

Abstract
A synthetic gene encoding human As(III) S-adenosylmethionine (SAM) methyltransferase (hAS3MT) was expressed, and the purified enzyme was characterized. The synthetic enzyme is considerably more active than a cDNA-expressed enzyme using endogenous reductants thioredoxin (Trx), thioredoxin reductase (TR), NADPH, and reduced glutathione (GSH). Each of the seven cysteines (the four conserved residues, Cys32, Cys61, Cys156, and Cys206, and nonconserved, Cys72, Cys85, and Cys250) was individually changed to serine. The nonconserved cysteine derivates were still active. None of the individual C32S, C61S, C156S, and C206S derivates were able to methylate As(III). However, the C32S and C61S enzymes retained the ability to methylate MAs(III). These observations suggest that Cys156 and Cys206 play a different role in catalysis than that of Cys32 and Cys61. A homology model built on the structure of a thermophilic orthologue indicates that Cys156 and Cys206 form the As(III) binding site, whereas Cys32 and Cys61 form a disulfide bond. Two observations shed light on the pathway of methylation. First, binding assays using the fluorescence of a single-tryptophan derivative indicate that As(GS)3 binds to the enzyme much faster than inorganic As(III). Second, the major product of the first round of methylation is MAs(III), not MAs(V), and remains enzyme-bound until it is methylated a second time. We propose a new pathway for hAS3MT catalysis that reconciles the hypothesis of Challenger ((1947) Sci. Prog., 35, 396–416) with the pathway proposed by Hayakawa et al. ((2005) Arch. Toxicol., 79, 183–191). The products are the more toxic and more carcinogenic trivalent methylarsenicals, but arsenic undergoes oxidation and reduction as enzyme-bound intermediates.
Introduction
Arsenic is the most ubiquitous toxic substance in the environment as the result of geochemical and anthropogenic exposure.3−6 In humans, arsenic is associated with a broad range of acute and chronic toxic effects that increase the lifetime risk of cancer and diabetes as well as cardiovascular and neurological diseases.7−9 Consequently, arsenic rates first on the United States Center for Disease Control and Prevention (CDC) and Environmental Protection Agency’s (EPA) priority list of hazardous substances (http://www.atsdr.cdc.gov/spl/).
In humans, S-adenosylmethionine As(III) methyltransferase (hAS3MT, EC 2.1.1.137) catalyzes transfer of methyl groups from SAM to As(III), producing MAs, DMAs, and traces of TMAs.10 Human AS3MT is primarily a liver enzyme that is a member of a large superfamily of methyltransferases that are involved in many physiological functions.11 In humans, arsenic methylation paradoxically both detoxifies arsenic and simultaneously transforms it into carcinogenic species. Biotransformation of small molecules into carcinogens is common,12 and arsenic biomethylation is associated with arsenic-related cancers by conversion of inorganic arsenic (As(III)) into carcinogenic trivalent MAs(III) and DMAs(III).2,10,13
To date, the pathway of methylation remains controversial.14 One hypothesis proposed by Challenger is that the enzyme catalyzes a series of alternating oxidative methylations and reductions, using S-adenosylmethionine as the methyl donor to generate the pentavalent products methylarsenate (MAs(V), dimethylarsenate (DMAs(V)) and a lesser amount of trimethylarsine oxide (TMAs(V)O).1 The trivalent species MAs(III), DMAs(III), and TMAs(III) are intermediates but not products. Most consistent with this hypothesis is that humans primarily excrete DMAs(V) and to a lesser extent MAs(V).15,16 However, with careful handling, trivalent methylated arsenicals can be detected in urine.17,18 More recently, Hayakawa and co-workers proposed an alternate pathway in which the preferred substrates of the methyltransferase are the glutathione (GSH) conjugates As(GS)3 and MAs(GS)2, and the products are the trivalent conjugates MAs(GS)2 and DMAs(GS).2 In this pathway, there is no change in the oxidation state of arsenic, which remains trivalent throughout the catalytic cycle. The conjugates dissociate to unstable MAs(III) and DMAs(III), which rapidly oxidize nonenzymatically in air to MAs(V) and DMAs(V), the primary observed urinary species.18,19
Knowledge of the enzymatic mechanism of AS3MT is critical for understanding its parallel roles in arsenic detoxification and carcinogenesis. There have been few biochemical studies with purified human AS3MT. AS3MT was first purified from rat liver cytosol by Thomas and co-workers, and the rat and human genes were subsequently cloned.20−22 The enzyme methylates inorganic As(III) using SAM as methyl donor and a variety of reductants, both artificial and natural, including thioredoxin (Trx) (with an NADPH and thioredoxin reductase (TR) regenerating system).17,21,23 Geng et al. expressed the hAS3MT cDNA in E. coli as a Trx tagged protein.24 However, their protein had such extremely low activity that an excess of enzyme (3 μM) over substrate (1 μM As(III)) was required for extended periods (2 h) to observe methylation. With an excess of enzyme, there is at most a single turnover and not real measurement of catalysis. Ding et al. also purified hAS3MT from a cDNA clone.17 This enzyme was capable of slowly producing both MAs(III) and DMAs(III), with either Trx or the artificial reductant tris(2-carboxyethyl)phosphine (TCEP) supplying reducing potential. In vitro, GSH is not sufficient but increases methylation in conjunction with Trx.23 The identity of the physiological electron donors is not clear. Perhaps Trx and GSH have different roles in the catalytic cycle of AS3MT. This purified hAS3MT methylates As(III) primarily to DMAs(V), but, like the other hAS3MT expressed from a cDNA clone,25 its activity is low. Equimolar enzyme and As(III) (each at 1 μM) for 2 h was required to transform As(III) to DMAs(V), so this may also represent only a single turn over and not sustained catalysis.
The in vitro properties of human AS3MT have not been systematically analyzed under catalytic conditions but only in assays with stoichiometric enzyme. We also expressed a cDNA clone of human AS3MT and purified hAS3MT by heterologous expression in Escherichia coli.(17) However, this preparation of recombinant enzyme was no more active than those of other laboratories. To produce a highly active form of human AS3MT functional in catalytic concentrations and to improve expression, the hAS3MT gene was chemically synthesized with codon optimization for E. coli. Codon optimization does not alter the amino acid sequence of the protein but generally improves translation, protein folding, and activity.26,27 The synthetic enzyme exhibited high activity in catalytic concentrations. In this study, we examined the catalytic properties of the synthetic enzyme. Binding of arsenicals to a single-tryptophan derivative of hAS3MT was estimated from the quenching of intrinsic protein fluorescence, and the enzyme appears to bind As(GS)3 preferentially over inorganic As(III). In addition, MAs(III) was the primary product observed within the first few minutes of catalysis but remained mostly enzyme-bound until it was methylated a second time. These results are consistent with the pathway proposed by Hayakawa and co-workers and are inconsistent with the Challenger pathway. The involvement of the four conserved cysteine residues, Cys32, Cys61, Cys156, and Cys206, was examined by site-directed mutagenesis. Substitution of any of the four resulted in loss of ability to methylate As(III), but both the C32S and C61S derivates could still methylate MAs(III). These results suggest that only Cys156 and Cys206 are required for all steps in the methylation pathway and that Cys32 and Cys61 play a different role in the catalysis. A homology model of hAS3MT based on the crystal structure of a thermophilic orthologue indicates that Cys156 and Cys206 form the binding site for As(III) and MAs(III), whereas Cys32 and Cys61 approach each other closely enough to form a disulfide bond. We propose that this disulfide bond forms during the catalytic cycle and that a role of Trx is to reduce the disulfide and regenerate the active form of AS3MT.
Materials and Methods
Reagents
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO). MAs(V) was reduced to MAs(III) as described.28 For DNA manipulation, E. coli TOP10 (F–mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139Δ(araA-leu)7697 galU galK rpsL endA1 nupG) was employed through out.
Synthesis of the Human AS3MT Gene and Cloning of hAS3MT cDNA
A hAS3MT gene corresponding to the sequence of the cDNA clone,22 which lacks the last nine residues of the hAS3MT sequence, was chemically synthesized with 5′ NcoI and 3′ SalI sites and with codon optimization for expression in E. coli and subcloned into the EcoRV site of pUC57-Kan (GenScript, NJ, USA). The synthetic hAS3MT gene was cloned as an NcoI/SalI digest from pUC57-Kan-hAS3MT into expression vector pET41a(+) that produces a fusion with the Schistosoma japonicum glutathione S-transferase (GST) gene at the 5′ end and eight histidine residues at the 3′ end.
To produce the natural protein, hAS3MT cDNA was generously provided by M. Styblo, University of North Carolina. The 1.1 kb fragment was amplified by PCR using forward primer 5′-CCAGCCATGGCTGCACTTCGTGACGCTGAGA-3′ (NcoI site underlined) and reverse primer 5′-CCTAGTCGACTCCAGCAGCATCAGGGACACATC-3′ (SalI site underlined). The PCR product was cloned into pBAD-Myc/His-A as an NcoI/SalI digest, generating plasmid pBAD-hAS3MT in which the gene was fused with a six-histidine tag at the 3′ end. All sequences were verified by sequencing the entire gene by Sequetech, CA, USA.
Mutant Construction
Mutations in the AS3MT were introduced by site-directed mutagenesis using a QuikChange mutagenesis kit (Stratagene, La Jolla, CA). The oligonucleotides used for mutagenesis are listed in Table S1. The codons for the conserved Cys32, Cys61, Cys156, and Cys206 and nonconserved Cys72, Cys85, and Cys250 residues were changed to serine codons, generating seven different single-cysteine mutants of the synthetic hAS3MT. The double C32S/C61S mutant was generated by mutating codon 32 in the C61S derivative to a serine codon. Each mutation was confirmed by sequencing the entire gene.
Protein Expression and Purification
Wild-type synthetic AS3MT and its mutants were expressed in E. coli BL21(DE3). Cells bearing plasmid pET41a-hAS3MT were cultured at 37 °C in 1 L of Luria Broth medium consisting of 10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl per liter containing 50 μg/mL kanamycin to mid log phase (A600nm ∼ 0.5–0.6) before induction with 0.3 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3 h.29 The induced culture was centrifuged at 5000 rpm at 4 °C for 15 min, suspended in 20 mL of buffer A consisting of 50 mM NaH2PO4, pH 8.0, containing 0.3 M NaCl and 1 mM TCEP to which 10 mM imidazole was added. The cells were lysed using a French press in the presence of diisopropyl fluorophosphate and centrifuged at 35 000 rpm for 1 h. The supernatant was loaded (0.7 mL/min) onto a 5 mL Ni-NTA agarose column, pre-equilibrated with 5 column volumes of buffer A. The column was washed (1 mL/min) with 10 column volumes of buffer A containing 20 mM imidazole, hAS3MT was eluted (0.7 mL/min) with 8 column volumes of buffer A containing 0.25 M imidazole. Imidazole was removed, and the protein was concentrated to 1 mL by centrifugation, washing three times with 4 mL of buffer A containing no imidazole, with Amicon ultra centrifugal filters with 30K membrane (Millipore, Billerica, MA). Glycerol was added to 20% (v/v), and the protein was stored in 25 μL aliquots at −80 °C until use. Protein concentrations were determined from calculated extinction coefficients at 280 nm.30 The cDNA gene was expressed from E. coli TOP10 bearing pBAD-hAS3MT. The cells were cultured at 37 °C in 1 L of Luria Broth medium containing 100 μg/mL ampicillin to mid log phase (A600nm ∼ 0.5–0.6) before induction with 0.2% (w/v) l-arabinose for 3 h. The natural hAS3MT was purified as described above. Trx and TR were purified by Ni-NTA chromatography as described above from E. coli BL21(DE3) carrying either pET14b-trxA or pET14b-trxB (kindly provided by J. Messens of VIB-Vrije Universiteit, Brussels, Belgium). All buffers were degassed by bubbling with argon for 30 min before use.
Arsenic Methylation
Methylation of As(III) and MAs(III) was assayed both in E. coli expressing AS3MT genes and by purified proteins. Individual colonies of E. coli strains BL21(DE3) or TOP10 bearing the appropriate plasmids were inoculated into 2 mL of Luria Broth medium supplemented with the appropriate antibiotics and incubated at 37 °C overnight. Late exponential phase cells were diluted 200-fold into 2 mL of Luria Broth medium containing 50 μg/mL kanamycin, 0.3 mM IPTG, and 20 μM sodium arsenite. After 12 h at 37 °C, the cells were removed by centrifugation, and the supernatant solution was immediately passed through a 3 kDa cutoff Amicon ultrafilter (Millipore, Billerica, MA). The filtrate was speciated by high-pressure liquid chromatography (HPLC) (PerkinElmer Series 2000) using a C18 reversed-phase column eluted with a mobile phase consisting of 3 mM malonic acid, 5 mM tetrabutylammonium hydroxide, and 5% (v/v) methanol (pH 5.9) with a flow rate of 1 mL/min, and arsenic content was determined by inductively coupled plasma mass spectrometry (ICP-MS) using an ELAN DRC-e spectrometer (PerkinElmer, Waltham, MA).
Methylation activity of purified AS3MTs was assayed at 37 °C in buffer B consisting of 50 mM NaH2PO4, pH 8.0, containing 0.3 M NaCl. Unless otherwise noted, the assays contained 5 mM GSH, 1 mM SAM, 10 μM Trx, 3 μM TR, and 0.3 mM NADPH, and the reactions were terminated by adding 10% (v/v) H2O2 to oxidize all arsenic species. Denatured protein was removed by centrifugation using a 3 kDa cutoff Amicon ultrafilter. The filtrate was speciated by HPLC-ICP-MS.
Where noted, H2O2 was not added to allow for determination of trivalent arsenicals. Without oxidation, the majority of the arsenic at early times was found to be bound to the enzyme. To determine the nature of AS3MT-bound arsenicals, purified synthetic hAS3MT (5 μM) was incubated at 37 °C with 20 μM As(III) as described above for 10 min. A portion of the assay mixture was passed through a Bio-Gel P-6 column pre-equilibrated with buffer B, and then portions (25 μL) were immediately diluted with 6 M guanidine HCl or 8 M urea to denature protein and release the bound arsenicals, which were determined as described above.
Fluorescence Assays
hAS3MT has three tryptophan residues at positions 73, 203, and 213 (numbering is according to the translated cDNA sequence) (Figure S3). To obtain a single-tryptophan derivative of hAS3MT, three double-tryptophan mutants were prepared by changing two of the three tryptophan residues at each position (Table S2), creating single-tryptophan derivatives Trp73 (W203L/W213Y), Trp203 (W73R/W213Y), and Trp213 (W73R/W203Y) by site-directed mutagenesis. For removal of the GST tag, the GST–Trp73 fusion protein was incubated with thrombin (0.7 U/mg fusion protein) in a buffer consisting of 0.14 M NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, and 1 mM TCEP, pH 7.3, at 23 °C for 18 h. Following the cleavage reaction, the Trp73 enzyme was purified at 4 °C using glutathione sepharose 4B (GE Healthcare, USA) following manufacturer’s instructions.
Fluorescence measurements were performed on a temperature-controlled QuantaMaster UV VIS QM-4 steady state spectrofluorometer (Photon Technology International, Birmingham, NJ) at 25 °C. For steady-state measurements, both emission and excitation monochromator slits were set at 1 nm. Samples were excited at 295 nm to excite tryptophan, and emission was set at 336 nm for time-based data acquisition. Spectra were corrected for background fluorescence and Raman scattering by subtracting buffer spectra. The buffer used was 50 mM MOPS and 0.5 M NaCl, pH 7.5. For determination of relative binding of free metalloids, As(III) and MAs(III), and preformed GSH conjugates, fluorescence spectra were acquired at the indicated concentrations of arsenicals in the presence and absence of GSH. To obtain binding rates, time-based fluorescence quenching data were fitted to a single exponential decay isotherm using Prism 6 (GraphPad Software Inc.).
Homology Model of the hAS3MT Structure
A homology model of hAS3MT was built on the structure of PhAs(III)-bound CmArsM (PDB ID: 4KU9) using a fully automated protein structure homology modeling server SWISS-MODEL (http://swissmodel.expasy.org/).31 The four conserved cysteine residues (Cys32, Cys61, Cys156, and Cys206) (residue numbers are based on the hAS3MT sequence) are present in the model structure. The model quality was estimated on the basis of the QMEAN scoring function.32 The model structure with residues 44–371 (residue numbers are based on the CmArsM sequence) from PhAs(III)-bound CmArsM was used as a template, and the final homology model incorporated 308 of those 328 residues. In silico docking with SAM was carried out using the PATCHDOCK server.33 The docked hAS3MT model with SAM was superimposed with the As(III)-bound structure of CmArsM (PDB ID: 4FSD) to acquire the arsenic atom in the As(III) binding site of hAS3MT.34 PyMOL v1.3 was used to visualize the structural models.35,36
Results
Chemical Synthesis of hAS3MT
In two prior studies, human AS3MT was expressed from a cDNA clone and purified after heterologous expression in E. coli.20,24 In both cases, methylation was assayed using molar ratios of AS3MT/As(III) of either 1:1 or 3:1 for extended periods of time.17,24 With stoichiometric or higher ratios of enzyme to substrate, not more than a single cycle of methylation is possible, so it is not know whether either construct could attain steady state, the most commonly used approach to enzyme kinetics.37 We took a different approach to the generation of purified AS3MT that could work at catalytic rather than stoichiometric concentrations.
The sequence of the cDNA that had been used by others to produce hAS3MT was chemically synthesized. The key feature of the chemical synthesis was an increase in the codon usage bias for E. coli by upgrading the codon adaptation index from 0.64 to 0.88. In addition, the GC content was optimized, and stem-loop structures were disrupted to increase ribosome binding and the half-life of the mRNA. The stem-loop structures, which impact ribosomal binding and stability of mRNA, were broken. The final product has a total of 662 residues with 366 of the AS3MT residues unchanged from the cDNA sequence, a fusion with glutathione S-transferase at the N-terminus to improve solubility including tandem thrombin and enterokinase sites, and eight histidines at the C-terminus for purification. The changes in the synthetic sequence from that of the cDNA sequence are highlighted in Figure S1, and the sequence of the protein is shown in Figure S2.
Binding of Trivalent Arsenicals to Synthetic hAS3MT
Intrinsic tryptophan fluorescence has been extensively exploited in examining arsenic binding in arsenic detoxification proteins.28,38−41 hAS3MT has three tryptophan residues at positions 73, 203, and 213 (Figure S3). To obtain a single-tryptophan derivative of hAS3MT, three double-tryptophan mutants were prepared by changing two of the three tryptophan residues at each position (Table S2), creating single-tryptophan derivatives Trp73 (W203L/W213Y), Trp203 (W73R/W213Y), and Trp213 (W73R/W203Y). The GST tag, which contains four tryptophan residues, was removed by thrombin cleavage, as described in the Materials and Methods. Of the three single-tryptophan derivatives, only the Trp73 derivative reported binding of arsenicals with a quenching of protein fluorescence. The observed rate of quenching by preformed As(GS)3 to hAS3MT was faster than could be measured accurately with our fluorometer (Figure 1A, curve 5). The fluorescence signal slowly increased, but addition of GSH to the buffer stabilized the quenching (Figure 1A, curve 6). We interpret this result as the As(GS)3 complex dissociating when diluted into the assay buffer, and that GSH in the buffer prevents dissociation. In contrast, the rate of quenching with inorganic As(III) was extremely slow (Figure 1A, curve 3), requiring approximately 10 min to produce the same quenching as that produced by As(GS)3 in 1 s, but the quenching by As(III) could be accelerated by addition of GSH to the buffer (Figure 1A, curve 4), which we interpret as conjugate formation in the assay buffer. When the quenching of tryptophan fluorescence was fitted to a single exponential decay isotherm, the rate with inorganic As(III) in buffer containing GSH (Figure 1A, curve 4) was approximately 130-fold faster than the rate in the absence of GSH (Figure 1A, curve 3).
Figure 1.

Effect of As(GS)3 and MAs(GS)2 on the fluorescence of Trp73 hAS3MT. Following removal of the GST tag, intrinsic protein fluorescence of Trp73 hAS3MT was assayed at 25 °C in degassed buffer, as described in the Materials and Methods. As noted, 5 mM GSH was added to the buffer. Excitation and emission wavelengths were 295 and 336 nm, respectively. Metalloids were added at a concentration of 1 mM to 1 μM Trp73 hAS3MT. (A) Quenching by inorganic arsenicals. Additions: curve 1, no addition; curve 2, As(V); curve 3, As(III); curve 4, As(III) with GSH in the buffer; curve 5, As(GS)3; curve 6, As(GS)3 with GSH in the buffer. (B) Quenching by methylated arsenicals. Additions: curve 1, no addition; curve 2, MAs(V); curve 3, MAs(III); curve 4, As(GS)3 with GSH in the buffer; curve 5, MAs(GS)2 with GSH in the buffer.
Similar results were observed with addition of either MAs(III) or MAs(GS)2. Fluorescence quenching with MAs(III) (Figure 1B, curve 3) was slow compared to that with MAs(GS)2, but it was faster than with inorganic As(III). Fluorescence quenching could be accelerated by addition of GSH to the buffer (Figure 1B, curve 4), which was similar to quenching with preformed MAs(GS)2 (Figure 1B, curve 5), but both were too fast to quantify. Clearly, binding of glutathione conjugates is extremely rapid, but detailed rate determinations will require stopped-flow analysis.
Catalytic Properties of Synthetic AS3MT
Initial studies investigated methylation of As(III) by hAS3MT in reaction mixtures in which physiological and nonphysiological reductants were employed. Under all conditions, As(III) was methylated to both MAs and DMAs, but the extent of methylation and the ratio of DMAs/MAs varied (Figure 2). In these assays, no differentiation was made between trivalent and pentavalent products because the reactions were terminated by addition of H2O2, which oxidizes all arsenicals to their pentavalent forms. As described below, this was necessary because a significant percentage of the total arsenic remained bound to the enzyme. Following oxidation with peroxide, all of the arsenic could be recovered. In human populations, individuals with higher conversion to DMAs have lower rates of bladder cancer and other arsenic-related diseases.42 MAs(III) is considered to be more toxic than DMAs(III), and a higher fraction of MAs excreted in urine correlates with arsenic-related diseases.16,19,42 People with more rapid methylation to DMAs may have more rapid clearance and be less susceptible to diseases. On the basis of those considerations, we define optimal hAS3MT activity as conditions that resulted (1) in the greatest total methylation and (2) produced the highest ratio of DMAs to MAs. Under the conditions employed, hAS3MT showed the highest activity with the combination of cysteine and the artificial reductant TCEP. Note that nearly all of the As(III) was methylated with an As(III)/hAS3MT ratio of 10, the first example of multiple rounds of catalysis by purified hAS3MT. With possible physiological reductants, GSH alone produced the lowest activity. High activity was observed with addition of purified Trx plus a Trx-regenerating system consisting of NAPH and TR (termed the Trx system). With the Trx system, the DMAs/MAs ratio was similar to that with TCEP alone (5.5–7.3). Addition of GSH to either TCEP or the Trx system both increased total methylation and the DMAs/MAs ratio (7.0–11.7). Thus, GSH is not a particularly good reductant but might play a different role than Trx, such as forming the As(GS)3 substrate.2
Figure 2.

Effect of reductants on methylation of As(III) by synthetic hAS3MT. The reaction containing 1 μM hAS3MT, 1 mM SAM, 10 μM As(III), and the indicated reductants was incubated for 90 min at 37 °C. The reactions were terminated by addition of 10% (v/v) H2O2 to oxidize all arsenic species, which were speciated by HPLC-ICP-MS. Reductants (final concentrations): 5 mM GSH, 5 mM cysteine, 1 mM TCEP, 10 μM Trx (+ 3 μM thioredoxin reductase and 0.3 mM NADPH), or mixtures of those as indicated. Open bars (left), inorganic arsenic; left-slanted bars (middle), MAs; right-slanted bars (right), DMAs. The data are the mean ± SE (n = 3).
Precursor–Product Relationship of hAS3MT Methylation
The time course of As(III) methylation over a 3 h period was determined using GSH and the Trx system (Figure 3A). The production of MAs was linear over the first 10 min, reaching a plateau at 30 min and decreasing to nearly zero after 180 min. At this point, MAs accounted for approximately about 15% of the total arsenic in the reaction mixture. The production of DMAs had a lag of approximately 10 min and then increased until 97% of the total arsenic was present as DMAs. These results are consistent with the Briggs–Haldane steady-state assumption of enzyme catalysis, which predicts a precursor–product relationship for an irreversible reaction.37 In this case, the immediate product is MAs, which disappears as it is converted to the major product DMAs. Under similar conditions, the native enzyme from the cDNA construct exhibited little methylation activity (Figure 3B). When both synthetic and native enzyme were present at catalytic concentrations (1 μM enzyme and 10 μM As(III)), little DMAs was produced by the native enzyme after 2 h, compared with 80% conversion with the synthetic enzyme. Thus, the synthetic enzyme is active when present in catalytic concentrations, whereas the purified native enzyme is not. Although we cannot eliminate other possibilities, a logical inference is that they differ in conformation, perhaps because the native enzyme does not fold as well as the synthetic enzyme, which was optimized for E. coli expression. One possible explanation is that the native gene has a total of 17 rare codons for Arg, Leu, Ile, and Pro that were corrected in the synthetic gene. That could slow the rate of translation and hence the rate of folding of the native protein.
Figure 3.

As(III) methylation by synthetic and native hAS3MTs. The reaction mixture (1.5 mL) containing 1 μM of either purified synthetic (A) or native (B) hAS3MT, 1 mM SAM, 5 mM GSH, 10 μM Trx, 3 μM TR, 0.3 mM NADPH, and 10 μM As(III) was incubated at 37 °C. Samples were withdrawn at the indicated times, and the reaction terminated by addition of 10% (v/v) H2O2, final concentration, and analyzed by HPLC-ICP-MS. The data are the mean ± SE (n = 3).
The Initial Product of the Methylation Reaction Is Trivalent and Remains Bound to the Enzyme
After 10 min at 37 °C, synthetic hAS3MT methylated As(III) to soluble MAs(III), with little MAs(V) observed (Figure 4A). These results support the postulate of Hayakawa et al. that the products of hAS3MT are trivalent, not pentavalent.2 However, at early times the majority of the arsenic appeared to be insoluble, and we predicted that it is bound to the enzyme. To examine this possibility, As(III) methylation was carried out for 10 min with concentrations of enzyme (5 μM) and As(III) (20 μM) higher than in the usual assay conditions to obtain sufficient enzyme-bound form. Unbound arsenic was removed, and the protein with bound arsenicals was denatured with guanidine HCl or urea to release the protein-bound arsenicals (Figure 4B). When a protein is denatured with guanidine HCl or urea, it is unfolded, disrupting the secondary and tertiary structures. Because substrate binding sites in enzymes are composed of amino acid residues that are brought together in precise distances and geometries, unfolding of the protein will destroy the binding site and release noncovalently bound ligands. Denatured protein was removed by filtration, and the filtrate was speciated by HPLC-ICP-MS. MAs(III) was the major bound arsenical form released from the enzyme. A small amount of MAs(V) was also observed, but in control assays, a similar amount of MAs(III) was nonenzymatically oxidized to MAs(V).
Figure 4.

MAs(III) forms an enzyme-bound intermediate. (A) Formation of soluble MAs(III). Purified hAS3MT (1 μM) was incubated at 37 °C with 10 μM As(III) containing 5 mM cysteine and 1 mM SAM for the indicated times. Protein was removed, and the soluble arsenicals were speciated by HPLC-ICP-MS. (B) Formation of enzyme-bound MAs(III). Purified hAS3MT (5 μM) was incubated at 37 °C with 20 μM As(III) containing 5 mM GSH, 1 mM SAM, 10 μM Trx, 3 μM TR, and 0.3 mM NADPH. After 10 min, samples were separated through a Bio-Gel P-6 spin column. Portions (25 μL) were diluted with 6 M guanidine HCl or 8 M urea to final concentrations of 4 M to denature the protein and release the enzyme-bound arsenicals, which were analyzed by HPLC-ICP-MS.
Effect of Substitution of Conserved Cysteine Residues on hAS3MT Activity
As(III) SAM methyltransferases have four conserved cysteine residues (Figure S3), Cys32, Cys61, Cys156, and Cys206, in hAS3MT.28,43 The cDNA clone lacks the last nine residues of the actual hAS3MT sequence, including three cysteine residues, but there are seven other nonconserved cysteines (Cys72, Cys85 Cys226, Cys250, Cys271, Cys334, and Cys360). Of these nonconserved residues, Cys72, Cys85, and Cys250 were altered to serine residues, and cells expressing the C72S, C85S, and C250S derivatives retained the ability to methylate As(III) (Figure 5A). When the conserved cysteines were altered to serine residues, and cells expressing the C32S, C61S, C156S, and C206S derivatives did not methylate As(III) (Figure 5A). The C32S, C61S, C156S, C206S, and C32S/C61S derivatives were expressed, and the activity of the purified enzymes examined. None of the altered proteins was able to methylate inorganic As(III) even after 3 h (Figure 5B). In contrast, C32S and C61S retained the ability to methylate MAs(III) (Figure 6), whereas neither C156S nor C206S were able to do so (data not shown). Although the results of mutagenesis are always open to interpretation, these observations indicate that Cys156 and Cys206 are essential for both the first and second cycles of methylation, whereas Cys32 and Cys61 are required for the first methylation but not the second. Thus, there appears to be two classes of cysteine residues: Cys32 and Cys61 in one class and Cys156 and Cys206 in the other.
Figure 5.

Role of cysteine residues in methylation of As(III). (A) In vivo methylation of As(III) by wild-type and mutant hAS3MTs. E. coli BL21(DE3) cells bearing vector pET41a(+) or plasmids with wild-type or cysteine mutant genes in pET41a-hAS3MT were grown in 2 mL of Luria Broth medium in the presence of 20 μM As(III), 0.3 mM IPTG, and 50 μg/mL kanamycin at 37 °C for 12 h. (B) Methylation of As(III) by purified wild-type and mutant hAS3MTs. Methylation of As(III) was assayed at 37 °C after 3 h with 10 μM As(III), 1 μM hAS3MT, 1 mM SAM, 5 mM GSH, 10 μM Trx, 3 μM TR, and 0.3 mM NADPH.
Figure 6.

Role of cysteine residues in methylation of MAs(III). Methylation of MAs(III) by cysteine mutants was assayed at the indicated times at 37 °C with 10 μM MAs(III), 1 μM hAS3MT, 1 mM SAM, 5 mM GSH, 10 μM Trx, 3 μM TR, and 0.3 mM NADPH. The reaction was terminated by addition of 10% (v/v) H2O2, final concentration. Protein was removed before analysis, and soluble arsenicals were analyzed by reversed-phase HPLC-ICP-MS. (A) Synthetic wild-type hAS3MT. (B) C32S mutant. (C) C61S mutant. (D) C32S/C61S double mutant. The C156S and C206S derivatives did not methylate MAs(III) (not shown).
A Homology Structural Model of hAS3MT
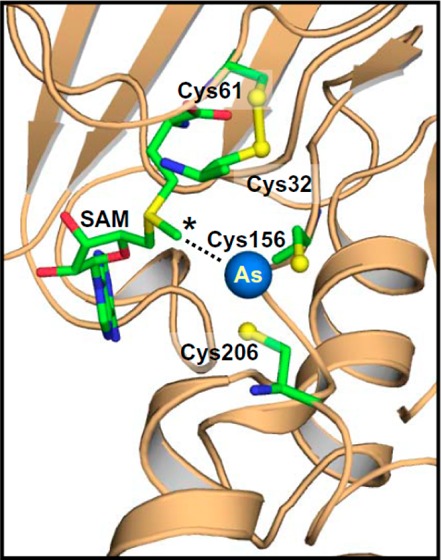
A homology model of hAS3MT model was built on the CmArsM structure with bound PhAs(III) (PDB ID: 4KU9) using the SWISS-MODEL fully automated protein structure homology modeling server (http://swissmodel.expasy.org/) (Figure 7).31 The model quality was estimated on the basis of a qualitative model energy analysis scoring function of 0.64, which is within the acceptable range.32 The homology model incorporated 308 of 328 residues from 4KU9. The secondary structural arrangement of hAS3MT was nearly the same as that of the CmArsM structure, with equivalent metalloid and SAM binding elements. In silico docking with SAM was carried out using the program PATCHDOCK.33 This model was superimposed with the As(III)-bound structure of CmArsM (PDB ID: 4FSD) to place an arsenic atom in the As(III) binding site of hAS3MT, and PyMOL v1.3 was used to visualize the structural model.36,43 Although only a model, it is instructive and provides testable predictions. The biochemical data demonstrate that the C-terminal pair of conserved cysteine residues (Cys156 and Cys206) are involved in methylation of both As(III) and MAs(III). The N-terminal conserved cysteine pair (Cys32 and Cys61) is involved in As(III) methylation but not MAs(III) methylation, suggesting a more subtle role in the catalytic cycle. In the model, Cys156 and Cys206 form an As(III) binding site, with the arsenic atom 2.2 and 2.3 Å from the sulfur atoms of Cys156 and Cys206, respectively. In contrast, Cys32 and Cys61 are oxidized in a disulfide bond. The methyl group of SAM was oriented toward the bound As(III) at approximately 3.9 Å distance.
Figure 7.

Homology structural model of human AS3MT. The cartoon diagram representation of human AS3MT model structure is colored in tan. The location of four conserved Cys residues are shown along with the bound SAM and As(III). The expanded view shows the four conserved cysteine residues (in ball-and-stick), with Cys32 and Cys61 in a disulfide bond and Cys156 and Cys206 binding the As (blue ball) atom. SAM (in ball-and-stick) occupies its binding site with its methyl group (*) poised to be donated to the arsenic atom.
A Disulfide Bond Cascade Reaction Mechanism for hAS3MT
On the basis of a combination of experimental data and structural modeling, we propose a novel reaction scheme involving a disulfide bond cascade with at least two sequential disulfide bonds: the first between Cys32–Cys61 and the second between Cys61–Cys156 (Figure 8), based on the disulfide bonds observed in the crystal structures of the Cyanidioschyzon merolae orthologue (PDB IDs: 4KU9 and 4FR0). The catalytic cycle for the first two rounds of methylation can be summarized in eight steps. (1) In the first round of methylation, hAS3MT binds As(III) in a series of three thiol transfer reactions from As(GS)3, which is the preferred substrate (Figure 1). (2) The methyl group of SAM is attacked by the arsenic lone pair, (3) which leads to formation of a transient positively charged pentavalent MAs(V) intermediate.44 (4) The role of Cys32 (or Cys61; at this time, we cannot distinguish between them) is to provide electrons to reduce enzyme bound MAs(V) to MAs(III) and to allow the next round of methylation. By donating electrons to the arsenic, Cys32 (or Cys61) becomes oxidized, forming a disulfide bond with Cys61. (5) The role of Trx is to reduce the disulfide bond, consistent with the traditional role of thioredoxins in facilitating reduction of protein cysteine disulfides by thiol–disulfide exchange. Oxidized Trx is reduced by TR with electrons from NADPH. MAs(III) remains strongly bound by the thiol pair Cys61–Cys156. The disulfide bond is reduced with Trx, and (6) the enzyme undergoes the next round of methylation, forming a transient positively charged pentavalent DMAs(V) intermediate, (7) which is reduced to DMAs(III) by Cys61, forming a Cys61–Cys156 disulfide. (8) The disulfide is reduced by Trx, regenerating the enzyme and releasing the major soluble product, DMAs(III). Finally, in urine, exposure to air nonenzymatically oxidizes DMAs(III) to DMAs(V). Thus, the substrates and products are all trivalent, as predicted by Hayakawa et al., but the arsenic undergoes a cycle of oxidation and reduction, as hypothesized by Challenger, but only when it is enzyme-bound. GSH and Trx are involved, but not as reductants of pentavalent intermediates. Instead, As(GS)3 serves as the arsenic donor to the enzyme, as hypothesized by Hayakawa et al., and Trx is a protein disulfide reductase.45 Advantages of this pathway are that it explains current results with As(III) SAM methyltranferases and reconciles the proposals of Challenger and Hayakawa et al.
Figure 8.

Proposed hAS3MT reaction scheme: (1) In the first round of methylation, hAS3MT binds As(III) in a series of three thiol transfer reactions from As(GS)3; (2) the methyl group of SAM is attacked by the arsenic lone pair; (3) a pentavalent MAs(V) intermediate is formed and (4) reduced to an enzyme-bound MAs(III) intermediate by Cys32 with formation of a Cys32–Cys61 disulfide; (5) the disulfide is reduced with Trx, and the enzyme undergoes the next round of methylation, (6) forming a pentavalent DMAs(V) intermediate, (7) which is reduced to DMAs(III) by Cys61. This form is a Cys61–Cys156 disulfide, which (8) is reduced by Trx, regenerating the enzyme and releasing the major soluble product, DMAs(III). Finally, in air, trivalent DMAs(III) is nonenzymatically oxidized to DMAs(V).
Discussion
Arsenic is clearly a health hazard, but is it inorganic or methylated arsenicals that produce the risk? The answer depends in large part on the products of AS3MT, the enzyme that methylates arsenic in liver and other organisms. There are two schools of thought on this matter. The original proposal by Challenger in the 1940s involved a series of oxidative methylations such that the substrates were trivalent arsenicals and the products were pentavalent.1 A more recent proposal by Hayakawa et al. suggests that both the substrates and products are trivalent, a key difference between the two major proposals.2 If Challenger is correct, then the products are relatively nontoxic and noncarcinogenic. If Hayakawa et al. is correct, then the products are more toxic and carcinogenic than the inorganic As(III) substrate.
Challenger was ahead of his time since nothing was known of the very large superfamily of methyltransferase enzymes in the 1940s. Members of that superfamily append a methyl group from SAM to acceptor groups by SN2 displacement mechanisms, involving attack of a nucleophile on the methyl group of SAM with inversion of configuration and concomitant release of SAH.11 Methyltransferases are categorized on the basis of the electron-rich, methyl-accepting atom, usually O, N, C, or S. These enzymes have been studied in exquisite detail for more than 50 years. They all have a conserved SAM binding fold and use a common enzymatic mechanism of methyltransfer, where SAM serves as a potent alkylating agent with destabilization of the sulfonium ion. While Challenger proposed a series of oxidations and reductions along with three rounds of methylations, none of the O-, N-, C- and S-methyltransferases oxidizes their substrates, and no one has demonstrated the presence of a pentavalent arsenic intermediate bound to AS3MT or its orthologues. The evidence that AS3MT directly generates As(V) metabolites is not strong. Lysine and arginine methyltransferases are good models for arsenic methyltransferases, as they carry out three very similar methyl transfers without oxidation of the nitrogen. In summary, methyltransferases methylate their substrates without oxidation. Nature is conservative, and there is no reason to consider that AS3MT would use a different enzymatic mechanism from that of any other member of the superfamily.
Yet, a major chemical difference between the metalloid arsenic and the nonmetal atoms (O, N, C, S) acceptors of the methyl group is its redox activity. For that reason, there is the possibility that As(III) in the metalloid binding site of hAS3MT becomes oxidized during the catalytic cycle and is reduced again by donation of electrons from the conserved cysteine residues. Thus, we propose the formation of the transient pentavalent intermediates shown in steps 3 and 6 of our proposed reaction cycle (Figure 8). However, it is not clear if pentavelent intermediates are obligatory components in the mechanism or side reactions that are the consequences of oxidation. Regardless of whether AS3MT catalyzes alternation between reduced substrates and oxidized intermediates, transient trivalent intermediates are likely to be easily oxidized, and there must be a mechanism to keep them reduced. In either case, the disulfide bond cascade that takes place using the conserved cysteine residues of AS3MT is a novel feature of As(III) SAM methyltransferases that differentiates it from other members of the methyltransferase superfamily.
Another prediction of Hayakawa et al. is that the substrate is As(GS)3 and not free inorganic arsenic. This is very reasonable because GSH, the major thiol in cells, is present at millimolar concentrations and reacts readily with As(III) to form the triglutathione complex inside cells.46 Some microorganisms use thiols other than GSH as their major intracellular reductant.47 Although we have no data on methylation in any of those organisms, we can predict that they will be able to use other As–thiol conjugates. Dithiols can readily abstract As(III) from As(GS)3, so it is also reasonable that As(GS)3 is able to donate As(III) to conserved cysteine residues in hAS3MT.48 The As(GS)3 complex is unstable in vitro, so it is challenging to observe its interaction with enzymes. Quenching of intrinsic protein fluorescence by arsenicals in single-tryptophan derivatives has been shown to reflect binding.28,41 Using single-tryptophan derivates, we examined the transfer of As(III) from As(GS)3 to the binding site in the ArsD metallochaperone and the CmArsM orthologue of hAS3MT. We took a similar approach with hAS3MT by introduction of a single tryptophan, Trp73, adjacent to the predicted As(III) binding site. The results are striking: free inorganic As(III) binds over a period of minutes, whereas preformed As(GS)3 binds in seconds. Similarly, MAs(GS)2 bound faster than MAs(III). An enzyme that takes minutes to bind its substrate is not a very useful catalyst. If As(III) was added to GSH-containing buffer, quenching was enhanced relative to that with GSH-free buffer, indicating formation of the conjugates during the course of the assay. Even As(GS)3 quenched fluorescence faster when the buffer contained GSH, suggesting that the conjugate dissociates when diluted into GSH-free buffer but is stabilized when the buffer contains GSH. These results are most consistent with the glutathione conjugates of inorganic and methylated arsenicals serving as the source of arsenic for hAS3MT.
Acknowledgments
We thank Joris Messens for the gift of plasmids and to Miroslav Styblo for the cDNA clone and for discussions on the nature of the bound intermediate.
Glossary
Abbreviations
- AS3MT
As(III) S-adenosylmethionine methyltransferase
- MAs(III)
methylarsenite
- MAs(V)
methylarsate
- DMAs(III)
dimethylarsenite
- DMAs(V)
dimethylarsenate
- TMAs(III)
trimethylarsine
- TMAs(V)O
trimethylarsine oxide
- SAM
S-adenosylmethionine
- SAH
S-adenosylhomocysteine
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- GuHCl
guanidine hydrochloride
- GSH
reduced glutathione
- As(GS)3
arsenic triglutathione
- TR
thioredoxin reductase
- Trx
thioredoxin
- IPTG
isopropyl β-d-1-thiogalactopyranoside
Supporting Information Available
Oligonucleotide primers for mutagenesis of cysteine and tryptophan residues, nucleotide sequence of the hAS3MT cDNA clone and the synthetic gene, amino acid sequence of synthetic hAS3MT, and multiple alignment of hAS3MT with orthologues. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health grant R37 GM55425.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Challenger F. (1947) Biological methylation. Sci. Prog. 35, 396–416. [PubMed] [Google Scholar]
- Hayakawa T.; Kobayashi Y.; Cui X.; Hirano S. (2005) A new metabolic pathway of arsenite: arsenic–glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch. Toxicol. 79, 183–191. [DOI] [PubMed] [Google Scholar]
- Zhu Y. G.; Yoshinaga M.; Zhao F. J.; Rosen B. P. (2014) Earth abides arsenic biotransformations. Annu. Rev. Earth Planet. Sci. 42, 443–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abernathy C. O.; Liu Y. P.; Longfellow D.; Aposhian H. V.; Beck B.; Fowler B.; Goyer R.; Menzer R.; Rossman T.; Thompson C.; Waalkes M. (1999) Arsenic: health effects, mechanisms of actions, and research issues. Environ. Health Perspect. 107, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1998) Arsenic in ground water of the United States, U.S. Geological Survey, http://co.water.usgs.gov/trace/arsenic/.
- Naujokas M. F.; Anderson B.; Ahsan H.; Aposhian H. V.; Graziano J. H.; Thompson C.; Suk W. A. (2013) The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ. Health Perspect. 121, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abernathy C. O.; Thomas D. J.; Calderon R. L. (2003) Health effects and risk assessment of arsenic. J. Nutr. 133, 1536S–1538S. [DOI] [PubMed] [Google Scholar]
- Tchounwou P. B.; Patlolla A. K.; Centeno J. A. (2003) Carcinogenic and systemic health effects associated with arsenic exposure—a critical review. Toxicol. Pathol. 31, 575–588. [DOI] [PubMed] [Google Scholar]
- States J. C.; Srivastava S.; Chen Y.; Barchowsky A. (2009) Arsenic and cardiovascular disease. Toxicol. Sci. 107, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. J., and Rosen B. P. (2013) Arsenic methyltransferases, in Encyclopedia of Metalloproteins (Kretsinger R. H., Uversky V. N., and Permyakov E. A., Eds.) pp 140–145, Springer New York, New York. [Google Scholar]
- Liscombe D. K.; Louie G. V.; Noel J. P. (2012) Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat. Prod. Rep. 29, 1238–1250. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. (2000) Metabolism of chemical carcinogens. Carcinogenesis 21, 345–351. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Drobna Z.; Jaspers I.; Lin S.; Thomas D. J. (2002) The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ. Health Perspect. 110, 767–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. J.; Li J.; Waters S. B.; Xing W.; Adair B. M.; Drobna Z.; Devesa V.; Styblo M. (2007) Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp. Biol. Med. 232, 3–13. [PMC free article] [PubMed] [Google Scholar]
- Engstrom K.; Vahter M.; Mlakar S. J.; Concha G.; Nermell B.; Raqib R.; Cardozo A.; Broberg K. (2011) Polymorphisms in arsenic (+III oxidation state) methyltransferase (AS3MT) predict gene expression of AS3MT as well as arsenic metabolism. Environ. Health Perspect. 119, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahter M. (1999) Methylation of inorganic arsenic in different mammalian species and population groups. Sci. Prog. 82, 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L.; Saunders R. J.; Drobna Z.; Walton F. S.; Xun P.; Thomas D. J.; Styblo M. (2012) Methylation of arsenic by recombinant human wild-type arsenic (+3 oxidation state) methyltransferase and its methionine 287 threonine (M287T) polymorph: role of glutathione. Toxicol. Appl. Pharmacol. 264, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le X. C.; Lu X.; Ma M.; Cullen W. R.; Aposhian H. V.; Zheng B. (2000) Speciation of key arsenic metabolic intermediates in human urine. Anal. Chem. 72, 5172–5177. [DOI] [PubMed] [Google Scholar]
- Drobna Z.; Del Razo L. M.; Garcia-Vargas G. G.; Sanchez-Pena L. C.; Barrera-Hernandez A.; Styblo M.; Loomis D. (2012) Environmental exposure to arsenic, AS3MT polymorphism and prevalence of diabetes in Mexico. J. Exposure Sci. Environ. Epidemiol. 23, 151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Shi Q.; Nix F. B.; Styblo M.; Beck M. A.; Herbin-Davis K. M.; Hall L. L.; Simeonsson J. B.; Thomas D. J. (2002) A novel S-adenosyl-l-methionine:arsenic(III) methyltransferase from rat liver cytosol. J. Biol. Chem. 277, 10795–10803. [DOI] [PubMed] [Google Scholar]
- Thomas D. J.; Waters S. B.; Styblo M. (2004) Elucidating the pathway for arsenic methylation. Toxicol. Appl. Pharmacol. 198, 319–326. [DOI] [PubMed] [Google Scholar]
- Wood T. C.; Salavagionne O. E.; Mukherjee B.; Wang L.; Klumpp A. F.; Thomae B. A.; Eckloff B. W.; Schaid D. J.; Wieben E. D.; Weinshilboum R. M. (2006) Human arsenic methyltransferase (AS3MT) pharmacogenetics: gene resequencing and functional genomics studies. J. Biol. Chem. 281, 7364–7373. [DOI] [PubMed] [Google Scholar]
- Waters S. B.; Devesa V.; Del Razo L. M.; Styblo M.; Thomas D. J. (2004) Endogenous reductants support the catalytic function of recombinant rat cyt19, an arsenic methyltransferase. Chem. Res. Toxicol. 17, 404–409. [DOI] [PubMed] [Google Scholar]
- Geng Z.; Song X.; Xing Z.; Geng J.; Zhang S.; Zhang X.; Wang Z. (2009) Effects of selenium on the structure and function of recombinant human S-adenosyl-l-methionine dependent arsenic (+3 oxidation state) methyltransferase in E. coli. J. Biol. Inorg. Chem. 14, 485–496. [DOI] [PubMed] [Google Scholar]
- Song X.; Geng Z.; Zhu J.; Li C.; Hu X.; Bian N.; Zhang X.; Wang Z. (2009) Structure–function roles of four cysteine residues in the human arsenic (+3 oxidation state) methyltransferase (hAS3MT) by site-directed mutagenesis. Chem.–Biol. Interact. 179, 321–328. [DOI] [PubMed] [Google Scholar]
- Hannig G.; Makrides S. C. (1998) Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 16, 54–60. [DOI] [PubMed] [Google Scholar]
- Nackley A. G.; Shabalina S. A.; Tchivileva I. E.; Satterfield K.; Korchynskyi O.; Makarov S. S.; Maixner W.; Diatchenko L. (2006) Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314, 1930–1933. [DOI] [PubMed] [Google Scholar]
- Marapakala K.; Qin J.; Rosen B. P. (2012) Identification of catalytic residues in the As(III) S-adenosylmethionine methyltransferase. Biochemistry 51, 944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular cloning, a laboratory manual, Cold Spring Harbor Laboratory, New York. [Google Scholar]
- Gill S. C.; von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326. [DOI] [PubMed] [Google Scholar]
- Kiefer F.; Arnold K.; Kunzli M.; Bordoli L.; Schwede T. (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37, D387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P.; Biasini M.; Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D.; Inbar Y.; Nussinov R.; Wolfson H. J. (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2001) PyMOL, DeLano Scientific, San Carlos, CA.
- Peters B.; Moad C.; Youn E.; Buffington K.; Heiland R.; Mooney S. (2006) Identification of similar regions of protein structures using integrated sequence and structure analysis tools. BMC Struct. Biol. 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs G. E.; Haldane J. B. (1925) A note on the kinetics of enzyme action. Biochem. J. 19, 338–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Rosen B. P. (1997) Tryptophan fluorescence reports nucleotide-induced conformational changes in a domain of the ArsA ATPase. J. Biol. Chem. 272, 19731–19737. [DOI] [PubMed] [Google Scholar]
- Zhou T.; Shen J.; Liu Y.; Rosen B. P. (2002) Unisite and multisite catalysis in the ArsA ATPase. J. Biol. Chem. 8, 23815–23820. [DOI] [PubMed] [Google Scholar]
- Liu J.; Rosen B. P. (1997) Ligand interactions of the ArsC arsenate reductase. J. Biol. Chem. 27, 21084–21089. [DOI] [PubMed] [Google Scholar]
- Yang J.; Rawat S.; Stemmler T. L.; Rosen B. P. (2010) Arsenic binding and transfer by the ArsD As(III) metallochaperone. Biochemistry 49, 3658–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. J.; Huang Y. L.; Huang Y. K.; Wu M. M.; Chen S. Y.; Hsueh Y. M.; Chen C. J. (2013) Urinary arsenic profiles and the risks of cancer mortality: a population-based 20-year follow-up study in arseniasis-endemic areas in Taiwan. Environ. Res. 122, 25–30. [DOI] [PubMed] [Google Scholar]
- Ajees A. A.; Marapakala K.; Packianathan C.; Sankaran B.; Rosen B. P. (2012) Structure of an As(III) S-adenosylmethionine methyltransferase: insights into the mechanism of arsenic biotransformation. Biochemistry 51, 5476–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen W. R. (2014) Chemical mechanism of arsenic biomethylation. Chem. Res. Toxicol. 27, 457–461. [DOI] [PubMed] [Google Scholar]
- Holmgren A. (1988) Thioredoxin and glutaredoxin: small multi-functional redox proteins with active-site disulphide bonds. Biochem. Soc. Trans. 16, 95–96. [DOI] [PubMed] [Google Scholar]
- Delnomdedieu M.; Basti M. M.; Styblo M.; Otvos J. D.; Thomas D. J. (1994) Complexation of arsenic species in rabbit erythrocytes. Chem. Res. Toxicol. 7, 621–627. [DOI] [PubMed] [Google Scholar]
- Ordonez E.; Van Belle K.; Roos G.; De Galan S.; Letek M.; Gil J. A.; Wyns L.; Mateos L. M.; Messens J. (2009) Arsenate reductase, mycothiol, and mycoredoxin concert thiol/disulfide exchange. J. Biol. Chem. 284, 15107–15116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delnomdedieu M.; Basti M. M.; Otvos J. D.; Thomas D. J. (1993) Transfer of arsenite from glutathione to dithiols: a model of interaction. Chem. Res. Toxicol. 6, 598–602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.