation of p21 protein was correlated with cleavage of PARP. (D) NRCMs were treated with DOX (1μM) for 24h without or with the addition of the 26S proteasome inhibitor MG132 (10μM) for 16h. Inhibition of the proteasome completely blocked DOX induced reduction of p21 protein levels.
3.6. Silencing of p21 exacerbated DOX-induced mitochondrial damage
While the precise mechanisms by which DOX-treatment induces cardiomyocyte apoptosis is not entirely clear, altered mitochondrial energetics likely plays a causal role[1, 2]. To test the hypothesis that p21 confers cardio-protection from DOX treatment by limiting stress-induced mitochondrial damage, control and p21 siRNA-treated NRCMs were subjected to DOX and mitochondrial membrane potential (ΔΨm) was assessed by staining with the mitochondria-specific cationic dye, JC-1. JC-1 accumulates and aggregates in energized mitochondria (wherein it fluoresces red) but fluoresces green in its cytosolic monomeric form. Confocal microscopy revealed that DOX treatment induced mitochondrial membrane depolarization, as measured by increased ratio of JC-1 monomers/J-aggregates (Figure 6A,B). Importantly, we found that p21 knockdown dramatically augmented DOX-induced mitochondrial membrane depolarization (Figure 6A,B). A major consequence of loss of ΔΨm is the release of cytochrome c into the cytosol which triggers the intrinsic apoptosis pathway via caspase activation. Accordingly, knockdown of p21 markedly increased DOX-dependent accumulation of cytosolic cytochrome c as assessed by sub-cellular fractionation (Figure 6C). Collectively, these data indicate that cytoplasmic p21 functions to protect cardiomyocytes from DOX-dependent activation of the intrinsic mitochondrial apoptotic pathway.
Figure 6.

Knockdown of p21 exacerbated DOX-induced mitochondrial damage in cardiomyocytes. NRCMs were transfected with si-Control or si-p21 before treatment with vehicle or DOX (1μM) for 24h. (A,B) Cells were stained with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1), which selectively enters mitochondria and forms J-aggregates (red) in healthy cells with high mitochondrial membrane potential (ΔΨm), but remains monomers (green) in apoptotic cells with low mitochondrial ΔΨm. (A) Representative images of JC-1 staining. Scale bar = 100μm. (B) Quantitative analysis of the ratio of JC-1 monomers / J-aggregates revealed that knockdown of p21 dramatically exacerbated DOX-induced mitochondrial depolarization. Results represent mean±SEM of 3 independent experiments.* P<0.05. (C) Subcellular fractionation revealed that depletion of p21 enhanced DOX-induced cytochrome c release into the cytosol. Tom20 served as a mitochondrial loading control and GAPDH as a cytosolic loading control. Blots are representative of 3 independent experiments.
3.7. Elevated expression of p21 mitigated DOX cardiotoxicity and repressed Bim
Knowing that p21 is a key survival factor in cardiomyocytes, we sought to further explore whether forced expression of exogenous p21 using an adenoviral vector promotes chemoresistance to DOX. Western blotting revealed that a moderate elevation in p21 levels inhibited DOX-induced caspase 3 cleavage (Figure 7A). Moreover, overexpression of p21 also suppressed cell death in response to DOX treatment as assessed by TUNEL assay and Calcein AM/ Ethidium homodimer-1 staining (Figure 7B,C). Measurement of ΔΨm using JC-1 revealed that DOX-induced mitochondrial membrane depolarization was limited by exogenous p21 (Figure 7D). In terms of mechanism, it was recently shown that DOX treatment led to increased expression of the proapoptotic BH3-only protein Bim [40], which was required for DOX cytotoxicity[41]. Therefore we tested the hypothesis that p21 might attenuate DOX cardiotoxicity by modulating Bim expression. Indeed, RT-PCR revealed that silencing of p21 enhanced DOX-induced expression of Bim (Figure 7E), and adenoviral mediated p21 overexpression suppressed Bim induction by DOX (Figure 7F). Together, these data provide strong evidence that p21 regulates cardiomyocyte survival by preserving mitochondrial membrane integrity.
Figure 7.

Elevated p21 expression attenuates DOX cardiotoxicity and represses Bim. (A-D) NRCMs were infected with LacZ or p21 adenoviruses prior to treatment with DOX (1μM) for 36h. Expression of exogenous p21 suppressed caspase 3 cleavage (A). Cell viability was assessed by staining with TUNEL (B) or calcein AM/Ethidium homodimer-1 (C) as described in the Methods. Mitochondrial membrane depolarization was assessed by staining with JC-1 (D). Data are mean±SEM of 3-4 independent experiments. * P<0.05 vs. LacZ. (E) NRCMs were transfected with control or p21 siRNA prior to treatment with DOX (1μM) for 2h. Transcript levels of Bim and p21 were assessed by semi-quantitative RT-PCR. GAPDH served as a loading control. (F) NRCMs were infected with LacZ or p21 adenoviruses prior to treatment with DOX (1μM) for 2h. Expression of exogenous p21 suppressed Bim expression following DOX treatment.
3.8. Knockdown of p21 compromised FAK-dependent DOX resistance
Finally, we sought to explore the extent to which elevated p21 levels account for FAK-dependent mitigation of DOX-induced cardiac toxicity. As shown in Figure 8A, DOX markedly induced caspase 3-dependent PARP cleavage, which was abrogated by SF overexpression. However, knockdown of p21 prevented SF-mediated protection from DOX-induced apoptosis, indicating that FAK promotes DOX resistance in cardiomyocytes, at least in part, by the up-regulation of p21.
Figure 8.
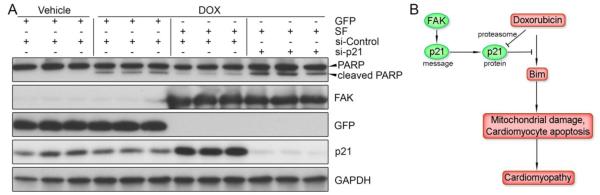
Cardiac FAK activation promotes DOX resistance via p21. (A) GFP- or SF-expressing NRCMs were transfected with control or p21 siRNA prior to treatment with DOX (1μM) for 16h. Knockdown of p21 reversed the protection of PARP cleavage conferred by SF expression, indicating that FAK-dependent DOX resistance is mediated by p21 in cardiomyocytes. (B) Schematic summary of the FAK/p21 signaling axis in resistance to DOX cardiotoxicity.
4. Discussion
To date, effective strategies for the prevention of DOX-induced cardiomyopathy and heart failure are unavailable [2]. Increased serum troponin levels (indicative of cardiomyocyte death) have been detected in patients within the first 3-5 days after administration of standard doses of anthracyclines and early troponin release is a reliable predictor of subsequent diastolic dysfunction [42]. Thus it has been postulated that multiple hits of acute anthracycline-induced apoptosis decreases cardiac reserve over time due to repeated acute loss of cardiomyocytes coupled with the limited regenerative capacity of this cell type. These findings highlight the importance of defining the mechanisms that control anthracycline-induced apoptosis as therapies that limit this response could be efficacious in preventing future development of refractive cardiomyopathies. Herein, for the first time, we identified a critical chemoresistant role of myocardial p21. We found that p21 represses expression of the BH3-only protein Bim and functions to protect mitochondrial integrity and cardiomyocyte viability and that DOX targets p21 for proteosomal degradation. Moreover, we found that dampened FAK activity exacerbated while elevated activity of FAK mitigated DOX-induced cardiomyopathy and cardiomyocyte apoptosis and that the ability of FAK to attenuate DOX toxicity was dependent on its ability to up-regulate p21 (see schematic, Figure 8C).
p21 is a universal cyclin-dependent kinase inhibitor and its overexpression can induce G1/S or G2/M cell cycle arrest. However, the continued expression and high abundance of this protein in post-mitotic cells such as cardiomyocytes supports its involvement in important biological processes apart from cell cycle withdrawal. Indeed, recent studies ascribe a pro-survival function to p21, as endogenous p21 serves to protect cancer cells[32, 43-46] and monocytes[47] from chemotherapy-dependent execution of apoptosis. Interestingly, this pro-survival function was correlated with its cytoplasmic localization and accumulation of p21 in the cytoplasm has been linked to increased tumor aggressiveness, metastasis, and poor prognosis [48]. Herein we found that p21 is almost exclusively cytoplasmic in neonatal cardiomyocytes and in the adult heart and showed that p21 depletion sensitized cardiomyocytes to DOX-dependent apoptosis, indicating that endogenous p21 also plays a major pro-survival role in these noncycling cells. While global p21 knock-out mice were reportedly modestly protected from DOX-dependent cardiomyopathy [49], future studies are warranted in mice with myocyte-restricted p21 depletion, as the former benefit was associated with a dampened inflammatory response, likely because p21 contributes to differentiation of monocytes to macrophages [50].
In some cancer cells, the chemoresistant effects of p21 expression has been linked to its capacity to arrest cell cycling, which when DNA damage is present, permits cells sufficient time for DNA repair[32-34]. We feel that this mechanism is not likely to explain the cardioprotective effects of p21 shown herein, because in our system cardiomyocyte cell cycle entry is extremely limited. Instead, we favor a mechanism by which cytoplasmic p21 antagonizes DOX-induced mitochondrial dysfunction. p21 has been reported to influence cell survival by acting both up- or down-stream of the intrinsic mitochondrial-mediated cell death pathway. p21 inhibits activation of CDK2/4/6 [51], kinases that induce translocation of Bax and Bim to the mitochondria [52, 53] and initiate mitochondrial membrane damage-induced cell death [54]. Pharmacological inhibition of CDK4/6 was recently shown to antagonize the cytotoxic effect of doxorubicin in several non-cardiac cells [55, 56]. As well, ectopic expression of dominant-negative CDK2 or p21 attenuated apoptotic cell death in cardiomyocytes subjected to prolonged hypoxia [57, 58]. Our results revealed that when combined with DOX treatment, knockdown of p21 resulted in a dramatic and synergistic depolarization of cardiomyocyte mitochondrial membranes and significantly increased the release of cytochrome c, indicating that endogenous p21 also functions to prevent initiation of the mitochondrial-death cascade in this setting. Importantly, we show that a modest up-regulation of p21 provides acute protection from DOX-induced mitochondrial membrane depolarization and apoptosis. Mechanistically, we found that myocyte p21 likely preserves mitochondrial integrity, at least in part, by antagonizing DOX-induced expression of the pro-apoptotic BH3-only Bim, a protein that is critical for initiation of mitochondrial membrane depolarization upon the induction of apoptosis [59-61] (also see Supplemental Discussion).
DOX, like many DNA damaging agents, has been reported to induce expression of p21 through a p53-dependent mechanism [62, 63]. In cardiomyocytes, we found that DOX regulated p21 levels in a bi-phasic fashion: an initial transient up-regulation which peaked 4 hr after DOX exposure, followed by rapid proteasome-dependent degradation that resulted in a significant depletion below basal levels by 16 hr following treatment. While others have reported that DOX treatment leads to activation of the myocardial ubiquitin-proteasome system [39], our studies are the first to reveal that degradation of p21 is an important consequence of this event (see Supplemental Discussion for possible ubiquitin ligases involved). Our findings also extend previous studies which revealed that UV irradiation-induced DNA damage signaling resulted in ubiquitin-dependent p21 degradation in osteosarcoma cells and normal human fibroblasts [37].
Herein we demonstrated that cardiac-specific SF transgenic mice displayed less mortality and better heart function accompanied by less apoptotic cardiomyocytes following DOX treatment, while the opposite was observed in MFKO mice. Moreover, overexpression of SF attenuated, and knockdown of FAK enhanced DOX-induced apoptosis in cultured cardiomyocytes. These results suggest for the first time that myocardial FAK activation promotes resistance to DOX cardiotoxicity. A potential limitation of the current study is that the cardiomyopathy in these animal models was induced by a single, maximum dose injection, which is distinct from repeated, lower dose administration in the clinic. However, it has been shown that chronic DOX cardiotoxicity also involves myocyte apoptosis [64], indicating the likelihood that similar pathways are involved. Our conclusion fits with the fact that FAK is frequently up-regulated in a variety of cancers, is associated with poor prognosis and patient survival, and that inactivation of FAK in breast cancer cells exaggerated DOX-induced apoptosis [11, 14-19, 65].
In conclusion, our findings indicate that levels of p21 in cardiomyocytes determine resistance to cardiotoxicity induced by the anthracycline DOX. Moreover, we provide evidence that treatments which preserve p21 levels (including those that activate FAK) may form the basis for the development of novel therapeutic strategies to prevent or alleviate genotoxic cardiotoxicities.
Supplementary Material
Highlights.
Cardiac FAK depletion exacerbated DOX-induced dysfunction and myocyte apoptosis.
Activation of cardiac FAK provided protection from DOX-induced toxicity.
FAK activation preserves mitochondrial integrity by inducing p21 and Bim.
Acknowledgments
The authors thank Devin Bailey for excellent technical assistance. This work was supported by grants from the National Heart, Lung, and Blood Institute, National Institutes of Health (HL-081844 and HL-071054 to J.M. Taylor) and the American Heart Association (AHA.0355776U to J.M. Taylor). Z. Cheng was supported by an American Heart Association Postdoctoral Fellowship (#11POST7600008).
Abbreviations
- CDK
cyclin-dependent kinase
- CDKI
cyclin-dependent kinase inhibitor
- DOX
doxorubicin
- FAK
focal adhesion kinase
- MFKO
myocyte-restricted FAK knockout mice
- NRCM
neonatal rat cardiomyocytes
- NTG
non-transgenic littermate control mice
- SF
Super activatible FAK variant, or SuperFAK
- SF2
cardiac-specific SF transgenic mice line 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures None.
References
- [1].Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–5. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- [2].Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–62. doi: 10.1159/000265166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–8. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- [4].Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- [5].Zhang XP, Liu F, Wang W. Two-phase dynamics of p53 in the DNA damage response. Proc Natl Acad Sci U S A. 2011;108:8990–5. doi: 10.1073/pnas.1100600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273:25–32. doi: 10.1007/s11010-005-5905-8. [DOI] [PubMed] [Google Scholar]
- [7].Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, et al. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009;119:99–106. doi: 10.1161/CIRCULATIONAHA.108.799700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, et al. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286:H933–9. doi: 10.1152/ajpheart.00759.2003. [DOI] [PubMed] [Google Scholar]
- [9].Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–9. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Naci D, El Azreq MA, Chetoui N, Lauden L, Sigaux F, Charron D, et al. alpha2beta1 Integrin Promotes Chemoresistance against Doxorubicin in Cancer Cells through Extracellular Signal-regulated Kinase (ERK) J Biol Chem. 2012;287:17065–76. doi: 10.1074/jbc.M112.349365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Eke I, Deuse Y, Hehlgans S, Gurtner K, Krause M, Baumann M, et al. beta(1) Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J Clin Invest. 2012;122:1529–40. doi: 10.1172/JCI61350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nam JM, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 2010;70:5238–48. doi: 10.1158/0008-5472.CAN-09-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Martinkova E, Maglott A, Leger DY, Bonnet D, Stiborova M, Takeda K, et al. alpha5beta1 integrin antagonists reduce chemotherapy-induced premature senescence and facilitate apoptosis in human glioblastoma cells. Int J Cancer. 2010;127:1240–8. doi: 10.1002/ijc.25187. [DOI] [PubMed] [Google Scholar]
- [14].Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivity. Biochem Biophys Res Commun. 2003;311:786–92. doi: 10.1016/j.bbrc.2003.10.060. [DOI] [PubMed] [Google Scholar]
- [15].Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67:10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- [16].Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8:2435–43. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith CS, Golubovskaya VM, Peck E, Xu LH, Monia BP, Yang X, et al. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Res. 2005;15:357–62. doi: 10.1097/00008390-200510000-00003. [DOI] [PubMed] [Google Scholar]
- [18].Chen YY, Wang ZX, Chang PA, Li JJ, Pan F, Yang L, et al. Knockdown of focal adhesion kinase reverses colon carcinoma multicellular resistance. Cancer Sci. 2009;100:1708–13. doi: 10.1111/j.1349-7006.2009.01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen Y, Wang Z, Chang P, Xiang L, Pan F, Li J, et al. The effect of focal adhesion kinase gene silencing on 5-fluorouracil chemosensitivity involves an Akt/NF-kappaB signaling pathway in colorectal carcinomas. Int J Cancer. 2010;127:195–206. doi: 10.1002/ijc.25025. [DOI] [PubMed] [Google Scholar]
- [20].DiMichele LA, Doherty JT, Rojas M, Beggs HE, Reichardt LF, Mack CP, et al. Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ Res. 2006;99:636–45. doi: 10.1161/01.RES.0000240498.44752.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Peng X, Kraus MS, Wei H, Shen TL, Pariaut R, Alcaraz A, et al. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–27. doi: 10.1172/JCI24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Clemente CF, Tornatore TF, Theizen TH, Deckmann AC, Pereira TC, Lopes-Cendes I, et al. Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ Res. 2007;101:1339–48. doi: 10.1161/CIRCRESAHA.107.160978. [DOI] [PubMed] [Google Scholar]
- [23].Hakim ZS, DiMichele LA, Rojas M, Meredith D, Mack CP, Taylor JM. FAK regulates cardiomyocyte survival following ischemia/reperfusion. J Mol Cell Cardiol. 2009;46:241–8. doi: 10.1016/j.yjmcc.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cheng Z, DiMichele LA, Hakim ZS, Rojas M, Mack CP, Taylor JM. Targeted focal adhesion kinase activation in cardiomyocytes protects the heart from ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2012;32:924–33. doi: 10.1161/ATVBAHA.112.245134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- [26].Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation. 2004;110:2869–74. doi: 10.1161/01.CIR.0000146889.46519.27. [DOI] [PubMed] [Google Scholar]
- [27].Gabarra-Niecko V, Keely PJ, Schaller MD. Characterization of an activated mutant of focal adhesion kinase: ‘SuperFAK’. Biochem J. 2002;365:591–603. doi: 10.1042/BJ20020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Spallarossa P, Altieri P, Aloi C, Garibaldi S, Barisione C, Ghigliotti G, et al. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am J Physiol Heart Circ Physiol. 2009;297:H2169–81. doi: 10.1152/ajpheart.00068.2009. [DOI] [PubMed] [Google Scholar]
- [29].Wang S, Basson MD. Protein kinase B/AKT and focal adhesion kinase: two close signaling partners in cancer. Anticancer Agents Med Chem. 2011;11:993–1002. doi: 10.2174/187152011797927661. [DOI] [PubMed] [Google Scholar]
- [30].Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Graham K, Moran-Jones K, Sansom OJ, Brunton VG, Frame MC. FAK deletion promotes p53-mediated induction of p21, DNA-damage responses and radio-resistance in advanced squamous cancer cells. PLoS One. 2011;6:e27806. doi: 10.1371/journal.pone.0027806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Koster R, di Pietro A, Timmer-Bosscha H, Gibcus JH, van den Berg A, Suurmeijer AJ, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010;120:3594–605. doi: 10.1172/JCI41939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Spierings DC, de Vries EG, Stel AJ, te Rietstap N, Vellenga E, de Jong S. Low p21Waf1/Cip1 protein level sensitizes testicular germ cell tumor cells to Fas-mediated apoptosis. Oncogene. 2004;23:4862–72. doi: 10.1038/sj.onc.1207617. [DOI] [PubMed] [Google Scholar]
- [34].Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–49. [PubMed] [Google Scholar]
- [35].Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–46. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- [36].Avitabile D, Bailey B, Cottage CT, Sundararaman B, Joyo A, McGregor M, et al. Nucleolar stress is an early response to myocardial damage involving nucleolar proteins nucleostemin and nucleophosmin. Proc Natl Acad Sci U S A. 2011;108:6145–50. doi: 10.1073/pnas.1017935108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- [38].Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22:33–41. doi: 10.1016/j.tcb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, et al. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. 2005;19:2051–3. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- [40].Hagenbuchner J, Kuznetsov A, Hermann M, Hausott B, Obexer P, Ausserlechner MJ. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J Cell Sci. 2012;125:1191–203. doi: 10.1242/jcs.092098. [DOI] [PubMed] [Google Scholar]
- [41].Lopez-Royuela N, Perez-Galan P, Galan-Malo P, Yuste VJ, Anel A, Susin SA, et al. Different contribution of BH3-only proteins and caspases to doxorubicin-induced apoptosis in p53-deficient leukemia cells. Biochem Pharmacol. 2010;79:1746–58. doi: 10.1016/j.bcp.2010.02.010. [DOI] [PubMed] [Google Scholar]
- [42].Cardinale D, Bacchiani G, Beggiato M, Colombo A, Cipolla CM. Strategies to prevent and treat cardiovascular risk in cancer patients. Semin Oncol. 2013;40:186–98. doi: 10.1053/j.seminoncol.2013.01.008. [DOI] [PubMed] [Google Scholar]
- [43].Le HV, Minn AJ, Massague J. Cyclin-dependent kinase inhibitors uncouple cell cycle progression from mitochondrial apoptotic functions in DNA-damaged cancer cells. J Biol Chem. 2005;280:32018–25. doi: 10.1074/jbc.M504689200. [DOI] [PubMed] [Google Scholar]
- [44].Sohn D, Essmann F, Schulze-Osthoff K, Janicke RU. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase-mediated caspase-9 activation. Cancer Res. 2006;66:11254–62. doi: 10.1158/0008-5472.CAN-06-1569. [DOI] [PubMed] [Google Scholar]
- [45].Li CH, Tzeng SL, Cheng YW, Kang JJ. Chloramphenicol-induced mitochondrial stress increases p21 expression and prevents cell apoptosis through a p21-dependent pathway. J Biol Chem. 2005;280:26193–9. doi: 10.1074/jbc.M501371200. [DOI] [PubMed] [Google Scholar]
- [46].Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. J Biol Chem. 2002;277:37949–54. doi: 10.1074/jbc.M204497200. [DOI] [PubMed] [Google Scholar]
- [47].Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–34. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19. doi: 10.1017/S1462399408000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Terrand J, Xu B, Morrissy S, Dinh TN, Williams S, Chen QM. p21(WAF1/Cip1/Sdi1) knockout mice respond to doxorubicin with reduced cardiotoxicity. Toxicol Appl Pharmacol. 2011;257:102–10. doi: 10.1016/j.taap.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Merched AJ, Chan L. Absence of p21Waf1/Cip1/Sdi1 modulates macrophage differentiation and inflammatory response and protects against atherosclerosis. Circulation. 2004;110:3830–41. doi: 10.1161/01.CIR.0000148681.01282.89. [DOI] [PubMed] [Google Scholar]
- [51].Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–44. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Choi JS, Shin S, Jin YH, Yim H, Koo KT, Chun KH, et al. Cyclin-dependent protein kinase 2 activity is required for mitochondrial translocation of Bax and disruption of mitochondrial transmembrane potential during etoposide-induced apoptosis. Apoptosis. 2007;12:1229–41. doi: 10.1007/s10495-006-0047-3. [DOI] [PubMed] [Google Scholar]
- [53].Chae HD, Kim BM, Yun UJ, Shin DY. Deregulation of Cdk2 causes Bim-mediated apoptosis in p53-deficient tumors following actin damage. Oncogene. 2008;27:4115–21. doi: 10.1038/onc.2008.46. [DOI] [PubMed] [Google Scholar]
- [54].Cerqueira A, Santamaria D, Martinez-Pastor B, Cuadrado M, Fernandez-Capetillo O, Barbacid M. Overall Cdk activity modulates the DNA damage response in mammalian cells. J Cell Biol. 2009;187:773–80. doi: 10.1083/jcb.200903033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11:2747–55. doi: 10.4161/cc.21127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120:2528–36. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Adachi S, Ito H, Tamamori-Adachi M, Ono Y, Nozato T, Abe S, et al. Cyclin A/cdk2 activation is involved in hypoxia-induced apoptosis in cardiomyocytes. Circ Res. 2001;88:408–14. doi: 10.1161/01.res.88.4.408. [DOI] [PubMed] [Google Scholar]
- [58].Hauck L, Hansmann G, Dietz R, von Harsdorf R. Inhibition of hypoxia-induced apoptosis by modulation of retinoblastoma protein-dependent signaling in cardiomyocytes. Circ Res. 2002;91:782–9. doi: 10.1161/01.res.0000041030.98642.41. [DOI] [PubMed] [Google Scholar]
- [59].Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–3. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–81. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–99. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest. 2000;105:533–9. doi: 10.1172/JCI8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Martinez LA, Yang J, Vazquez ES, Rodriguez-Vargas Mdel C, Olive M, Hsieh JT, et al. p21 modulates threshold of apoptosis induced by DNA-damage and growth factor withdrawal in prostate cancer cells. Carcinogenesis. 2002;23:1289–96. doi: 10.1093/carcin/23.8.1289. [DOI] [PubMed] [Google Scholar]
- [64].Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol. 2009;47:698–705. doi: 10.1016/j.yjmcc.2009.07.024. [DOI] [PubMed] [Google Scholar]
- [65].van Nimwegen MJ, Huigsloot M, Camier A, Tijdens IB, van de Water B. Focal adhesion kinase and protein kinase B cooperate to suppress doxorubicin-induced apoptosis of breast tumor cells. Mol Pharmacol. 2006;70:1330–9. doi: 10.1124/mol.106.026195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.