

Abstract
Fusarium avenaceum is a fungus commonly isolated from soil and associated with a wide range of host plants. We present here three genome sequences of F. avenaceum, one isolated from barley in Finland and two from spring and winter wheat in Canada. The sizes of the three genomes range from 41.6–43.1 MB, with 13217–13445 predicted protein-coding genes. Whole-genome analysis showed that the three genomes are highly syntenic, and share>95% gene orthologs. Comparative analysis to other sequenced Fusaria shows that F. avenaceum has a very large potential for producing secondary metabolites, with between 75 and 80 key enzymes belonging to the polyketide, non-ribosomal peptide, terpene, alkaloid and indole-diterpene synthase classes. In addition to known metabolites from F. avenaceum, fuscofusarin and JM-47 were detected for the first time in this species. Many protein families are expanded in F. avenaceum, such as transcription factors, and proteins involved in redox reactions and signal transduction, suggesting evolutionary adaptation to a diverse and cosmopolitan ecology. We found that 20% of all predicted proteins were considered to be secreted, supporting a life in the extracellular space during interaction with plant hosts.
Introduction
Fusarium is a large, ubiquitous genus of ascomycetous fungi that includes many important plant pathogens, as well as saprophytes and endophytes. The genomes of sixteen Fusarium spp. have been sequenced during the past decade with a focus on species that either display a narrow host plant range or which have a saprophytic life style. Fusarium avenaceum is a cosmopolitan plant pathogen with a wide and diverse host range and is reported to be responsible for disease on>80 genera of plants [1]. It is well-known for causing ear blight and root rot of cereals, blights of plant species within genera as diverse as Pinus and Eustoma [2], as well as post-harvest storage rot of numerous crops, including potato [3], broccoli [4], apple [5] and rutabaga [6]. Fusarium avenaceum has also been described as an endophyte [7], [8] and an opportunistic pathogen of animals [9], [10]. The generalist pathogen nature of F. avenaceum is supported by several reports on isolates that lack host specificity. One example of this is the report of F. avenaceum isolates from Eustroma sp. (aka Lisianthus) being phylogenetically similar to isolates from diverse geographical localities or which have been isolated from other hosts [11].
Fusarium avenaceum is often isolated from diseased grains in temperate areas, but an increased prevalence has also been reported in warmer regions throughout the world [12], [13]. The greatest economic impact of F. avenaceum is associated with crown rot and head blight of wheat and barley, and the contamination of grains with mycotoxins [12]. Co-occurrence of multiple Fusarium species in head blight infections is often observed, and several studies covering the boreal and hemiboreal climate zones in the northern hemisphere have revealed that F. avenaceum is often among the dominating species [14]. Previously, F. avenaceum has been shown to produce several secondary metabolites, including moniliformin, enniatins, fusarin C, antibiotic Y, 2-amino-14,16-dimethyloctadecan-3-ol (2-AOD-3-ol), chlamydosporol, aurofusarin [12], [15] and recently also fusaristatin A [16].
The genus Fusarium includes both broad-host pathogenic species, utilizing a generalist strategy, and narrow-host pathogenic species, which are specialized to a limited number of plant species. The F. oxysporum complex is a well-documented example of the specialist strategy, as each forma specialis displays a narrow host range. The genetic basis for this host specialization is dictated by a limited number of transferable genes, encoded on dispensable chromosomes [17]. However, the genetic foundation that allows F. avenaceum to infect such a wide range of host plant species and cope with such a diverse set of environmental conditions is currently not well understood. In an effort to shed light on the genetic factors that separates generalists from specialists within Fusarium, we sequenced the genomes of three different F. avenaceum strains isolated from two geographical locations, Finland and Canada, and from three small grain host plants: barley, spring and winter wheat. Comparison with existing Fusarium genomes would further explore pathogenic strategies.
Results and Discussion
Fusarium avenaceum genome sequences
We have sequenced three F. avenaceum genomes, one Finnish isolate from barley (Fa05001) and two Canadian isolates from spring (FaLH03) and winter wheat (FaLH27). Assembly of the 454 pyrosequencing based genomic sequence data from Fa05001 resulted in a total genome size of 41.6 Mb, while assembly of the Illumina HiSeq data for FaLH03 and FaLH27 resulted in genome sizes of 42.7 Mb and 43.1 Mb, respectively. Additional details on the assemblies can be found in (Table 1). Gene calling of the three F. avenaceum strains resulted in 13217 (Fa05001, gene naming convention FAVG1_XXXXX), 13293 (FaLH03, genes named FAVG2_XXXXX) and 13445 (FaLH27, genes named FAVG3_XXXXX) unique protein coding gene models. Previous comparative genomics studies of filamentous fungi have identified 69 core genes that are found ubiquitously across all fungal clades [18]. All three gene sets included the 69 core genes, suggesting a good assembly and reliable protein-coding gene prediction. Genome sequence data has been deposited at NCBI GenBank in the Whole Genome Shotgun (WGS) database as accession no. JPYM00000000 (Fa05001), JQGD00000000 (FaLH03) and JQGE00000000 (FaLH27), within BioProject PRJNA253730. The versions described in this paper are JPYM01000000, JQGD01000000, and JQGE01000000.
Table 1. Main assembly summary and annotation features of the three F. avenaceum genomes.
| Strain | Fa05001 | FaLH03 | FaLH27 |
| Sequencing technology | 454 | Hiseq | Hiseq |
| Genome size (Mb) | 41.6 | 42.7 | 43.2 |
| Sequencing coverage* | 21.6x | 426.6x | 986.2x |
| Number of contigs | 110 | 180 | 169 |
| Number of scaffolds | 83 | 104 | 77 |
| Number of Large Scaffolds (>100 Kb) | 40 | 22 | 18 |
| Number of Large Scaffolds (>1 Mb) | 17 | 14 | 11 |
| N50 scaffold length (Mb) | 1.43 | 4.11 | 4.14 |
| L50 scaffold count | 10 | 5 | 5 |
| GC content (%) | 48% | 48% | 48% |
| Number of predicted genes | 13217 | 13293 | 13445 |
| Average no of genes per Mb | 317.7 | 311.6 | 311.8 |
| Mean gene length (base pairs) | 1554 | 1557 | 1552 |
*Post- removal of mitochondrial genome.
The mitochondrial genome sequence was contained within a single assembled contig for each strain (Fa05001, 49075 bp; FaLH03, 49402 bp; FaLH27, 49396 bp), supporting sufficient coverage and a high quality assembly. Prior to trimming, the FaLH03 and FaLH27 mitochondrial contigs contained 39 and 53 bp, respectively, of sequence duplicated at each end, as expected with the acquisition of a circular sequence. As found in other Fusarium mitochondrial genomes [19], [20], the F. avenaceum mitochondrial genome sequences contain a low G+C content (about 33%) and encode 26 tRNAs and the ribosomal rRNAs rnl and rns. In addition, the 14 expected core genes (cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, atp6, atp8, atp9) involved in oxidative phosphorylation and ATP production are present and in the same order as other Fusarium mitochondrial genomes.
Genome structure in F. avenaceum
Electrophoretic karyotyping was performed to resolve the number of chromosomes in Fa05001. Previous karyotyping via fluorescence in situ hybridization has suggested that F. avenaceum isolated from wheat had 8–10 chromosomes [21]. Our attempt to determine the chromosome number in F. avenaceum Fa05001 strain by electrophoretic karyotyping was hampered due to the large size of several of the chromosomes. Southern analysis using a telomeric probe did however result in the detection of four distinct bands ranking from 1 to 5 MB, and several diffuse bands above the detection limit of the method (∼5 Mb) (Figure S1 in File S1). A high order reordering of the scaffolds from the three sequenced genomes resulted in 11 supercontigs ranging from 0.8 Mb to 6.5 Mb in size, likely corresponding to entire chromosomes or chromosome arms (Figure S2 in File S1). The three genomes display a high level of microcolinearity and only a single putative large genome rearrangement was observed in an internal region of Supercontig 1 between Fa5001 and Canadian isolates (Figure S3 in File S1).
Sequence comparisons between the three genomes revealed a 91–96% nucleotide alignment, with the two Canadian isolates having the fewest unaligned bases. In addition, approximately 1.4–3.2% of the aligned nucleotides exhibited single nucleotide polymorphisms (SNPs), insertions, or deletions between isolates; these were also fewer between the Canadian isolates (Figure 1). These genetic differences were unevenly distributed across the genomes and were largely concentrated at the ends of the supercontigs, while centrally located regions remained relatively conserved. This is similar to what has been previously observed between chromosomes of other Fusarium spp. [22]. BLASTn analysis indicated that more than 95% of predicted genes had a significant hit within the two other F. avenaceum genomes (Figure S4 in File S1). Together, the results suggest that, despite the large geographical distance between the collection sites, there is a high level of similarity between the three F. avenaceum genomes, both in genome structure and gene content. However, some instances of poorly conserved or missing genes were observed in either one or two isolates out of the three (Figure S4 in File S1, File S2, File S3). For example, the three isolates contained some unique polyketide synthases and non-ribosomal peptide synthases. This suggests that there may be some differences in secondary metabolism between the isolates.
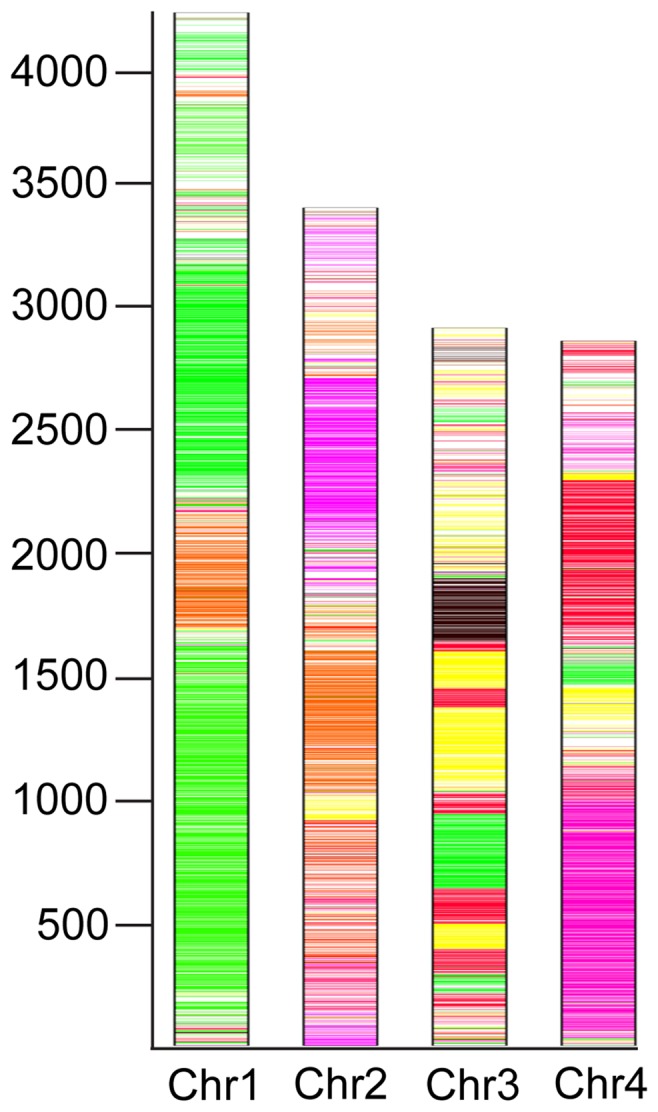
Figure 1. Sliding window map with numbers of SNP's and indels per 20 kb in the three F. avenaceum strains Fa05001, FaLH03 and FaLH27 on the 11 supercontigs.

Locations of the polyketide synthase and non-ribosomal peptide synthetase genes in the strain FaLH27 are plotted on the supercontigs.
Comparison of genome structure to other Fusarium species
Phylogenetic analysis of genome-sequenced Fusaria based on RPB1, RPB2, rDNA cluster (18S rDNA, ITS1, 5.8S rDNA and 28S rDNA), EF-1a and Lys2 suggest that F. avenaceum is more closely related to F. graminearum, with greater phylogenetic distance to F. verticillioides, F. oxysporum and F. solani [23], [24]. Phylogeny using β-tub alone [24] suggested that F. avenaceum is more closely related to F. verticillioides than the other three species. The genome data for F. avenaceum allowed us to reanalyse the evolutionary history within the Fusarium genus based on the 69 conserved proteins, initially identified by Marcet-Houben and Gabaldón [18]. The Maximum Likelihood analysis was based on 25,535 positions distributed on six super-proteins and showed that F. graminearum and F. avenaceum clustered together in 93% of 500 iterations, with F. oxysporum and F. verticillioides as sister taxa in 100% of the cases (Figure 2). Fusarium avenaceum scaffolds have good alignment with supercontigs of both F. graminearum and F. verticillioides with long, similar stretches of syntenic regions (Figure S5 in File S1). The synteny of F. avenaceum with F. graminearum is visualized in Figure 3, in which long stretches of genes from the same F. avenaceum supercontig have orthologs to neighbouring genes on F. graminearum chromosomes, indicating a shared genomic architecture. F. graminearum genes lacking orthologs in F. avenaceum are not distributed uniformly across the supercontig, and are mostly confined to telomeric regions, except for some at interstitial chromosomal sites. Such chromosome regions in F. graminearum have been shown to have a higher SNP density [22] and are influencing host specific gene expression patterns [25].
Figure 2. Molecular phylogenetic analysis of Fusarium species based on 69 orthologous proteins.

The evolutionary history was inferred by using the Maximum Likelihood method and the tree with the highest log likelihood (−152577,9625) is shown. Bootstrap values, as percentages, are shown next to the individual branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated prior to the ML analysis and the final data set contained 25535 positions.
Figure 3. Shared gene homology map between Fa05001 and F. graminearum PH-1 has been created using the four defined F. graminearum chromosomes as templates.
Genes in F. graminearum are coloured according to whether genes have corresponding orthologs in Fa05001, with one gene being one strip. Regions of the same color match to the same supercontigs (Figure S2 in File S1) in F. avenaceum. White regions represent lack of orthologs in Fa05001.
Fusarium avenaceum possesses the genetic hallmarks of a heterothallic sexual life cycle
The observation of two mating-type idiomorphs was another dissimilarity between the F. avenaceum isolates [26], [27]. The Finnish F. avenaceum isolate is of the mating-type MAT1-1, possessing the three genes MAT1-1-1, MAT1-1-2, and MAT1-1-3 (FAVG1_07020, FAVG1_07021, and FAVG1_07022), while the two Canadian isolates are of mating-type MAT1-2, containing the genes MAT1-2-1 and MAT1-2-3 (FAVG2_03853 and FAVG2_03854 or FAVG3_03869 and FAVG3_03870). Such idiomorphs with different sets of genes has been observed previously in Fusarium spp. [26], [27], and surveys of F. avenaceum populations often find isolates evenly split between mating types [28]. The sexual stage of F. avenaceum has been observed [29], [30], and both MAT1-1 and MAT1-2 transcripts have been detected in this species under conditions favorable for perithecial production in other Fusaria [31], suggesting that F. avenaceum is likely capable of heterothallism. This is further supported by our data, in which a single mating-type is present in a given F. avenaceum isolate. This is characteristic for heterothallic fungal species, differing from homothallic species such as F. graminearum, which contain both mating-types in a single nucleus [32].
Occurrence of few repetitive elements supports the hypothesis that F. avenaceum is sexually active
A search for repetitive elements in the Fa05001 genome (using RepeatMasker [33] with CrossMatch) identified 1.0% of the Fa05001 genome as being repetitive or corresponding to transposable elements (Table S1 in File S1). This value is comparable to the 1.12% found for F. graminearum, although there were differences in the distributions of the various types of genetic elements. Tad1 and MULE-MuDR transposons were more enriched in F. avenaceum while F. graminearum had higher proportions of the TcMar-Ant1 transposon and small RNA. The low level of repeats supports the hypothesis that F. avenaceum is sexually active in nature, as such low levels are typical for species with an active sexual cycle, such as F. graminearum, F. verticillioides [1.21% repeats] and F. solani [3.8% repeats], while species which rely on asexual reproduction, such as F. oxysporum, have higher levels [10.6% repeats]. These results could be somewhat influenced by the fact that the Fusaria genomes are generated with different technologies.
Gene families enriched in F. avenaceum
Analysis of the predicted function of the 13217 Fa05001 gene models showed that Fa05001 contains a greater diversity of InterPro families than the other four analyzed Fusaria (Table 2, Figure 4, and File S4). Two highly enriched InterPro categories stand out in Fa05001; “Polyketide synthase, enoylreductase” (IPR020843) and “Tyrosine-protein kinase catalytic domain” (IPR020635), involved in secondary metabolism and signal transduction, respectively. With the exception of F. solani, Fa05001 also has the highest number of predicted transcription factors (Table S2 in File S1). Sixty-eight InterPro domains were predicted to be unique to Fa05001, including four tryptophan dimethylallyltransferase (IPR012148) proteins, commonly found in alkaloid biosynthesis. A comparison of Gene Ontology (GO) terms [34] indicated additional differences between the analyzed Fusarium species. Functional categories in which Fa05001 had higher numbers of proteins than the other genome-sequenced Fusaria were: “Cellular response to oxidative stress”, “Branched-chain amino acid metabolic process”, “Toxin biosynthetic process”, “Oxidoreductase activity”, “rRNA binding” and “Glutathione transferase activity” (Table S3, S4, S5 in File S1).
Table 2. InterproScan analysis and comparison of Fa05001 with other Fusaria.
| InterPro analysis | Families | Total domains |
| Fa05001 | 5,710 | 27,474 |
| F. graminearum | 5,602 | 23,560 |
| F. oxysporum | 5,609 | 29,873 |
| F. verticillioides | 5,545 | 25,393 |
| F. solani | 5,582 | 30,538 |
Figure 4. Functional analysis of Fa05001 and other sequenced Fusaria based on InterPro visualizing similarities and differences between the fungi.

Categories with most differences between Fa05001 and others are presented with the number of proteins in each category. All details are listed in File S4. FA = F. avenaceum Fa05001, FG = F. graminearum PH-1, FO = F. oxysporum f. sp. lycopersici 4287, FV = F. verticillioides, FS = F. solani.
Reciprocal BLAST revealed that about ¾ of the predicted proteins in Fa05001 have orthologs in F. graminearum (76.7%), F. verticillioides (76.9%), F. oxysporum (78.8%) and F. solani (74.1%) (File S5). The F. avenaceum proteins for which no ortholog (no hits or e-value>10−10) was found in the other Fusarium genomes were especially enriched in GO biological categories “Oxidation-reduction process”; “Toxin biosynthetic process”, “Alkaloid metabolic process”; “Cellular polysaccharide catabolic process” and “Transmembrane transport” (Table 3, Table 4, Figure S6 in File S1).
Table 3. Enriched biological processes in Fa05001 proteins without orthologs in other genome sequenced Fusaria (P<0.05).
| F. avenaceum vs F. graminearum | |||
| GO-ID | Term | FDR | P-Value |
| GO: 0055114 | Oxidation-reduction process | 2.16E-06 | 9.72E-10 |
| GO: 0009820 | Alkaloid metabolic process | 0.35 | 6.33E-04 |
| GO: 0003333 | Amino acid transmembrane transport | 0.37 | 0.001 |
| GO: 0009403 | Toxin biosynthetic process | 0.71 | 0.004 |
| GO: 0016998 | Cell wall macromolecule catabolic process | 1 | 0.02 |
| GO: 0006366 | Transcription from RNA polymerase II promoter | 1 | 0.03 |
| GO: 0044247 | Cellular polysaccharide catabolic process | 1 | 0.03 |
| GO: 0006573 | Valine metabolic process | 1 | 0.03 |
See also Figure S6 in File S1.
Table 4. Genes annotated as reduction-oxidation process in Fa05001 proteins without orthologs in other genome sequenced Fusaria (with expect>1e-10).
| Reduction-oxidation function | Genes |
| 4-carboxymuconolactone decarboxylase | FAVG1_07738 |
| ABC multidrug | FAVG1_08208 |
| Acyl dehydrogenase | FAVG1_08563, FAVG1_04763 |
| Alcohol dehydrogenase | FAVG1_04699, FAVG1_12680 |
| Aryl alcohol dehydrogenase | FAVG1_08747 |
| Bifunctional p-450: nadph-p450 reductase | FAVG1_11632 |
| Transcription factor | FAVG1_09453, FAVG1_07648 |
| Choline dehydrogenase | FAVG1_12183 |
| Cytochrome p450 monooxygenase | FAVG1_08576, FAVG1_13151, FAVG1_07923, FAVG1_10161, FAVG1_08627, FAVG1_02807, FAVG1_08721, FAVG1_10699 |
| Delta-1-pyrroline-5-carboxylate dehydrogenase | FAVG1_04749 |
| Dimethylaniline monooxygenase | FAVG1_11196 |
| Ent-kaurene synthase | FAVG1_10701 |
| Glutaryl- dehydrogenase | FAVG1_02842 |
| Homoserine dehydrogenase | FAVG1_09776 |
| l-lactate dehydrogenase a | FAVG1_08604 |
| Mitochondrial 2-oxoglutarate malate carrier protein | FAVG1_08564 |
| Monooxygenase fad-binding protein | FAVG1_10705 |
| Nadp-dependent alcohol dehydrogenase | FAVG1_10174, FAVG1_12103, FAVG1_06950 |
| Nitrilotriacetate monooxygenase component b | FAVG1_07690 |
| Pyoverdine dityrosine biosynthesis | FAVG1_12703 |
| Salicylate 1-monooxygenase | FAVG1_09825 |
| Salicylaldehyde dehydrogenase | FAVG1_09676 |
| Short-chain dehydrogenase | FAVG1_07710, FAVG1_12690, FAVG1_04239, FAVG1_10281, FAVG1_08519 |
See also Figure S6 in File S1.
The secretome of F. avenaceum
The interplay between the invading fungus and the host plant occurs mainly in the extra-cellular space. The proportion of genes encoding predicted secreted proteins in the Fa05001 genome (File S6, Table S6 in File S1) is remarkably high (∼20%; 2,580 proteins) as compared to plant pathogens such as F. graminearum (11%) and Magnaporthe grisea (13%), saprophytes such as Neurospora crassa (9%) and Aspergillus nidulans (9%) [22], and the insect pathogen Cordyceps militaris (16%) [35]. The secretome appears particularly enriched in proteins involved in redox reactions (Figure S7 in File S1). Inspecting the F. avenaceum proteins with no orthologs in other Fusaria (1223 proteins from Figure S6 in File S1), we found that 36% were predicted to be secreted.
Small cysteine-rich proteins (CRPs) can exhibit diverse biological functions, and some have been shown to play a role in virulence, including Avr2 and Avr4 in Cladosporium fulvum [36] and the Six effectors in F. oxysporum f. sp. lycopersici [37]. Other reported functions for CRPs have been adherence [38], antimicrobiosis [39] and carbohydrate binding activity that interferes with host recognition of the pathogen [40]. We found 19 candidates in Fa05001 containing more than four cysteine residues, and an additional 55 containing less than four cysteine residues, but with significant similarity to CRP HMM models (File S7, Table S7 in File S1). Of the predicted F. avenaceum CRPs, several are also found in other sequenced Fusarium species, but only two were noted with a putative function: CRP5760, with similarity to lectin-B, and CRP5810, a putative chitinase 3.
Metabolic profiling of F. avenaceum
A determination of secondary metabolites produced by these three F. avenaceum strains was performed on agar media, additionally Fa05001 was also grown in planta (barley and oat). Extraction of F. avenaceum cultures grown on PDA and YES solid media revealed the presence of 2-amino-14,16-dimethyloctadecan-3-ol, acuminatopyrone, antibiotic Y, fusaristatin A, aurofusarin, butenolide, chlamydosporols, chrysogine, enniatin A, enniatin B, fusarin C, and moniliformin (Table 5). These metabolites have previously been reported from a broad selection of F. avenaceum strains [5], [16], [41], [42] whereas the following metabolites previously reported from F. avenaceum were not detected in the present study: beauvericin [43], fosfonochlorin [44], diacetoxyscirpenol, T-2 toxin and zearalenone [45]–[50]. These reports are based on single observations using insufficient specific chemical methods that could yield false positive detection and/or poor fungal identification and no deposition of the strain in a collection for verification. The lack of zearalenone production agrees with the finding that none of the three F. avenaceum genomes described here contains the genes required for its production [51], [52].
Table 5. Metabolic profiling of the three F. avenaceum strains.
| FaLH03 | FaLH27 | Fa05001 | Other strains | ||||||
| YES | PDA | YES | PDA | YES | PDA | Barley | OAT | F. avenaceum | |
| PKS | |||||||||
| 2-Amino-14,16-dimethyloctadecan-3-ol | + | + | + | + | + | + | ND | ND | + |
| Acuminatopyrone | + | + | ND | ND | + | + | ND | ND | + |
| Antibiotic Y | + | + | + | + | + | + | ND | ND | + |
| Aurofusarin | + | + | + | + | + | + | + | + | + |
| Fuscofusarin | + | + | + | + | + | + | ND | ND | NA |
| Moniliformin | + | + | + | + | + | + | NA | NA | + |
| Chlamydosporols | + | + | ND | ND | + | + | ND | ND | + |
| NRPS and mixed NRPS-PKS | |||||||||
| Butenolide | ND | ND | ND | ND | + | + | + | ND | + |
| Chrysogine | + | + | + | + | + | + | + | + but 10 × lower than barley | + |
| Visoltricin | ND | ND | ND | ND | ND | ND | ND | ND | + (by UV-Vis) |
| Fusarins C and A | + | ND | + | ND | + | + | ND | ND | + |
| Enniatins A's and B's | + | + | + | + | + | + | + | + but 100 × lower than barley | + |
| Beauvericin | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Apicidin | ND | ND | ND | ND | ND | ND | ND | ND | + |
| Fusaristatins | + | + | + | + | + | + | + | Trace | + |
| Fusarielin A | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Ferricrocin | + | ND | + | ND | + | + | ND | ND | NA |
| Fusarinines | ND | ND | ND | ND | ND | ND | ND | ND | NA |
| JM-47 | + | + | + | + | + | + | + | + | NA |
| Malonichrome | + | ND | + | ND | + | ND | ND | ND | NA |
| Other | |||||||||
| Fosfonochlorin | ND | ND | ND | ND | ND | ND | ND | ND | NA |
| Unknown 26 (NRPS) | + | + | + | + | |||||
| Fusarium unknown 31 (NRPS) | + | + | + | + | |||||
| ND not detected | |||||||||
| NA not analyzed | |||||||||
Preliminary genomic analysis had predicted the production of ferricrocin, malonichrome, culmorin, fusarinine and gibberellins [23], but chemical analysis only verified the production of the first two metabolites. The polyketide fuscofusarin (an aurofusarin analogue or intermediate in the biosynthetic pathway) was detected for the first time in F. avenaceum. Furthermore, fusaristatin A was found for the first time to be produced in planta during climate chamber experiments, and was generally found in F. avenaceum-infected barley, but only in trace amounts in oat. In addition, the tetrapeptide JM-47, an HC-toxin analogue reported from an unidentified Fusarium strain [53], was detected from all three strains in all cultures, including in barley and oats in climate chamber experiments. Apicidins have been detected in other strains of F. avenaceum cultured on YES agar (unpublished results), whereas these compounds were not detected in cultures of the three sequenced strains. Further characterization of the metabolomes on PDA and YES media revealed three major peaks in the ESI+ chromatograms, which were also detected in the barley and oat extracts, that could not be matched to known compounds in Antibase [54]. Since these novel compounds are produced in planta, they are candidates for novel virulence factors.
Large potential for secondary metabolite production
The three F. avenaceum genomes encode 75 (Fa05001), 77 (FaLH03) and 80 (FaLH27) key enzymes for the production of secondary metabolites, exhibiting a far greater biosynthetic potential than the known secondary metabolites produced by the species. These numbers include genes for 25–27 iterative type I polyketide synthases (PKSs), 2–3 type III PKSs, 25–28 non-ribosomal peptide synthases (NRPSs), four aromatic prenyltransferases (DMATS), 12–13 class I terpene synthases (head-to-tail incl. cyclase activity), two class I terpene synthase (head-to-head) and four class II terpene synthases (cyclases for the class I terpene synthase head-to-head type) (Table 6).
Table 6. The number of identified signature genes for secondary metabolism in the three F. avenaceum strains compared to the other public Fusarium genome sequences.
| PKS I | PKS III | NRPS | TS I HT* | TS I HH** | TS II*** | DMATS | |
| Fa05001 | 25 (12) | 2 (1) | 25 (9) | 13 (2) | 2(0) | 4(0) | 4(2) |
| FaLH03 | 25 (12) | 3 (2) | 26 (10) | 12 (1) | 2(0) | 4(0) | 4(2) |
| FaLH27 | 27 (14) | 2 (1) | 28 (12) | 13 (1) | 2(0) | 4(0) | 4(2) |
The number of genes that are unique for F. avenaceum is given in the parentheses. Gene classes: type I iterative PKS (PKS I), type III PKS (PKS III), non-ribosomal peptide synthetases (NRPS), terpene synthase class I head-to-tail type (TS I HT), terpene synthase class I head-to-head type (TS I HH), terpene synthases class II (TS II) and aromatic prenyltransferases (DMATS).
*incl. ERG20, COQ1, BTS1,
**incl. ERG9,
***incl. ERG7.
The number of type I PKSs is surprisingly high, considering that the six other public Fusarium genomes (F. graminearum, F. verticillioides, F. oxysporum, F. solani, F. pseudograminearum and F. fujikuroi) only encode between 13 and 18 type I PKSs. The three F. avenaceum genomes share a core set of 24 type I PKS, and the individual isolates also encode unique type I PKS: oPKS47 (FAVG1_08496) is unique to Fa05001, while the two Canadian isolates share a single oPKS53 (FAVG2_01811, FAVG3_01846) and FaLH27 has two additional PKSs, oPKS54 (FAVG3_02030) and oPKS55 (FAVG3_06174). Of the 55 different type I PKSs described in the seven Fusarium genomes, only two (oPKS3 and oPKS7) are found in all species. Orthologs for 12 of the type I PKSs found in F. avenaceum could be identified in one or several of the publicly available Fusarium genomes, and hence 16 are new to the genus (Figure 5). None of these 16 have obvious characterized orthologs in other fungal genomes or in the GenBank database. The PKSs with characterized orthologs within the Fusarium genus includes the PKSs responsible for formation of fusarubin (oPKS3), fusaristatins (oPKS6), fusarins (oPKS10), aurofusarin (oPKS12) and fusaristatin A (oPKS6) [16], [55]–[57]. Prediction of domain architecture of the 28 PKSs found in F. avenaceum showed that 17 belong to the reducing subclass, four to the non-reducing subclass, two are PKSs with a carboxyl terminal Choline/Carnitine O-acyltransferase domain, four are PKS-NRPS hybrids and one is a NRPS-PKS hybrid. The iterative nature of this enzyme class makes it impossible to predict the products of these enzymes without further experimental data; this research has been initiated. The F. avenaceum genomes also encode type III PKSs, a class that among fungi was first described in Aspergillus oryzae [58], and recently in F. fujikuroi [59]. The FaLH03 isolate possesses three proteins (oPKSIII-1 to oPKSIII-3), while the two other isolates only have two (oPKSIII-1 and oPKSIII-3).
Figure 5. Shared and unique polyketide synthase (PKS), non-ribosomal peptide synthetases (NRPS) and terpene cyclase (TC) encoding genes in public available Fusaria genomes.

Green and yellow boxes are the number of shared and unique genes, respectively. Fg = F. graminearum, Fv = F. verticillioides, Fo = F. oxysporum, Fs = F. solani, Fp = F. pseudograminearum, Ff = F. fujikuroi and Fa05001, FaLH03 and FaLH27 = F. avenaceum.
NRPSs provide an alternative to ribosomal-based polypeptide synthesis and in addition allow for the joining of proteinous amino acids, nonproteinous amino acids, α-hydroxy acids and fatty acids as well as cyclization of the resulting polypeptide [60]. The non-ribosomal peptide group of metabolites includes several well characterized bioactive compounds, such as HC-toxin (pathogenicity factor) and apicidin (histone deacetylase inhibitor) [61], [62]. Of the 30 unique NRPSs encoded by the three F. avenaceum isolates, 16 are novel to the Fusarium genus, and include seven mono-modular and nine multi-modular NRPS, with between 2 and 11 modules. Of these, NRPS41 (FAVG1_08623, FAVG2_11354 and FAVG3_11434) is a likely ortholog to gliP2 (similar to gliotoxin synthetase) from Neosartorya fischeri (Genbank accession no. EAW21276), sharing 75% amino acid identity. The other 14 NRPSs are orthologs to previously reported proteins in other Fusarium species [63], and include the three NRPSs responsible for the formation of the siderophores malonichrome (oNRPS1), ferricrocin (sidC, oNRPS2) and fusarinine (sidA, oNRPS6).
All three strains encoded oNRPS31 which shows a significant level of identity to the apicidin NRPS (APS1) described in F. incarnatum and F. fujikuroi [59], [64], and the HC-toxin NRPS (HTS1) from Cochliobolus carbonum SB111, Pyrenophora tritici-repentis and Setosphaeria turcica [65], [66]. It has not yet been investigated whether NRPS31 is involved in the production of JM-47. Alignment of the genomic regions surrounding oNRPS31 with the APS1 and HTS1 clusters, showed that the three F. avenaceum strains encode nine of the twelve APS proteins found in the other two Fusaria, but lack clear orthologs encoding APS3 (pyrroline reductase), APS6 (O-methyltransferase) and APS12 (unknown function). The APS-like gene cluster in F. avenaceum has undergone extensive rearrangements, resulting in the loss of the three APS genes and gain of three new ones (APS13-15) (Figure 6 and Table S8 in File S1). Of these, APS14 (FAVG1_08581, FAVG2_02887 and FAVG3_02926) encodes a fatty acid synthase β subunit, which shares 60% identity with the FAS β subunit (encoded by FAVG1_03575, FAVG2_06952 and FAVG3_07032) involved in primary metabolism. APS14 likely interacts with APS5 (fatty acid synthase α subunit) to form a functional fungal FAS (α6β6) responsible for the formation of the decanoic acid core of (S)-2-amino-8-oxodecanoic acid, proposed by Jin and co-workers [64] to be incorporated into apicidin by APS1. It has previously been hypothesized that APS5 (FAS α) interacts with the FAS α unit from primary metabolism to fulfill this role [64], however, the new model implies that the F. incarnatum genome encodes an unknown APS14 ortholog (no genome sequence available). The F. fujikuroi genome does not contain an APS14 ortholog (TBLASTn against the genome), but apicidin F has been detected in this species [59], [67]. Alignment of the apicidin and HC-toxin gene clusters confirmed the observations made by Manning et al. [66] and Condon et al. [65], regarding the similarities of the two types of gene clusters, which yield very different products (Figure 6).
Figure 6. Apicidin-like gene cluster (oNRPS31) in the three F. avenaceum strains (Fa05001_Scaffold14, FaLH03_contig11, FaLH27_contig13) compared to the characterized apicidin gene cluster from F. incarnatum and the HC-toxin gene clusters from Cochliobolus carbonum, Pyrenophora tritici-repentis and Setosphaeria turcica (A).
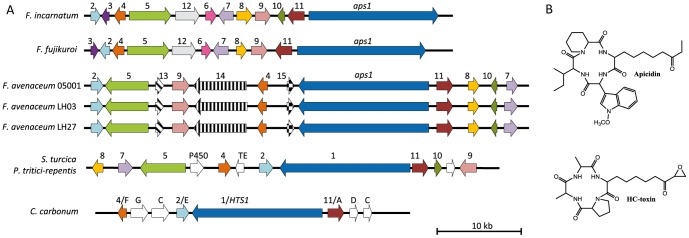
The genes are colored based on homology across the species. Chemical structure of apicidin and HC-toxin (B). See Table S8 in File S1 for further details.
Dimethylallyltransferase and indole-diterpene biosynthesis proteins are common in the production of bioactive compounds in endophytes [68], [69]. The three F. avenaceum genomes encoded four tryptophan dimethylallyltransferase (DMATS), aromatic prenyltransferases, typically involved in alkaloid biosynthesis or modification of other types of secondary metabolites [68]. This is one more than is found in F. verticillioides and F. oxysporum, and two more than found in F. fujikuroi. One putative indole-diterpene biosynthesis gene was found in all three F. avenaceum genomes (FAVG1_08136, FAVG2_00906 and FAVG3_00934).
The three F. avenaceum genomes are also rich in genes involved in terpene biosynthesis. In the case of the class I terpene synthase, responsible for the head-to-tail joining of isoprenoid and extended isoprenoid units and eventually cyclization, the three genomes encode 12–13 enzymes, of which three were putatively identified as being involved in primary metabolism (ERG20, COQ1, BTS1). In the case of the head-to-head class I systems, F. avenaceum encodes two enzymes, similar to the other fully genome-sequenced Fusaria, of which one gene encodes ERG9 and the other is involved in carotenoid biosynthesis. Cyclization of the formed head-to-head type product, if such a reaction occurs, is probably catalyzed by a class II terpene synthase/cyclase, of which F. avenaceum encodes four, including an ERG7 ortholog. This is more than the other Fusaria spp. genomes, as F. graminearum, F. pseudograminearum and F. verticillioides only have ERG7, while F. solani, F. oxysporum and F. fujikuroi have two.
One of the four Type II terpene synthase encoding genes (TS_II_01: FAVG1_10701, FAVG2_04190 and FAVG3_04223) shared by all three F. avenaceum strains was found to be orthologous to the gibberellic acid (GA) copalyldiphosphate/ent-kaurene synthase (cps/ks) from F. fujikuroi (Table S9 in File S1). Previously, the ability to synthesize the GA group of diterpenoid plant growth hormones in fungi has only been found in F. fujikuroi mating population A, Fusarium proliferatum, Sphaceloma manihoticola and Phaeosphaeria sp. strain L487 [70]–[74]. Biosynthesis of GA was first thoroughly characterized in F. fujikuroi and depends on the coordinated activity of seven enzymes, encoded by the GA gene cluster, that convert dimethylallyl diphosphate (DMAPP) to various types of gibberellic acids, with the main end-products being GA1 and GA3 [70]. S. manihoticola's GA biosynthesis ends at the intermediate GA4 due to the lack two of genes (DES and P450-3), compared to F. fujikuroi, responsible for converting GA4 to GA7, GA3 and GA1 [73]. Analysis of the genes surrounding the F. avenaceum CPS/KS encoding gene showed that six of the seven genes from the F. fujikuroi GA gene cluster are also found in all three F. avenaceum genomes, with only P450-3 (C13-oxidase) missing (Figure 7). The architecture of the GA cluster in F. avenaceum is highly similar to the F. fujikuroi cluster, and a single inversion in five of the six genes can explain the relocation of the desaturase (des) encoding gene. The inversion could potentially have involved the P450-3 gene and resulted in the disruption of its coding sequence, however the shuffle-LAGAN analysis (Figure 7) and dot plot showed that this has not been the case. The missing gene is not found elsewhere in the genome based on a tblastn search. The presence of six of the seven GA biosynthesis genes suggest that F. avenaceum has the potential to produce all the GA's up to G4 and G7, but lack the ability to convert these into the GA1 and GA3. None of the three F. avenaceum isolates have been reported to produce this plant growth hormone.
Figure 7. Architecture of the gibberellic acid (GA) gene clusters from F. fujikuroi MP-A, F. avenaceum and Sphaceloma manihoticola.

The gene cluster and surrounding genes are identical in the three F. avenaceum strains and only Fa05001 is shown (FaLH03 cluster: FAVG2_04186 - FAVG2_04192 and FaLH27 cluster: FAVG3_04219 - FAVG3_04224). The mVista trace shows the similarity over a 100 bp sliding windows (Shuffle-LAGAN plot) between the F. avenaceum and F. fujikuroi clusters, bottom line = 50% and second line = 75% identity. Genes: gss2 = geranylgeranyldiphosphate synthase, cps/ks = copalyldiphosphate/ent-kaurene synthase, P450-4 = ent-kaurene oxidase, P450-1 = GA14 synthase, P450-2 = C20-oxidase, P450-3 = 13-hydroxylase and DES = desaturase. Note that the intergenic regions are unknown for the S. manihoticola GA cluster, while the size of these regions is not to drawn to size.
In summary, F. avenaceum has a very large potential for producing secondary metabolites belonging to the PKS, NRPS and terpene classes, with a total of 75–80 key enzymes, see File S8 that summarizes all orthology groups. However, it is expected that multiple enzymes will participate in a single biosynthetic pathway, thereby reducing the potential number of final metabolites. It is possible that some of the metabolites function as virulence factors during infection; however systematic deletion of all PKS encoding genes in F. graminearum showed that none of the 15 PKSs in this fungus had significant effect on virulence [75]. It is therefore more likely that at least some of the secondary metabolites function as antibiotics towards competing microorganisms in the diverse set of niches that the species inhabits. When plotting the PKSs and NRPSs on the 11 supercontigs, areas with higher numbers of SNPs, insertions, or deletions between the three strains, were also more populated with secondary metabolite genes (Figure 1), as seen in other Fusaria [22].
Transcriptomics of F. avenaceum in barley
To increase our understanding of F. avenaceum behaviour in planta, we performed RNA-seq on F. avenaceum-inoculated barley. Table S10 in File S1 shows a list of F. avenaceum genes with the most stable and significant expression (FDR<0.05). Due to putative false positive genes expressed in the host, we applied high stringency in the analysis, and only genes found expressed at 14 days post inoculation (dpi), and which were absent in control, were considered. Genes involved in stress related responses, especially oxidative stress (as defined in the fungal stress response database [76]) were highly represented. This strongly supports our hypothesis formulated from the comparative genomic analysis that the broad host range of F. avenaceum is likely due to a generalized mechanism allowing it to cope with and overcome the innate immune response of plants, such as the generation of reactive oxygen species (ROS) [77].
Genes involved in signal transduction were also overrepresented in the transcriptome, including GO categories for GTP binding, ATP binding, calcium ion binding, and membrane activity (Figure S8 in File S1, File S8). Approximately 33% of all proteins predicted in the genome with Interpro “Tyrosine-protein kinase, catalytic domain” (IPR020635) were found in the transcriptome. This was one of the most highly enriched F. avenaceum categories when compared to other Fusarium genome sequences, and the in planta transcriptome hence supports the comparative results from the genome analysis. During plant infection, fungi need to monitor the nutrient status and presence of host defenses, and respond to or tolerate osmotic or oxidative stress, light and other environmental variables [78]. Stress-signaling/response genes of fungal pathogens are known to play important roles in virulence, pathogenesis and defense against oxidative burst (rapid production of ROS) from the host [79], [80]. It is plausible to predict that tyrosine-protein kinases assist in the stress related response. There is a tendency that F. avenaceum isolates from one host can be pathogenic on other distantly related plants [11]. This is in contrast to, for example, F. oxysporum, a pathogen with a remarkably broad host range at the species level, but where individual isolates often cause disease only on one or a few plant species [81]. Our results support the chameleon nature of F. avenaceum, as it is capable of adapting to diverse hosts and environments. This lack of host specialization is likely to be a driving force in F. avenaceum evolution. Apart from the general functional categories, the in planta transcriptome of F. avenaceum also revealed many orthologs of F. graminearum pathogenicity and virulence factors, especially those involved in signal transduction and metabolism (Table S11 in File S1, [82]).
Conclusions
In summary, the comparative genomic analyses of F. avenaceum to other Fusaria point out several functional categories that are enriched in this fungal genome, and which indicate a great potential for F. avenaceum to sense and transduce signals from the surroundings, and to respond to the environment accordingly. Fusarium avenaceum has a large potential for redox, signaling and secondary metabolite production, and 20% of all predicted proteins were considered to be secreted. This could suggest that interaction with plant hosts is predominantly in the extracellular space. These genome sequences provide a valuable tool for the discovery of genes and mechanisms for bioactive compounds, and to increase our knowledge of the mechanisms contributing to a fungal lifestyle on diverse plant hosts and in different environments.
Materials and Methods
Sequencing, assembly, gene prediction and annotation
Fusarium avenaceum isolate Fa05001 (ARS culture collection: NRRL 54939, Bioforsk collection: 202103, DTU collection: IBT 41708) was isolated from barley in 2005 in Finland [83]. The strain was grown on complete medium [84] at room temperature for three days on a shaker, before mycelium was vacuum filtered and harvested for storage at −80°C. DNA was isolated using the Qiagen DNeasy Plant Maxi. The sequencing and assembly was performed by Eurofins MGW, using a combination of shotgun (1.5 plate) and paired-end (¼ plate of 3 kb, and ¼ plate of 20 kb) 454 pyrosequencing. Newbler 2.6 (www.roche.com) was used for automatic assembly, and gap closure and further assembly was performed using GAP4 (Version 4.4; 2011, http://www.gap-system.org), a total of 50 primer pairs, and manual editing. PCR products from gaps were Sanger sequenced in both directions, and 38 PCR products were successfully integrated into the assembly. These approaches significantly improved the results, starting from 502 contigs and 89 scaffolds after automatic annotation to 110 contigs and 83 scaffolds (Table 1).
Two Canadian F. avenaceum strains, FaLH03 (spring wheat, New Brunswick) and FaLH27 (winter wheat, Nova Scotia), were isolated from wheat samples harvested in 2011 (Canadian Grain Commission, Winnipeg, MB) and deposited in the Canadian Collection of Fungal Cultures (AAFC, Ottawa, ON) with the strain designations DAOM242076 and DAOM242378, respectively. Species identification was confirmed by sequencing the tef1-α gene [85]. Strains were single-spored prior to any analysis and confirmed to retain virulence towards durum wheat. After growth for 3 days in glucose-yeast extract-peptone liquid culture, mycelia was collected and freeze-dried. Genomic DNA was extracted using the Nucleon Phytopure genomic DNA extraction kit (GE Healthcare, Baie d'Urfe, Québec) and then used to prepare an Illumina TruSeq library. Each library was sequenced in a single lane on an Illumina HiSeq platform (100 bp paired-end) at the Génome Québec Innovation Centre (Montréal, Québec), yielding 99,386,445 and 233,211,138 reads for FaLH03 and FaLH27, respectively. Reads were assembled in CLC Genomics Workbench 6.0.1. The higher order of the obtained scaffolds in the three isolates was resolved through comparison between the strains. The contigs from the Canadian isolates were ordered to each other by ABACAS [86]. A reiterative reordering approach was used to generate a stable order of the contigs. The successful reordering is illustrated in the alignment of the ordered contigs by MUMMER [87] (Figure S2 in File S1). The scaffolds from the Fa05001 were then ordered according to the Canadian isolates by ABACAS, and aligned to the Canadian isolates by ‘MUMMER’ (Figure S2 in File S1). Contig/scaffold overlaps and boundaries in the ‘MUMMER’ alignments were used to determine the higher order assembly.
Gene prediction was performed with Augustus v2.5.5 (Fa05001) and v2.6 (FaLH03, FaLH27) [88], using default settings and F. graminearum as a training set. Protein sequences were annotated and enrichment analysis of gene ontology categories were compared using Blast2GO [89]. Table 1 shows the general statistics of the three genome sequences.
The species phylogenies were constructed based on 69 orthologous proteins from the included species. The 69 protein sets were first aligned individually using MUSCLE with default parameters [18], then manually inspected and concatenated. These super-protein alignments were then analyzed using MEGA6.0 by first identifying the best substitution model and then inferring the evolutionary history of the species using the Maximum Likelihood method, Nearest-Neighbour-Interchange, [90] using the LG+(F) substitution model, uniform substitution rate, removal of all positions with gaps and missing data and bootstrapping with 500 iterations.
Functional analysis and orthology prediction
For comparative genomics analysis, the previously sequenced genomes of F. graminearum, F. verticillioides, F. oxysporum f. sp. lycopersici 4287 (Fusarium Comparative Sequencing Project, Broad Institute of Harvard and MIT, http://www.broadinstitute.org/) and F. solani [91] were re-annotated using Blast2GO [89] concurrently with Fa05001. A functional comparison was performed using the Gene Ontology (GO) categories Biological Process, Molecular Function, and Cellular Compartments at level six, and Interpro. To compare transcription factors, we used the Interpro list from the Fungal Transcription Factor Database [92]. To identify orthologs, protein sets were compared in both directions with BLASTp to identify the best reciprocal hit for each individual protein with expectation values less than 1e-10. RepeatMasker [11] was used on all species to find repetitive sequences in the Fusarium genomes, using CrossMatch (http://www.phrap.org/phredphrap/general.html). BLASTn was used to compare gene sequences between the three F. avenaceum genomes.
Electrophoretic karyotyping
Plugs containing 4×108/ml protoplasts were loaded on a CHEF gel (1% FastLane agarose [FMC BioProducts, Rockland, Maine] in 0.5×TBE) and ran for 255 hours, using switch times between 1200–4800 s at 1.8 V/cm. Chromosomes of Schizosaccharomyces pombe and Hansenula wingei were used as a molecular size marker (BioRad). Chromosomes separated in the gel were blotted to Hybond-N+. Southern hybridization was done overnight at 65°C using the CDP star method (GE Healthcare). A 350 bp HindIII-EcoRI fragment from plasmid pNLA17 was used as a probe, containing the F. oxysporum telomeric repeat TTAGGG 18 times.
Prediction of putative secretome
To determine the putative secretome, we employed a pipeline consisting of the following: A combination of WolfPsort (http://wolfpsort.org/), IPsort (http://ipsort.hgc.jp/) and SignalP4.1 [93] to identify subcellular localization and/or signalP motifs of all F. avenaceum Fa05001 proteins. Then, we used TMHMM [94] to predict all transmembrane domains. The secretome fasta file with the sequences was created with GPRO [95], with proteins predicted by either the PSORT tools or SignalP that does not contain transmembrane domains. Of the whole set of 13217 predicted proteins in Fa05001, a subset of 2580 sequences has been determined as the putative secretome of F. avenaceum (File S6).
Prediction of cysteine-rich proteins
Prediction of putative cysteine-rich proteins CRPs was based on a previously described approach [96]. In brief, this method is based on their expected sequence characteristics, with predicted small open reading frames (ORFs) (20 to 150 amino acids), containing at least four cysteine residues and a predicted signal peptide from the secretory pathway. The 83 Fa05001 scaffolds were used as input against GETORF available in the EMBOSS package [97]. We obtained 565.652 ORFs, which were translated to proteins using the tool Transeq [97]. All proteins were separated into two files (more/less than four cysteine residues). We downloaded a collection of 513 HMM profiles based on CRP models [98], and then created a single HMM database file that was formatted with HMMER3 [99] and used as subject against a HMMER comparison with the two files obtained. Nineteen candidates with more than four cysteine residues were predicted as CRP, and 55 additional sequences with less than four cysteine residues but with significant similarity to particular CRP HMM models were also included (Table S7 in File S1, File S7). We compared these to the supercontigs and scaffolds of the genome sequenced Fusarium spp. using BLASTn and TBLASTx, bit-score>50, and BLASTp in NCBI.
Identification of secondary metabolites
The three F. avenaceum strains were grown at 25°C in darkness for 14 days as triple point inoculations on Potato Dextrose Agar (PDA, [100]) and Yeast Extract Sucrose agar (YES, [100]). The metabolites of the strains were extracted using a modified version of the micro-scale extraction procedure for fungal metabolites [101]. Six 5-mm plugs from each plate taken across the colonies were transferred to 2 mL HPLC vials and extracted with 1.2 mL methanol:dichloromethane:ethyl acetate (1∶2∶3 v/v/v) containing 1% (v/v) formic acid. After 1 hr in an ultrasonication bath, extracts were evaporated with nitrogen, the residue dissolved in 150 µL acetonitrile:water (3/2 v/v) and filtered through a standard 0.45-µm PTFE filter.
Barley and oat samples (all in biological triplicates), including none-inoculated samples from climate chamber experiments described below were ground in liquid nitrogen and 50 mg extracted with 1 mL of 50% (vol) acetonitrile in water in a 2 mL Eppendorf tube. Samples were placed in an ultrasonication bath for 30 min, centrifuged at 15,000 g, and the supernatant was transferred to a clean 2 mL vial that was loaded onto the auto sampler prior to analysis. UHPLC-TOFMS analysis of 0.3–2 µL extracts were conducted on an Agilent 1290 UHPLC equipped with a photo diode array detector scanning 200–640 nm, and coupled to an Agilent 6550 qTOF (Santa Clara, CA, USA) equipped with a dual electrospray (ESI) source [102]. Separation was performed at 60°C at a flow rate of 0.35 mL/min on a 2.1 mm ID, 250 mm, 2.7 µm Agilent Poroshell phenyl hexyl column using a water-acetonitrile gradient solvent system, with both water and acetonitrile containing 20 mM formic acid. The gradient started at 10% acetonitrile and was increased to 100% acetonitrile within 15 min, maintained for 4 min, returned to 10% acetonitrile in 1 min. Samples were analyzed in both ESI+ and ESI− scanning m/z 50 to 1700, and for automated data-dependent MS/MS on all major peaks, collision energies of 10, 20 and 40 eV for each MS/MS experiment were used. An MS/MS exclusion time of 0.04 min was used to get MS/MS spectra of less abounded ions.
Data files were analyzed in Masshunter 6.0 (Agilent Technologies) in three different ways: i) Aggressive dereplication [103] using lists of elemental composition and the Search by Formula (10 ppm mass accuracy) of all described Fusarium metabolites as well as restricted lists of only F. avenaceum and closely related species; ii) Searching the acquired MS/MS spectra in an in-house database of approx. 1200 MS/MS spectra of fungal secondary metabolites acquired at 10, 20 and 40 eV [102]; iii) all major UV/Vis and peaks in the base peak ion chromatograms not assigned to compounds (and not present in the media blank samples) were also registered. For absolute verification, authentic reference standards were available from 130 Fusarium compounds and additional 100 compounds that have been tentatively identified based on original producing strains using UV/Vis, LogD and MS/HRMS [102]–[104].
Identification of secondary metabolite genes
Type I iterative polyketide synthase (PKS), type III PKS, non-ribosomal peptide synthase (NRPS), aromatic prenyltranferase (DMATS) and class I & II terpene synthase encoding genes were identified by BLASTp using archetype representatives for the six types of genes [105]. Identification of orthologous genes was further supported by comparison to the genomic DNA, using the shuffle LAGAN algorithm with default settings [106], [107]. Functional protein domains were identified using the NCBI CDD and pfam databases [108]. Domains specific to non-reducing PKSs, e.g. ‘Product template’ (PT) and ‘Starter Acyl-Transferase’ (SAT), were inferred via multiple sequence alignment with the bikaverin PKS (PKS16), which was one of the founding members of the domain group [109]. The nomenclature for PKS and NPS follows that which was introduced by Hansen et al. [63], as indicated by the use of the oPKSx and oNRPSx name, where the prefix ‘o’ signals that it refers to orthology-groups rather than the original overlapping names schemes used previously in each species. Following the idea regarding transparency in the names, introduced by Hansen and co-works, we applied a similar nomenclature scheme to the type III PKSs (oPKSIII_x) and the various enzyme classes involved in terpene biosynthesis: Terpene Synthase class I head-to-tail (oTS-I-HT_x), Terpene Synthase class I head-to-head (oTS-II-HH_x) and Terpene Synthase class II (oTS-II-x).
Climate chamber infection experiment
Fa05001 was grown on mung bean agar [110] for three weeks at room temperature under a combination of white and black (UVA) light with a 12 h photoperiod. Macroconidia were collected by washing the agar plate with 5 mL sterile distilled water, and diluted with 1.5% carboxymethylcellulose solution to a concentration of 5×104 conidia/mL for inoculation. Conidial concentration was determined using a Bürker hemacytometer.
Barley (Hordeum vulgare), cultivar Iron, and oat (Avena sativa) cultivar Belinda were grown in a climate chamber under the following conditions: Two weeks at 10°C/8°C 17 h/7 h 70%RH/60%RH, two weeks 15°C/12°C 18 h/6 h 70%RH/60%RH, three weeks 18°C/15°C 18 h/6 h 70%RH/60%RH and three weeks 20°C/15°C 17 h/6 h 70%RH/60%RH. During anthesis, approximately 1 mL of conidial solution was sprayed on each panicle, a bag was placed over the panicle and removed after 4 days. We used 6 plants per pot, 2 panicles per plant and 3 replicate pots per treatment. At sampling, panicles were immediately stored at −80°C.
Transcriptomics of Fa05001 on barley heads
Panicles from one pot grown in climate chamber experiment were mixed and ground in liquid nitrogen. RNA was extracted from 50 mg subsample from three biological replicates (pots) of untreated control (0 dpi) and F. avenaceum-inoculated tissue (14 dpi), using Spectrum plant total RNA kit (Sigma-Aldrich, Steinheim, Germany), with slight modifications. Due to the high amount of starch in barley heads at 14 dpi, the volume of lysis buffer and binding solution were increased from 500 µL to 750 µL per sample, and samples were incubated for 5 min at room temperature and the lysates were filtered 2 times for 10 minutes. On-column DNase digestion (Sigma-Aldrich, Steinheim, Germany) was used.
PolyA purification and fragmentation, cDNA synthesis, library preparation and 1×100 bp single read module (half a lane Hi-seq 2500) sequencing were done by Eurofins MGW. The resulting fastaq files were trimmed (quality score limit: 0.05, maximum number of ambiguities: 2), and RNA-seq was performed with predicted F. avenaceum genes using CLC Genomics Workbench 6.05, with stringent settings (minimum similarity fraction: 0.95, minimum length fraction: 0.9, maximum number of hits for a read: 10) to subtract host-specific transcripts. Gene expression was calculated using reads per kilobase per million (RPKM) values. A T-test was used to determine significant expression levels in the biological replicates, comparing F. avenaceum inoculated samples against a control. Transcripts found solely in the F. avenaceum-inoculated plant were used to limit the amount of false positives coming from the host.
Supporting Information
Supplementary figures and tables.
(DOCX)
BLASTn results of the three F. avenaceum isolates Fa05001, FaLH03 and FaLH27.
(XLSX)
Venn diagram of BLASTn results corresponding to Figure S4.
(XLSX)
Interpro results of F. avenaceum Fa5001, F. graminearum , F. verticillioides, F. oxysporum and F. solani .
(XLSX)
Reciprocal blast of F. avenaceum Fa5001 vs F. graminearum, F. verticillioides, F. oxysporum and F. solani .
(XLSX)
Secretome of F. avenaceum Fa5001.
(XLSX)
Cysteine rich proteins in F. avenaceum Fa5001.
(TXT)
Summary of secondary metabolite genes.
(XLSX)
Transcriptome of F. avenaceum Fa5001 on barley heads.
(XLSX)
Acknowledgments
We thank Päivi Parikka, MTT, Finland for the F. avenaceum Fa05001 strain and Tom Gräfenhan, Canadian Grain Commission, for the two Canadian isolates FaLH03 and FaLH27 used for genome sequencing. We thank Danielle Schneiderman and Catherine Brown for technical support and Philippe Couroux for bioinformatics support.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files, and genome sequences are deposited at NCBI GenBank in the Whole Genome Shotgun (WGS) database as accession no. JPYM00000000 (Fa05001), JQGD00000000 (FaLH03) and JQGE00000000 (FaLH27), within BioProject PRJNA253730. The versions described in this paper are JPYM01000000, JQGD01000000, and JQGE01000000.
Funding Statement
Nordisk komité for jordbruks- og matforskning (NKJ), Project number: “NKJ 135 - Impact of climate change on the interaction of Fusarium species in oats and barley”, and Agriculture and Agri-Food Canada's Genomics Research & Development Initiative funded this work. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Leach MC, Hobbs SLA (2013) Plantwise knowledge bank: delivering plant health information to developing country users. Learned Publ 26: 180–185. [Google Scholar]
- 2. Desjardins AE (2003) Gibberella from a (venaceae) to z (eae). Annu Rev Phytopathol 41: 177–198. [DOI] [PubMed] [Google Scholar]
- 3. Satyaprasad K, Bateman GL, Read PJ (1997) Variation in pathogenicity on potato tubers and sensitivity to thiabendazole of the dry rot fungus Fusarium avenaceum . Potato Res 40: 357–365. [Google Scholar]
- 4. Mercier J, Makhlouf J, Martin RA (1991) Fusarium avenaceum, a pathogen of stored broccoli. Can Plant Dis Surv 71: 161–162. [Google Scholar]
- 5. Sørensen JL, Phipps RK, Nielsen KF, Schroers HJ, Frank J, et al. (2009) Analysis of Fusarium avenaceum metabolites produced during wet apple core rot. J Agricult Food Chem 57: 1632–1639. [DOI] [PubMed] [Google Scholar]
- 6. Peters RD, Barasubiye T, Driscoll J (2007) Dry rot of rutabaga caused by Fusarium avenaceum . Hortscience 42: 737–739. [Google Scholar]
- 7. Crous PW, Petrini O, Marais GF, Pretorius ZA, Rehder F (1995) Occurrence of fungal endophytes in cultivars of Triticum aestivum in South Africa. Mycoscience 36: 105–111. [Google Scholar]
- 8. Varvas T, Kasekamp K, Kullman B (2013) Preliminary study of endophytic fungi in timothy (Phleum pratense) in Estonia. Acta Myc 48: 41–49. [Google Scholar]
- 9. Yacoub A (2012) The first report on entomopathogenic effect of Fusarium avenaceum (Fries) Saccardo (Hypocreales, Ascomycota) against rice weevil (Sitophilus oryzae L.: Curculionidae, Coleoptera). J Entomol Acarol R 44: 51–55. [Google Scholar]
- 10. Makkonen J, Jussila J, Koistinen L, Paaver T, Hurt M, et al. (2013) Fusarium avenaceum causes burn spot disease syndrome in noble crayfish (Astacus astacus). J Invert Pat 113: 184–190. [DOI] [PubMed] [Google Scholar]
- 11. Nalim FA, Elmer WH, McGovern RJ, Geiser DM (2009) Multilocus phylogenetic diversity of Fusarium avenaceum pathogenic on lisianthus. Phytopathology 99: 462–468. [DOI] [PubMed] [Google Scholar]
- 12. Uhlig S, Jestoi M, Parikka P (2007) Fusarium avenaceum - The North European situation. Int J Food Microbiol 119: 17–24. [DOI] [PubMed] [Google Scholar]
- 13. Kulik T, Pszczolkowska A, Lojko M (2011) Multilocus phylogenetics show high intraspecific variability within Fusarium avenaceum . Int J Mol Sci 12: 5626–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kohl J, de Haas BH, Kastelein P, Burgers SLGE, Waalwijk C (2007) Population dynamics of Fusarium spp. and Microdochium nivale in crops and crop residues of winter wheat. Phytopathology 97: 971–978. [DOI] [PubMed] [Google Scholar]
- 15. Sørensen JL, Giese H (2013) Influence of carbohydrates on secondary metabolism in Fusarium avenaceum . Toxins 5: 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sørensen L, Lysøe E, Larsen J, Khorsand-Jamal P, Nielsen K, et al. (2014) Genetic transformation of Fusarium avenaceum by Agrobacterium tumefaciens mediated transformation and the development of a USER-Brick vector construction system. BMC Mol Biol 15: 15 10.1186/1471-2199-15-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, et al. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium . Nature 464: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marcet-Houben M, Gabaldón T (2009) The tree versus the forest: The fungal tree of life and the topological diversity within the yeast phylome. Plos One 4: e4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al-Reedy RM, Malireddy R, Dillman CB, Kennell JC (2012) Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fung Genet Biol 49: 2–14. [DOI] [PubMed] [Google Scholar]
- 20. Fourie G, van der Merwe NA, Wingfield BD, Bogale M, Tudzynski B, et al. (2013) Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genom 14: 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T, Taga M, Saitoh H, Nakayama T, Takehara T (1998) Karyotypic analysis of five Fusarium spp. causing wheat scab by fluorescence microscopy and fluorescence in situ hybridization. Int Con Plant Pat 2.2.48.
- 22. Cuomo CA, Guldener U, Xu JR, Trail F, Turgeon BG, et al. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317: 1400–1402. [DOI] [PubMed] [Google Scholar]
- 23. O'Donnell K, Rooney AP, Proctor RH, Brown DW, McCormick SP, et al. (2013) Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fung Genet Biol 52: 20–31. [DOI] [PubMed] [Google Scholar]
- 24. Watanabe M, Yonezawa T, Lee K, Kumagai S, Sugita-Konishi Y, et al. (2011) Molecular phylogeny of the higher and lower taxonomy of the Fusarium genus and differences in the evolutionary histories of multiple genes. Bmc Evolutionary Biology 11: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lysøe E, Seong KY, Kistler HC (2011) The transcriptome of Fusarium graminearum during the infection of wheat. Mol Plant Microbe Interact 24: 995–1000. [DOI] [PubMed] [Google Scholar]
- 26. Ma LJ, Geiser DM, Proctor RH, Rooney AP, O'Donnell K, et al. (2013) Fusarium pathogenomics. Annu Rev Microbiol 67: 399–416. [DOI] [PubMed] [Google Scholar]
- 27. Martin SH, Wingfield BD, Wingfield MJ, Steenkamp ET (2011) Structure and evolution of the Fusarium mating type locus: New insights from the Gibberella fujikuroi complex. Fung Genet Biol 48: 731–740. [DOI] [PubMed] [Google Scholar]
- 28. Holtz MD, Chang KF, Hwang SF, Gossen BD, Strelkov SE (2011) Characterization of Fusarium avenaceum from lupin in central Alberta: genetic diversity, mating type and aggressiveness. Can J Plant Pathol 33: 61–76. [Google Scholar]
- 29. Cook RJ (1967) Gibberella avenacea sp. n., perfect stage of Fusarium roseum f. sp. cerealis ‘avenaceum’. Phytopathology 57: 732–736. [Google Scholar]
- 30. Booth C, Spooner BM (1984) Gibberella avenacea, teleomorph of Fusarium avenaceum, from stems of Pteridium aquilinum . T Brit Mycol Soc 82: 178–180. [Google Scholar]
- 31. Kerényi Z, Moretti A, Waalwijk C, Oláh B, Hornok L (2004) Mating type sequences in asexually reproducing Fusarium species. Appl Environ Microbiol 70: 4419–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee J, Lee T, Lee YW, Yun SH, Turgeon BG (2003) Shifting fungal reproductive mode by manipulation of mating type genes: obligatory heterothallism of Gibberella zeae . Mol Microbiol 50: 145–152. [DOI] [PubMed] [Google Scholar]
- 33. Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 4: 4–10. [DOI] [PubMed] [Google Scholar]
- 34. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng P, Xia YL, Xiao GH, Xiong CH, Hu X, et al. (2011) Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional chinese medicine. Genome Biol 12: R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thomma BPHJ, Van Esse HP, Crous PW, De Wit PJGM (2005) Cladosporium fulvum (syn. Passalora fulva), a highly specialized plant pathogen as a model for functional studies on plant pathogenic Mycosphaerellaceae . Mol Plant Pathol 6: 379–393. [DOI] [PubMed] [Google Scholar]
- 37. Rep M, van der Does HC, Meijer M, van Wijk R, Houterman PM, et al. (2004) A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol Microbiol 53: 1373–1383. [DOI] [PubMed] [Google Scholar]
- 38. Farman ML, Eto Y, Nakao T, Tosa Y, Nakayashiki H, et al. (2002) Analysis of the structure of the AVR1-CO39 avirulence locus in virulent rice-infecting isolates of Magnaporthe grisea . Mol Plant Microbe Interact 15: 6–16. [DOI] [PubMed] [Google Scholar]
- 39. Marx F (2004) Small, basic antifungal proteins secreted from filamentous ascomycetes: a comparative study regarding expression, structure, function and potential application. Appl Microbiol Biotechnol 65: 133–142. [DOI] [PubMed] [Google Scholar]
- 40. de Jonge R, Thomma BPHJ (2009) Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol 17: 151–157. [DOI] [PubMed] [Google Scholar]
- 41. Hershenhorn J, Park SH, Stierle A, Strobel GA (1992) Fusarium avenaceum as a novel pathogen of spotted knapweed and its phytotoxins, acetamido-butenolide and enniatin B. Plant Sci 86: 155–160. [Google Scholar]
- 42. Thrane U (1988) Screening for Fusarin C production by European isolates of Fusarium species. Mycotox Res 4: 2–10. [DOI] [PubMed] [Google Scholar]
- 43. Morrison E, Kosiak B, Ritieni A, Aastveit AH, Uhlig S, et al. (2002) Mycotoxin production by Fusarium avenaceum strains isolated from Norwegian grain and the cytotoxicity of rice culture extracts to porcine kidney epithelial cells. J Agricult Food Chem 50: 3070–3075. [DOI] [PubMed] [Google Scholar]
- 44. Takeuchi M, Nakajima M, Ogita T, Inukai M, Kodama K, et al. (1989) Fosfonochlorin, a new antibiotic with spheroplast forming activity. J Antibiot (Tokyo) 42: 198–205. [DOI] [PubMed] [Google Scholar]
- 45. Hussein HM, Baxter M, Andrew IG, Franich RA (1991) Mycotoxin production by Fusarium species isolated from New Zealand maize fields. Mycopathologia 113: 35–40. [DOI] [PubMed] [Google Scholar]
- 46. Chelkowski J, Manka M (1983) The ability of Fusaria pathogenic to wheat, barley and corn to produce zearalenone. Phytopathol Z 106: 354–359. [Google Scholar]
- 47. Chelkowski J, Visconti A, Manka M (1984) Production of trichothecenes and zearalenone by Fusarium species isolated from wheat. Nahrung 28: 493–496. [DOI] [PubMed] [Google Scholar]
- 48. Chelkowski J, Golinski P, Manka M, Wiewiórowska M, Szebiotko K (1983) Mycotoxins in cereal grain. Part IX. Zearalenone and Fusaria in wheat, barley, rye and corn kernels. Die Nahrung 27: 525–531. [DOI] [PubMed] [Google Scholar]
- 49. Ishii K, Sawano M, Ueno Y, Tsunoda H (1974) Distribution of zearalenone-producing Fusarium species in Japan. Appl Microbiol 27: 625–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marasas W. F O., Nelson P E., Tousson T A. (1984) Toxigenic Fusarium species. Identity and mycotoxicology. University Park, Pennsylvania, U.S.A., 328p: Pennsylvania State University Press. [Google Scholar]
- 51. Lysøe E, Klemsdal SS, Bone KR, Frandsen RJN, Johansen T, et al. (2006) The PKS4 gene of Fusarium graminearum is essential for zearalenone production. Appl Environ Microbiol 72: 3924–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim YT, Lee YR, Jin JM, Han KH, Kim H, et al. (2005) Two different polyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae . Mol Microbiol 58: 1102–1113. [DOI] [PubMed] [Google Scholar]
- 53. Jiang Z, Barret MO, Boyd KG, Adams DR, Boyd ASF, et al. (2002) JM47, a cyclic tetrapeptide HC-toxin analogue from a marine Fusarium species. Phytochemistry 60: 33–38. [DOI] [PubMed] [Google Scholar]
- 54.Laatch H. (2012) Antibase 2012: The natural compound identifier. Wiley-VCH Verlag GmbH. [Google Scholar]
- 55. Studt L, Wiemann P, Kleigrewe K, Humpf HU, Tudzynski B (2012) Biosynthesis of fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl Environ Microbiol 78: 4468–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song ZS, Cox RJ, Lazarus CM, Simpson TJ (2004) Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum . Chembiochem 5: 1196–1203. [DOI] [PubMed] [Google Scholar]
- 57. Malz S, Grell MN, Thrane C, Maier FJ, Rosager P, et al. (2005) Identification of a gene cluster responsible for the biosynthesis of aurofusarin in the Fusarium graminearum species complex. Fung Genet Biol 42: 420–433. [DOI] [PubMed] [Google Scholar]
- 58. Seshime Y, Juvvadi PR, Fujii I, Kitamoto K (2005) Discovery of a novel superfamily of type III polyketide synthases in Aspergillus oryzae . Biochem Biophys Res Commun 331: 253–260. [DOI] [PubMed] [Google Scholar]
- 59. Wiemann P, Sieber CMK, Von Bargen KW, Studt L, Niehaus EM, et al. (2013) Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. Plos Pathog 9: e1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. von Döhren H (2004) Biochemistry and general genetics of nonribosomal pepticle synthetases in fungi. Adv Biochem Engin/Biotechnol 88: 217–264. [DOI] [PubMed] [Google Scholar]
- 61. Panaccione DG, Scottcraig JS, Pocard JA, Walton JD (1992) A cyclic peptide synthetase gene required for pathogenicity of the fungus Cochliobolus carbonum on maize. Proc Natl Acad Sci U S A 89: 6590–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jose B, Oniki Y, Kato T, Nishino N, Sumida Y, et al. (2004) Novel histone deacetylase inhibitors: cyclic tetrapeptide with trifluoromethyl and pentafluoroethyl ketones. Bioorg Med Chem Lett 14: 5343–5346. [DOI] [PubMed] [Google Scholar]
- 63. Hansen FT, Sørensen JL, Giese H, Sondergaard TE, Frandsen RJN (2012) Quick guide to polyketide synthase and nonribosomal synthetase genes in Fusarium . Int J Food Microbiol 155: 128–136. [DOI] [PubMed] [Google Scholar]
- 64. Jin JM, Lee S, Lee J, Baek SR, Kim JC, et al. (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum . Mol Microbiol 76: 456–466. [DOI] [PubMed] [Google Scholar]
- 65. Condon BJ, Leng YQ, Wu DL, Bushley KE, Ohm RA, et al. (2013) Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. Plos Genet 9: e1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Manning VA, Pandelova I, Dhillon B, Wilhelm LJ, Goodwin SB, et al. (2013) Comparative genomics of a plant-pathogenic fungus, Pyrenophora tritici-repentis, reveals transduplication and the impact of repeat elements on pathogenicity and population divergence. G3-Genes Genom Genet 3: 41–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Niehaus EM, Janevska S, von Bargen KW, Sieber CMK, Harrer H, et al. (2014) Apicidin F: Characterization and genetic manipulation of a new secondary metabolite gene cluster in the rice pathogen Fusarium fujikuroi . Plos One 9: e103336 doi: 10.1371/journal.pone.0103336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee SL, Floss HG, Heinstein P (1976) Purification and properties of dimethylallylpyrophosphate - tryptophan dimethylallyl transferase, first enzyme of ergot alkaloid biosynthesis in Claviceps. sp. SD 58. Arch Biochem Biophys 177: 84–94. [DOI] [PubMed] [Google Scholar]
- 69. Young CA, Tapper BA, May K, Moon CD, Schardl CL, et al. (2009) Indole-diterpene biosynthetic capability of Epichloe endophytes as predicted by ltm gene analysis. Appl Environ Microbiol 75: 2200–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bomke C, Tudzynski B (2009) Diversity, regulation, and evolution of the gibberellin biosynthetic pathway in fungi compared to plants and bacteria. Phytochemistry 70: 1876–1893. [DOI] [PubMed] [Google Scholar]
- 71. Rim SO, You YH, Yoon H, Kim YE, Lee JH, et al. (2013) Characterization of gibberellin biosynthetic gene cluster from Fusarium proliferatum . J Microbiol Biot 23: 623–629. [DOI] [PubMed] [Google Scholar]
- 72. Malonek S, Rojas MC, Hedden P, Gaskin P, Hopkins P, et al. (2005) Functional characterization of two cytochrome P450 monooxygenase genes, P450-1 and P450-4, of the gibberellic acid gene cluster in Fusarium proliferatum (Gibberella fujikuroi MP-D). Appl Environ Microbiol 71: 1462–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bomke C, Rojas MC, Gong F, Hedden P, Tudzynski B (2008) Isolation and characterization of the gibberellin biosynthetic gene cluster in Sphaceloma manihoticola . Appl Environ Microbiol 74: 5325–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kawaide H (2006) Biochemical and molecular analyses of gibberellin biosynthesis in fungi. Biosci Biotechnol Biochem 70: 583–590. [DOI] [PubMed] [Google Scholar]
- 75. Gaffoor I, Brown DW, Plattner R, Proctor RH, Qi WH, et al. (2005) Functional analysis of the polyketide synthase genes in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot Cell 4: 1926–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Karányi Z, Holb I, Hornok L, Pócsi I, Miskei M (2013) FSRD: fungal stress response database. Database (Oxford) 2013: bat037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Plancot B, Santaella C, Jaber R, Kiefer-Meyer MC, Follet-Gueye ML, et al. (2013) Deciphering the responses of root border-like cells of Arabidopsis and flax to pathogen-derived elicitors. Plant Physiol 163: 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kosti I, Mandel-Gutfreund Y, Glaser F, Horwitz BA (2010) Comparative analysis of fungal protein kinases and associated domains. BMC Genom 11: 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hamilton AJ, Holdom MD (1999) Antioxidant systems in the pathogenic fungi of man and their role in virulence. Med Mycol 37: 375–389. [DOI] [PubMed] [Google Scholar]
- 80. de Dios CH, Roman E, Monge RA, Pla J (2010) The role of MAPK signal transduction pathways in the response to oxidative stress in the fungal pathogen Candida albicans: Implications in virulence. Curr Protein Pept Sci 11: 693–703. [DOI] [PubMed] [Google Scholar]
- 81. Dean R, van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, et al. (2012) The Top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13: 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Urban M, Hammond-Kosack KE (2013) Molecular genetics and genomic approaches to explore Fusarium infection of wheat floral tissue. In: Brown DW, Proctor RH, editors.Fusarium: Genomics, Molecular and Cellular Biology.Norfolk, UK: Caister Academic Press. pp.43–79. [Google Scholar]
- 83. Kokkonen M, Ojala L, Parikka P, Jestoi M (2010) Mycotoxin production of selected Fusarium species at different culture conditions. Int J Food Microbiol 143: 17–25. [DOI] [PubMed] [Google Scholar]
- 84. Harris SD, Morrell JL, Hamer JE (1994) Identification and characterization of Aspergillus nidulans mutants defective in cytokinesis. Genetics 136: 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Geiser DM, Jimenez-Gasco MD, Kang SC, Makalowska I, Veeraraghavan N, et al. (2004) FUSARIUM-ID v. 1.0: A DNA sequence database for identifying Fusarium. Eur J Plant Pathol 110: 473–479. [Google Scholar]
- 86. Assefa S, Keane TM, Otto TD, Newbold C, Berriman M (2009) ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25: 1968–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5. [DOI] [PMC free article] [PubMed]
- 88. Stanke M, Diekhans M, Baertsch R, Haussler D (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644. [DOI] [PubMed] [Google Scholar]
- 89. Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 90. Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25: 1307–1320. [DOI] [PubMed] [Google Scholar]
- 91. Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, et al. (2009) The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. Plos Genet 5: e1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Park J, Park J, Jang S, Kim S, Kong S, et al. (2008) FTFD: an informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 24: 1024–1025. [DOI] [PubMed] [Google Scholar]
- 93. Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- 94. Krogh A, Larsson B, von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 305: 567–580. [DOI] [PubMed] [Google Scholar]
- 95.Futami R, Muñoz-Pomer L, Viu JM, Dominguez-Escriba L, Covelli L, et al. (2011) GPRO: The professional tool for annotation, management and functional analysis of omics databases. Biotechvana Bioinformatics 2011-SOFT3 2011.
- 96. Marcet-Houben M, Ballester AR, de la Fuente B, Harries E, Marcos JF, et al. (2012) Genome sequence of the necrotrophic fungus Penicillium digitatum, the main postharvest pathogen of citrus. BMC Genom 13: 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- 98. Silverstein KAT, Moskal WA, Wu HC, Underwood BA, Graham MA, et al. (2007) Small cysteine-rich peptides resembling antimicrobial peptides have been under-predicted in plants. Plant J 51: 262–280. [DOI] [PubMed] [Google Scholar]
- 99. Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Samson RA, Houbraken J, Thrane U, Frisvad JC, Andersen B (2010) Food and indoor fungi. Utrecht: CBS-KNAW Fungal Biodiversity Centre.
- 101. Smedsgaard J (1997) Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J Chromatogr A 760: 264–270. [DOI] [PubMed] [Google Scholar]
- 102. Kildgaard S, Månsson M, Dosen I, Klitgaard A, Frisvad JC, et al. (2014) Accurate dereplication of bioactive secondary metabolites from marine-derived fungi by UHPLC-DAD-QTOFMS and a MS/HRMS Library. Mar Drugs 12: 3681–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Klitgaard A, Iversen A, Andersen MR, Larsen TO, Frisvad JC, et al. (2014) Aggressive dereplication using UHPLC-DAD-QTOF: screening extracts for up to 3000 fungal secondary metabolites. Anal Bioanal Chem 406: 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nielsen KF, Månsson M, Rank C, Frisvad JC, Larsen TO (2011) Dereplication of microbial natural products by LC-DAD-TOFMS. J Nat Prod 74: 2338–2348. [DOI] [PubMed] [Google Scholar]
- 105. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32: W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Brudno M, Malde S, Poliakov A, Do CB, Couronne O, et al. (2003) Global alignment: finding rearrangements during alignment. Bioinformatics 19: i54–i62. [DOI] [PubMed] [Google Scholar]
- 108. Marchler-Bauer A, Lu SN, Anderson JB, Chitsaz F, Derbyshire MK, et al. (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39: D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Crawford JM, Dancy BCR, Hill EA, Udwary DW, Townsend CA (2006) Identification of a starter unit acyl-carrier protein transacylase domain in an iterative type I polyketide synthase. Proc Natl Acad Sci U S A 103: 16728–16733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dill-Macky R (2003) Inoculation methods and evaluation of Fusarium head blight resistance in wheat. In: Leonard KJ, Bushnell WR, editors.Fusarium head blight of wheat and barley. pp.184–210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures and tables.
(DOCX)
BLASTn results of the three F. avenaceum isolates Fa05001, FaLH03 and FaLH27.
(XLSX)
Venn diagram of BLASTn results corresponding to Figure S4.
(XLSX)
Interpro results of F. avenaceum Fa5001, F. graminearum , F. verticillioides, F. oxysporum and F. solani .
(XLSX)
Reciprocal blast of F. avenaceum Fa5001 vs F. graminearum, F. verticillioides, F. oxysporum and F. solani .
(XLSX)
Secretome of F. avenaceum Fa5001.
(XLSX)
Cysteine rich proteins in F. avenaceum Fa5001.
(TXT)
Summary of secondary metabolite genes.
(XLSX)
Transcriptome of F. avenaceum Fa5001 on barley heads.
(XLSX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files, and genome sequences are deposited at NCBI GenBank in the Whole Genome Shotgun (WGS) database as accession no. JPYM00000000 (Fa05001), JQGD00000000 (FaLH03) and JQGE00000000 (FaLH27), within BioProject PRJNA253730. The versions described in this paper are JPYM01000000, JQGD01000000, and JQGE01000000.