

Abstract
Aims
We propose and test an efficient and rapid protocol for the detection of toxigenic Fusarium isolates producing three main types of Fusarium-associated mycotoxins (fumonisins, trichothecenes and zearelanone).
Methods and Results
The novel approach utilizes partially multiplexed markers based on genes essential for mycotoxin biosynthesis (fumonisin—fum6, fum8; trichothecenes—tri5, tri6; zearalenone, zea2) in Fusarium spp. The protocol has been verified by screening a collection of 96 isolates representing diverse species of filamentous fungi. Each Fusarium isolate was taxonomically identified through both molecular and morphological techniques. The results demonstrate a reliable detection of toxigenic potential for trichothecenes (sensitivity 100%, specificity 95%), zearalenone (sensitivity 100%, specificity 100%) and fumonisins (sensitivity 94%, specificity 88%). Both presence and identity of toxin biosynthetic genes were further confirmed by direct sequencing of amplification products.
Conclusions
The cross-species-specific PCR markers for key biosynthetic genes provide a sensitive detection of toxigenic fungal isolates, contaminating biological material derived from agricultural fields.
Significance and Impact of the Study
The conducted study shows that a PCR-based assay of biosynthetic genes is a reliable, cost-effective, early warning system against Fusarium contamination. Its future use as a high-throughput detection strategy complementing chemical assays enables effective targeted application of crop protection products.
Keywords: fungi, markers, mycotoxins, plant pathology
Introduction
The numerous plant pathogens of the genus Fusarium are responsible for significant losses in crop yield due to both loss of biomass and accumulation of mycotoxins in infiltrated parts. The major toxic compounds synthesized by divergent Fusarium isolates include the following: zearalenone, fumonisins, trichothecenes and their derivatives (D'Mello et al. 1999). While there is a growing body of work documenting biological significance of additional, emergent toxins (e.g. butenolide, fusarins, equisetin, beauvericin and enniatins), their estimated economic and biomedical importance is considerably lower (Desjardins and Proctor 2007).
Notably, the above-mentioned major toxins (fumonisins, trichothecenes, zearalenone and derivative compounds) are frequently not inactivated during food/feed processing and can be present in a masked form (plant-formed conjugates, i.e. glucosides, which can be activated by mammal gut microbiota—e.g. Berthiller et al. 2013; Dall'Erta et al. 2013), increasing health risks to farm animals and humans (Creppy et al. 2002). As more research results are collected, the estimates of health and economical risks (associated with long-term masked mycotoxin exposure) are revised upwards. The updated estimates lead to increasingly restrictive norms for toxin content for food and feed (e.g. European Commission Recommendation 2006/576/EC proposing norms for ochratoxin A, T-2 and HT-2 toxins as well as deoxynivalenol and zearalenone). This only serves to increase a need for efficient and quick methods of assessing possible sources of contamination, preferably by preventing losses in crop yield, via good farming practices including effective fungicide treatments.
The genetic determinants of fumonisin, trichothecene and zearalenone biosynthesis have been characterized in multiple plant pathogenic taxa. Characterization of both core biosynthetic genes (polyketide synthases, trichodiene synthase) and key accessory genes (such as transcription factors or key processing enzymes) enables construction of toxigenicity assays directly targeting the genetic basis of toxin production and accumulation. At the same time (Stepien et al. 2011), the biosynthetic gene alleles exhibit significant interspecific differences, which makes them useful for precise identification of infectious species/populations.
The zearalenone biosynthetic cluster spanning 25 kb of the genomic sequence has been characterized in Fusarium graminearum (Kim et al. 2005), with four principal genes required for toxin biosynthesis (zea1, zea2, zeb1, zeb2) and 3 other genes regulated in conjunction with zeb2 expression patterns (FG02394, FG02399 and FG012015—uncovered by qRT-PCR experiments described by Lysøe et al. (2009)).
Conversely, trichothecene biosynthesis constitutes a multistage process, controlled by at least 12 essential genes, forming a 25-kb-long cluster in F. graminearum (Brown et al. 2001; Kimura et al. 2003). The trichothecene cluster is linked to a key tri5 gene encoding trichodiene synthase, however, four genes segregate at separate loci (notably tri13 and tri14 controlled by a transcription factor encoded by tri10—Tag et al. (2001)). To date, the main cluster has been extensively characterized with numerous studies targeted especially at F. graminearum and F. sporotrichioides species (Kimura et al. 2007), as well as some members of the genus Trichoderma (Cardoza et al. 2011). There is considerable evidence for complex gene relocation scenarios underlying chemotype diversification leading to extant trichothecene type-A- and type-B-producing species (Proctor et al. 2009).
In the past decade, the fumonisin cluster structure (16 gene cluster spanning 42 kb length) has been determined for three toxigenic Fusarium species: F. verticillioides, F. oxysporum (FRC O-1890 strain) and F. proliferatum (Proctor et al. 2008). The interspecies differences between individual biosynthesis-related sequences encompass up to 20% of constituent residues. Notably larger differences are found in gene-flanking regions, an observation which suggests divergent evolutionary paths for cluster copies in different species. Here, the difference in species history and gene phylogeny has been attributed to complex birth/death evolution of the cluster (with independent sorting of copies) and/or horizontal gene transfer events (Proctor et al. 2013). During fumonisin biosynthesis, substitutions of polyketide synthase and/or termination factor can lead to significant changes in the specificity of polyketide condensation for fumonisin analogs (Zhu et al. 2008; Li et al. 2009).
As the broad, genetic basis of the biosynthetic pathway for three major Fusarium mycotoxins is known and multiple exemplar sequences are readily available, it is now possible to develop targeted diagnostic solutions. Through utilizing knowledge about disparate species for the design of degenerate cross-species-specific primers, it is possible to target well-conserved parts of coding sequence (corresponding to conserved parts of protein sequence). Especially for core, secondary metabolite biosynthetic genes, these regions of the coding sequence are unlikely to change in toxin-producing isolates (corroborated by recent evidence for purifying selection in secondary metabolism genes—e.g. Baker et al. 2012).
Current studies on the variability and diversity of the fungal populations make use of various genetic markers, such as the translation elongation factor (tef-1α) and internal transcribed spacer (ITS1/2), employed in assays of the genus Trichoderma (Chaverri et al. 2003; Blaszczyk et al. 2011) and conservative fragments of the genome such as a calmodulin gene (CaM) in Trichoderma and Fusarium populations (Chaverri et al. 2003; Mulè et al. 2004). Also, mitochondrial DNA (mtDNA) is used as a marker of genetic variation. Its relatively short length and the presence of conserved and variable regions allow the identification of closely related species (Ma and Michailides 2007). The sequence of the large subunit of the RNA polymerase II (Hibbett et al. 2007) can also be used to distinguish between divergent phytopathogenic species. Among so many molecular markers, the translation elongation factor (tef-1α) appears to be the most useful in taxonomic studies of fungi, especially in the genus Fusarium (Geiser et al. 2004; Kristensen et al. 2005). Recently, more attention is devoted to markers directly involved in the secondary metabolism (Proctor et al. 2009). Many researchers use genes from the FUM cluster as a good additional marker for phylogenetic and taxonomic studies of the fumonisin-producing Fusarium species (González-Jaén et al. 2004; Baird et al. 2008; Stepien et al. 2011).
The current line of research for the detection of toxigenic species involves simultaneous use of multiple genes belonging to different clusters responsible for toxin production, for example mPCR assays detecting aflatoxigenic, trichothecene- and fumonisin-producing and ochratoxigenic fungal isolates (Rashmi et al. 2013). The recent studies also aim to combine qualitative and quantitative methods for detecting the toxigenic potential. One of the approaches, based on multiplex real-time PCR, is able to detect and quantify mycotoxigenic species in cereal grains with the use of markers targeting the trichothecene synthase (tri5) gene in trichothecene-producing Fusarium sp. isolates, the rRNA gene in Penicillium verrucosum and the polyketide synthase gene (Pks) in Aspergillus ochraceus (Vegi and Wolf-Hall 2013).
The problem addressed in the proposed work was to design and standardize a diagnostic tool allowing the identification of toxigenic Fusarium isolates producing fumonisin B1, trichothecenes and zearalenone. The new protocol is applicable for both in vitro and field samples, with resolution sufficient for direct sequencing of amplified sequences.
Materials and methods
Fungal isolates and field samples
Fungal isolates originated from the culture collections of the Institute of Plant Genetics (Polish Academy of Sciences, Poznan, Poland). The isolates originated from soil, infected cereal grain samples and buildings infested by fungal pathogens. To avoid contamination of fungal cultures with cryptic species, which are hard to distinguish with traditional morphological methods, isolates were purified using single-spore culturing (Leslie and Summerell 2006). Scabby kernels were plated on small nutrient agar (SNA) medium in Petri dishes, and taxa were morphologically identified using an optical microscope (Olympus, Center Valley, PA) at 400–500 × magnification, according to the manual of Leslie and Summerell (2006). Mycelia of isolates cultivated on potato dextrose agar (PDA) were used for DNA isolation. All 96 isolates were identified with at least one molecular marker (ITS 1/2 and/or tef-1α marker), and species assignment was carried out through comparison with reference sequences in NCBI/GenBank and Fusarium-ID (Geiser et al. 2004). Assignment of species to monophyletic complexes was based on the recent taxonomic and phylogenetic research conducted by O'Donnell et al. (2013).
DNA extraction from fungal cultures and field samples
Fungal cultures
Mycelium used for DNA extraction was grown in Czapek-Dox broth (Sigma-Aldrich, St Louis, MO) with yeast extract (Oxoid, Waltham, MA) and streptomycin sulphate (50 mg l−1; AppliChem, Darmstadt, Germany) and after incubation at 25°C for 21 days on a rotary shaker (100 g). Mycelium was collected on filter paper in a Büchner funnel and freeze-dried. Total DNA was extracted using the CTAB method (Doohan et al. 1998). The quality of DNA was estimated by NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, NC) and via Experion Automated Electrophoresis System (Bio-Rad, Hercules, CA).
Field samples
Infected wheat chaffs and kernels (2012, Parabola cultivar) were ground to fine powder, and DNA was obtained using the DNase kit (Qiagen, Hilden, Germany).
Primer design
The degenerate, cross-species-specific primers were designed on the basis of backtranslated codon alignments created from protein sequence alignments of homologous genes from NCBI/RefSeq release ver. 56 (Pruitt et al. 2012) and NCBI/GenBank release ver. 194 (Benson et al. 2013) and Ensembl/Fungi (Flicek et al. 2012) release 18. Protein alignments for fum8, fum6, zea2, tri5 and tri6 genes were obtained with MAFFT-LINSI (Katoh and Toh 2010), subsequently backtranslated and screened for primers with Python scripts. Primer sequences were screened against propensity for homodimer and heterodimer formation on the basis of nearest-neighbour energy/melting temperature calculations with both IDT OligoAnalyzer and in-house Python scripts implementing nearest-neighbour enthalpy/entropy calculations described by SantaLucia (1998) with corrections based on Owczarzy et al. 2008.
PCR amplification
The PCR was carried out in a 25 μl reaction mixture containing the following: 1 μl of DNA (50 ng μl−1), 12·5 μl PCR buffer (50 mmol l−1 KCl, 1·5 mmol l−1 MgCl2, 10 mmol l−1 Tris-HCl, pH 8·8, 0·1% TritonX-100), 1U polymerase (Sigma-Aldrich), l0 mmol l−1 dNTP (Invitrogen, Carlsbad, CA), 0·5 μl 100 mmol l−1 of each primer and 11·5 μl H2O. Amplifications were performed in C1000 Touch™ Thermal Cycler (Bio-Rad) under the following conditions: initial denaturation 5 min at 94°C, 35 cycles of 45 s at 94°C, 45 s at 53–56°C (Table1), 1 min at 72°C and for the final extension 10 min at 72°C. Amplification products were separated on a 1·5% agarose gel (Invitrogen) in 1 × TBE buffer (0·178 mol l−1 Tris-borate, 0·178 mol l−1 boric acid, 0·004 mmol l−1 EDTA) and stained with ethidium bromide. The 10 μl PCR products were combined with 2 μl of loading buffer (0·25% bromophenol blue, 30% glycerol). A 100-bp DNA LadderPlus (Fermentas, St. Leon-Rot, Germany) was used as a size standard. PCR products were electrophoresed at 3 Vcm−1 for about 2 h, visualized under UV light and photographed (Gel DOC EZ Imager; Bio-Rad).
Table 1.
The sequences of the primers used for amplification and sequencing
| Gene targeted | Primer name | Sequences (5′–3′) | Estimated product length (base pairs) |
|---|---|---|---|
| Trichodiene synthase (tri5) | T5_am_fA1 | CTY MRR ACM ATY GTN GGC ATG | 468 |
| T5_am_rA1 | AVA CCA TCC AGT TYT CCA TYT G | ||
| Zinc finger transcription factor (tri6) | TRI6_dm_fA2 | TAT GAA TCA CCA ACW TTC GA | 526 |
| TRI6_dm_rA1 | CGC CTR TAR TGA TCY CKC AT | ||
| Zearalenone polyketide synthase (zea2) | ZEA2_dm_fA1 | ACM TCA CCA TCM AAR TTC TG | 340 |
| ZEA2_dm_rA1 | GCR TCY CKG TAR TCR CTC AT | ||
| Oxygenase (fum6) | FUM6_dm_fA2 | CRA CMG AGA TCA TGG TGA C | 672 |
| FUM6_dm_rA1 | GTY TCR TGT CCK GCA ATG AG | ||
| Oxoamine synthase (fum8) | F8_am_fA1 | GGY TCK TTT GAG TGG TGG C | 350 |
| F8_am_rA1 | CRA CWG GAA ARC AKA YRA YGG |
Sequencing
The 3-μl PCR products were purified with exonuclease I and shrimp alkaline phosphatase according to Chelkowski et al. (2003). Sequencing reactions were prepared using the ABI Prism BigDye Terminator Cycle Sequencing ReadyReaction Kit in 5 μl volume (Applied Biosystems, Grand Island, NY). DNA sequencing was performed on an ABI PRISM3100 GeneticAnalyzer (Applied Biosystems).
Sequences were edited and assembled using Chromasv.1.43 (Applied Biosystems). clustal w (Thompson et al. 1994) and MUSCLE (Edgar 2004) were used to align the sequences; the resulting alignments were inspected and refined manually. All positions containing gaps and missing data were eliminated from the data set.
Multiplex PCR
The multilplex PCR was carried out in a 25 μl reaction mixture containing the following: 1 μl 50 ng μl−1 of DNA, 4 μl PCR buffer (20 mmol l−1 Tris-HCl, 0·1 mmol l−1 EDTA, 1 mmol l−1 DTT, 100 mmol l−1 KCl, stabilizers, 200 μg/ml BSA and 50% glycerol), 1U polymerase (Thermo Scientific), l0 mmol l−1 dNTP (Invitrogen), 0·5 μl 100 mmol l−1 of each primer and 14·5 μl H2O. To each reaction mixture, 3 μl of Q-Solution was added to avoid primer dimerization. Amplifications were performed in Touch™ Thermal Cycler (Bio-Rad) under the following conditions: initial denaturation 30 s at 98°C, 35 cycles of 5 s at 98°C, 5 s at 55°C, 15 s at 72°C, with the final extension of 1 min at 72°C. Amplification products were separated on 2% agarose gel (Invitrogen) in 1 × TBE buffer (0·178 mol l−1 Tris-borate, 0·178 mol l−1 boric acid, 0·004 mol l−1 EDTA) and stained with Midori Green (Nippon Genetics, Dueren, Germany). A 100-bp DNA LadderPlus (Fermentas) was used as a size standard. PCR products were electrophoresed at 3 Vcm−1 for about 2 h, visualized under ultraviolet (UV) light and photographed (Gel DOC EZ Imager; Bio-Rad).
Determination of trichothecenes concentration
Determination of trichothecenes was performed in solid PDA culture. Briefly, subsamples (1 g of mycelium with medium) were extracted with acetonitrile/water (82:18) and cleaned-up on a Myco Sep 227 Trich + column. The group B trichothecenes (DON, NIV, 3AcDON, 15AcDON, FUS-X) were analysed as trimethylsilylsilyl ethers derivatives. After sililation, samples were extracted with isooctane and 1 μl of sample was injected on a GC/MS system. The analyses were run on a gas chromatograph (Hewlett Packard GC 6890, Waldbronn, Germany) hyphenated to a mass spectrometer (Hewlett Packard 5972 A, Waldbronn, Germany), using an HP-5MS, 0·25 mm × 30 m capillary column. The injection port temperature was 280°C, the transfer line temperature was 280°C, and the analyses were performed with programmed temperature. Initial temperature was 80°C held for 1 min, from 80 to 200°C at 15°C min−1 held 6 min and from 200 to 280°C at 10°C min−1, the final temperature being maintained for 3 min. The helium flow rate was held constant at 0·7 ml min−1. Quantitative analysis was performed in single ion monitored mode, and qualitative analysis was performed in SCAN mode (100–700 amu). Recoveries for analysed toxins were as follows: DON 84 ± 3·8%; 3AcDON 78 ± 4·8%; 15AcDON 74 ± 2·2%; FUS X 87%±5·9%; NIV 81 ± 3·8%. Limit of detection was 0·01 mg kg−1.
Determination of zearalenone concentration
Determination of zearalenone was performed in solid PDA medium. Subsamples (1 g of mycelium with medium) were extracted with acetonitrile/water (82:18) and cleaned-up on Zearala test affinity columns. Prepared samples were analysed by HPLC consisting of a Waters HPLC 2695 apparatus with a Waters 2475 Multi λ Fluorescence Detector and a Waters 2996 Array Detector (Waters, Milford, MA). Separation was achieved on a 150 mm length × 3·9 mm diameter Nova Pak C-18, 4-μm particle size column and eluted with acetonitrile–water–methanol (46:46:8, v/v/v) at a flow rate of 0·5 ml min−1. ZEA was detected with a Waters 2475 Multi λ Fluorescence Detector, and the excitation and emission wavelengths were 274 and 440 nm, respectively. Estimation of ZEA was performed by a comparison of peak areas with those of an external standard (>95%; Sigma-Aldrich) or by co-injection with the standard. The detection limit of ZEA was 3 ng g−1. The similar process was used to determine zearalenone concentration in a wheat bioassay (Gromadzka et al. 2009).
Determination of fumonisin B1 concentration
The samples (5 ml of liquid culture) were filtered through Whatman No. 5 (Whatman, Piscataway, NJ) filter paper and dried under nitrogen stream. The residues were dissolved into methanol water (3:1, v/v), adjusted to the pH value of 5·8–6·5 by 0·1 mol l−1 KOH water solution and cleaned using a SAX cartridge. The cartridge was conditioned with 5 ml of methanol followed by 5 ml of methanol–water (3:1, v/v). FB1 was eluted from the column to a glass collection vial with 10 ml of 1% acetic acid in methanol. The eluate was evaporated to dryness at 40°C under a stream of nitrogen. The cleaned sample was derivatized with OPA reagent (20 mg 0·5 ml−1 methanol diluted with 2·5 ml 0·1 mol l−1 disodium tetraborate (Na2B4O7 × 10H2O), then combined with 25 μl 2-mercaptoethanol) immediately before HPLC analysis by mixing the OPA reagent and the sample in ratio 4:1 v/v. After 2 min, the reaction mixture (10 μl) was injected in a HPLC C18 Nova Pak column (3·9 × 150 mm). Methanol–sodium dihydrogen phosphate (0·1 mol l−1 in water) solution (77:23; v/v) was adjusted to pH 3·35 with o-phosphoric acid and used as the mobile phase with the flow rate of 0·6 ml min−1. A Waters 2695 HPLC with a fluorescence detector (λEx = 335 nm and λEm = 440 nm, Waters 2475; Waters) was used for analysis.
Benchmarking
Diagnostic quality parameters (sensitivity, specificity and comparison with a naive predictor) were assessed with the open-source software r (ver. 2.15.2) using the caret package (Kuhn 2008). Results of chemotype identification and marker testing were visualized using in-house R scripts dependent on ggplot2 (Wickham 2009).
Results
Sensitivity and specificity of diagnostic markers
Sensitivity and specificity of designed markers (Table1) were tested on a collection of 96 fungal isolates. The tests took into account divergent Fusarium species (72 isolates) as well as multiple non-Fusarium filamentous fungi (24 isolates) (Fig.2). The individual performance of markers was as follows: trichothecene biosynthesis (tri5 + tri6, sensitivity 100%, specificity 95%, P-value vs naive classifier: 1·42e-09), zearalenone (zea2, sensitivity 100%, specificity 100%, P-value vs naive classifier: 7·48e-09) and fumonisin (fum6 + fum8, sensitivity 89%, specificity 89%, P-value vs naive classifier: 3·53e-07). The final results show that the protocol can reliably identify the toxigenic potential for all three toxin groups (trichothecenes, zearalenone and fumonisins) with a sensitivity and specificity of over 90%, excepting fumonisin production within the F. oxysporum complex (see also Discussion).
Figure 2.

Results of chemical analyses and molecular diagnostics tested for 96 isolates. The results are grouped by monophyletic complexes within the genus Fusarium reported by O'Donnell et al. (2013). For trichothecene type-A producers (Fusarium sporotrichioides), zearalenone and trichothecene-associated chemotype was qualitatively assayed on rice kernels. Non-specific marker amplification denotes a non-specific band at different height. Marker: ( ) n/a; (
) n/a; ( ) present; (
) present; ( ) non-specific. Toxin: (
) non-specific. Toxin: ( ) n/a; (
) n/a; ( ) present.
) present.
Confirmation of results via chemical analysis and sequencing
Taxonomic identification of all isolates was confirmed by sequencing and analysis of rDNA internal transcribed spacer (ITS; 95 isolates) and/or translation elongation factor 1 α (tef-1α; 41 isolates) partial sequences (Table2). The identified Fusarium chemotypes are consistent with the recent knowledge on species-related compounds (Moretti et al. 2013), and in case of all positive markers, those results were confirmed by direct sequencing of the PCR product (Fig.1; Table S4).
Table 2.
Fungal isolates from the collection of the Institute of Plant Genetics PAS (Functional Evolution of Biological Systems Team) used to develop a multiplex PCR. The naming of monophyletic complexes within Fusarium sp. derived from (O'Donnell et al. 2013)
| Complex | Species | Collection number | Source | Year of isolation | Chemotype | Molecular identification | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fumonisin B1 | Trichothecene A | Trichothecene B | Zearalenone | ITS1/2 | tef-1α | |||||
| F. fujikuroi | F. proliferatum | 1 | Italy | 1984 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 3 | Canada | 1982 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 7 | Poland | 1986 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 18 | Norway | 2006 | + | − | − | − | + | − |
| F. fujikuroi | F. proliferatum | 21 | Italy | 1986 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 44 | Poland | 1993 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 58 | USA | 1993 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 59 | Poland | 1999 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 66 | Poland | 1999 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 82 | Poland | 2006 | + | − | − | − | + | − |
| F. fujikuroi | F. proliferatum | 84 | Poland | 2006 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 85 | Norway | 2006 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 99 | Italy | 1993 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 111 | Poland | 2008 | + | − | − | − | + | − |
| F. fujikuroi | F. proliferatum | 113 | Poland | 2008 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 141 | Poland | 2010 | + | − | − | − | + | + |
| F. fujikuroi | F. proliferatum | 142 | Poland | 2011 | + | − | − | − | + | + |
| F. fujikuroi | F. subglutinans | 60 | Poland | 1984 | + | − | − | − | + | − |
| F. fujikuroi | F. succisae | 4 | Poland | 1996 | − | − | − | − | + | + |
| F. fujikuroi | F. temperatum | 151 | Poland | 1987 | + | − | − | − | + | − |
| F. fujikuroi | F. proliferatum | 13 | Poland | 1988 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 16 | Poland | 1987 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 17 | Poland | 1987 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 23 | Poland | 1982 | + | − | − | − | + | + |
| F. fujikuroi | F. verticillioides | 29 | Poland | 1985 | + | − | − | − | + | + |
| F. fujikuroi | F. verticillioides | 43 | Poland | 1986 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 45 | Poland | 1982 | + | − | − | − | + | + |
| F. fujikuroi | F. verticillioides | 71 | Poland | 1986 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 75 | Poland | 2010 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 79 | Poland | 2010 | + | − | − | − | + | − |
| F. fujikuroi | F. verticillioides | 88 | Poland | 1982 | + | − | − | − | + | − |
| F. fujikuroi | F. xyllarioides | 67 | Guinea | 1985 | − | − | − | − | + | + |
| F. incarnatum-equiseti | F. equiseti | 72 | Poland | 2010 | − | + | − | + | + | − |
| F. oxysporum | F. oxysporum | 19 | Poland | 2010 | + | − | − | − | + | + |
| F. oxysporum | F. oxysporum | 55 | Poland | 1997 | + | − | − | − | + | − |
| F. oxysporum | F. oxysporum | 57 | Poland | 2010 | + | − | − | − | + | + |
| F. oxysporum | F. oxysporum | 62 | Poland | 2010 | + | − | − | − | + | − |
| F. oxysporum | F. oxysporum | 65 | Poland | 1984 | + | − | − | − | + | − |
| F. oxysporum | F. oxysporum | 69 | Poland | 2010 | + | − | − | − | + | − |
| F. oxysporum | F. oxysporum | 115 | Poland | 2010 | + | − | − | − | + | − |
| F. oxysporum | F. oxysporum | 131 | Poland | 2010 | + | − | − | − | + | − |
| F. sambucinum | F. cerealis | 33 | Poland | 1998 | − | − | + | + | + | − |
| F. sambucinum | F. cerealis | 41 | Poland | 1986 | − | − | + | + | + | − |
| F. sambucinum | F. cerealis | 87 | Poland | 1987 | − | − | + | + | + | − |
| F. sambucinum | F. culmorum | 48 | Poland | 1984 | − | − | + | + | + | − |
| F. sambucinum | F. culmorum | 49 | Poland | 1997 | − | − | + | + | + | − |
| F. sambucinum | F. culmorum | 70 | Poland | 2010 | − | − | + | + | + | − |
| F. sambucinum | F. culmorum | 90 | Poland | 1982 | − | − | + | + | + | − |
| F. sambucinum | F. culmorum | 93 | Poland | 1986 | − | − | + | + | + | − |
| F. sambucinum | F. graminearum | 52 | Poland | 1986 | − | − | + | + | + | − |
| F. sambucinum | F. graminearum | 76 | Poland | 1986 | − | − | + | + | + | − |
| F. sambucinum | F. graminearum | 144 | Poland | 2011 | − | − | + | + | + | − |
| F. sambucinum | F. graminearum | 149 | Poland | 1986 | − | − | + | + | + | − |
| F. sambucinum | F. poae | 12 | Poland | 1987 | − | − | + | − | + | − |
| F. sambucinum | F. sporotrichioides | 8 | Poland | 1999 | − | + | − | + | + | − |
| F. sambucinum | F. sporotrichioides | 32 | Poland | 2010 | − | + | − | + | + | − |
| F. sambucinum | F. sporotrichioides | 39 | Poland | 2010 | − | + | − | + | + | − |
| F. sambucinum | F. sporotrichioides | 54 | Poland | 1993 | − | + | − | + | + | − |
| F. sambucinum | F. sporotrichioides | 106 | Poland | 2010 | − | + | − | + | + | − |
| F. sambucinum | F. sporotrichioides | 116 | Poland | 2010 | − | + | − | + | + | − |
| F. sambucinum | F. sporotrichioides | 119 | Poland | 2010 | − | + | − | + | + | − |
| F. tricinctum | F. avenaceum | 68 | Poland | 2010 | − | − | − | − | + | + |
| F. tricinctum | F. avenaceum | 105 | Poland | 2010 | − | − | − | − | + | − |
| F. tricinctum | F. avenaceum | 108 | Poland | 2010 | − | − | − | − | + | + |
| F. tricinctum | F. avenaceum | 114 | Poland | 2010 | − | − | − | − | + | + |
| F. tricinctum | F. avenaceum | 117 | Poland | 2010 | − | − | − | − | + | − |
| F. tricinctum | F. avenaceum | 120 | Poland | 2011 | − | − | − | − | + | + |
| F. tricinctum | F. avenaceum | 132 | Poland | 2011 | − | − | − | − | + | − |
| F. tricinctum | F. tricinctum | 14 | Poland | 2011 | − | − | − | − | + | − |
| F. tricinctum | F. tricinctum | 31 | Poland | 1986 | − | − | − | − | + | − |
| F. tricinctum | F. tricinctum | 109 | Poland | 2010 | − | − | − | − | + | − |
| F. solani | F. solani | 6 | Poland | 1997 | − | − | − | − | + | − |
| NA | Alternaria alternata | 129 | Poland | 2010 | − | − | − | − | + | + |
| NA | A. alternata | 139 | Poland | 2010 | − | − | − | − | + | + |
| NA | Alternaria brassicicola | 128 | Poland | 2010 | − | − | − | − | + | + |
| NA | Alternaria sp. | 97 | Poland | 2010 | − | − | − | − | + | + |
| NA | Aspergillus niger | 148 | Poland | 2010 | + | − | − | − | + | − |
| NA | Clonostachys rosea | 20 | Poland | 2010 | − | − | − | − | + | − |
| NA | Clonostachys sp. | 104 | Poland | 2010 | − | − | − | − | + | − |
| NA | Penicillium commune | 136 | Poland | 2010 | − | − | − | − | + | − |
| NA | P. commune | 138 | Poland | 2010 | − | − | − | − | + | − |
| NA | P. herbarum | 137 | Poland | 2010 | − | − | − | − | + | − |
| NA | Trichoderma aggressivum | 100 | Poland | 2009 | − | − | − | − | + | + |
| NA | Trichoderma atroviride | 98 | Poland | 2009 | − | − | − | − | + | + |
| NA | T. atroviride | 158 | Poland | 2010 | − | − | − | − | − | + |
| NA | Trichoderma hamatum | 95 | Poland | 2010 | − | − | − | − | + | − |
| NA | T. hamatum | 133 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. harzianum | 5 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. harzianum | 25 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. harzianum | 123 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. harzianum | 125 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. harzianum | 153 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. harzianum | 154 | Poland | 2010 | − | − | − | − | + | + |
| NA | Trichoderma longibrachiatum | 124 | Poland | 2010 | − | − | − | − | + | + |
| NA | Trichoderma viridescens | 24 | Poland | 2010 | − | − | − | − | + | + |
| NA | T. viridescens | 96 | Poland | 2009 | − | − | − | − | + | + |
Some of F. oxysporum isolates have the capacity to produce small amounts of fumonisin B1.
Figure 1.
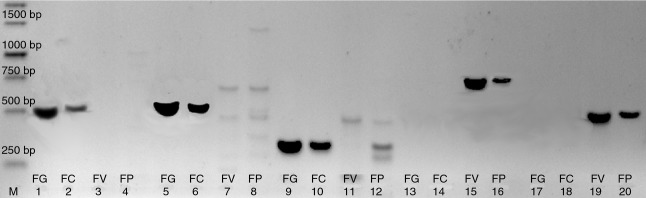
Markers used for diagnostics of the toxigenic potential. (Lane M—DNA marker, line 1–4 tri5 marker; line 5–8 tri6 marker; line 9–12 zea2 marker; line 13–16 fum6 marker; line 17–20 fum8 marker; FC—Fusarium culmorum, FG—F. graminearum, FV—F. verticilioides, FP—F. proliferatum).
Chemical analyses have shown that the analysed isolates produce highly varying amounts of toxin. Additionally, in case of F. sporotrichioides, no chemotype (zearalenone/trichothecene biosynthesis) was detected on potato dextrose agar (PDA medium); however, we were able to qualitatively observe accumulation of toxins in a wheat bioassay (Tables S1–S3). This is consistent with reported influence of different carbon sources on toxin production (Jiao et al. 2008) and suggests differentially regulated expression in F. sporotrichioides compared to F. graminearum.
Although no quantitative assay of the type-A trichothecene accumulation was conducted, the genes present in F. sporotrichioides isolates are highly similar to the model F. sporotrichioides counterparts. Notably, polymorphisms in both tri5 and tri6 partial sequences, obtained from direct sequencing, can unambiguously differentiate between trichothecene type-A and type-B producers (F. graminearum, F. culmorum, F. cerealis, F. poae) within the F. sambucinum complex (Fig.2).
False-positive results observed in Fusarium xyllarioides and F. succisae were identified as coding sequences corresponding to unrelated genes. In practice, the resulting product band was also visibly different (very weak and of different height) and easily told apart from specific product. In case of zea2, the product of amplification in a single Phoma herbarum isolate was identified as a related gene (reducing polyketide synthase, highly similar to hypothemycin-reducing polyketide synthase from Hypomyces subiculosus; Reeves et al. (2008)). Again, the band was of visibly different height; however, the strength and quality of amplification suggests that zea2 marker could be adapted towards the recognition of different reducing polyketide synthases involved in resorcyclic acid biosynthesis.
Multiplex PCR
Following the assessment of the individual marker performance, the assay was tested and optimized towards multiplexing the PCR. Multiplexing attempts have shown best results for assays over two separate sets of multiplexed markers: zea2 + tri5 + tri6 for the trichothecene/zearalenone and fum6 + fum8 for the fumonisine toxigenic potential (Fig.3). In case of both detection sets, the amplification of at least one product was taken to confirm the toxigenic potential of pathogens infecting the tested sample.
Figure 3.
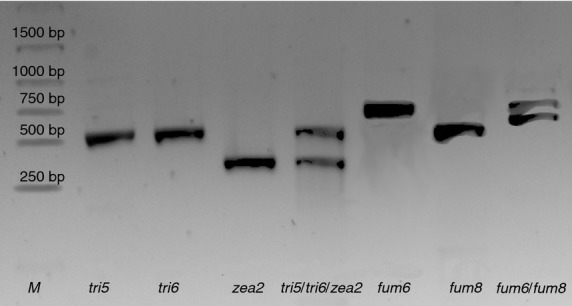
Multiplexed PCRs detecting the toxigenic potential of isolates (Lane M––DNA marker, Fusarium graminearum tri5, tri6, zea2, Fusarium verticilioides fum6, fum8).
Field samples
In addition to the standard PCRs conducted on DNA obtained from cultivated isolates, the toxigenic potential was also examined on genetic material obtained directly from infected tissue samples. This resulted in the positive identification of infected wheat kernel samples (Fig.4); however, this was not successful in samples of the wheat chaff. Multiplex PCRs conducted on diluted samples (500, 50, 5 ng, 500 pg) gave distinct, specific signatures even at the lowest DNA concentration level of 500 pg.
Figure 4.
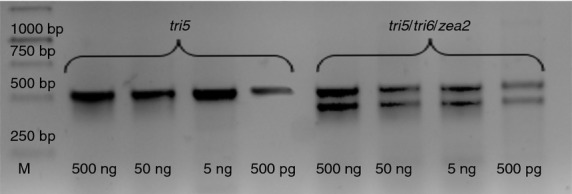
PCRs detecting the toxigenic potential of diluted environmental samples (tri5––trichothecene marker; tri5/tri6/zea2––trichothecene and zearalenone markers). DNA concentration 500, 50, 5 ng, 500 pg (Lane M––DNA marker).
Discussion
In this study, we present a novel approach to detect the toxigenic potential of various phytopathogenic fungi by partially multiplexed, degenerate primers based on the genes essential for biosynthesis of major Fusarium sp. mycotoxins (fumonisins, trichothecenes and zearalenone). Such tools are especially valuable when updated risk assessments concerning fungal toxin contamination lead to more restrictive norms regulating their acceptable levels in food and/or feed. These trends result in an increase in demand for efficient and rapid methods for the detection and assessment of potential sources of contamination which can be used also as a part of decision support systems (DSS). At the moment, DSS are primarily focused on the observation of the occurrence of pathogens on host plants (Evans et al. 2008), spores in the air (Kaczmarek et al. 2009) or the impact of weather conditions on the life cycles of pathogens (Dawidziuk et al. 2012).
The isolates of the F. oxysporum complex constitute a remarkable outlier in the results obtained for fumonisin-producing species. In this case, the trace amounts of fumonisin were found in cultures of several isolates, but mPCR markers were consistently absent. Previous works by Proctor et al. (2008, 2013) demonstrate possible divergent origins of the fumonisin clusters in distinct member species of F. oxysporum and F. fujikuroi complexes. Past research also shows that synthesis of the long reduced polyketide mycotoxins is controlled by accessory genes (i.e. fum8) under a scheme which permits complementation by different core/accessory genes (Zhu et al. 2008; —fum8 complementation for control of biosynthesis). As F. oxysporum is a species with high supernumerary chromosome content (c. 25%; Ma et al. 2010) likely stemming from past horizontal transfers, there is a possibility of different/highly divergent genetic basis complementing biosynthesis of low amounts of fumonisins and/or fumonisin-like compounds in the F. oxysporum complex. Notably, the molecular and morphological identification of isolates can be a grey area in some cases (e.g. newly characterized cryptic species like F. temperatum—Scauflaire et al. 2011; low resolution of broad barcode markers in complexes of related species—Blaszczyk et al. 2011). Current and future research is poised to demonstrate finer splits in the complexes of closely related species, previously characterized as monophyletic species (O'Donnell et al. 2013). The taxonomic identification is supplemented and supported by differences in chemotype and sequence of biosynthesis-related genes from closely related taxa—a process made easier by markers designed for direct sequencing of amplification products. Nevertheless, the problematic results do not apply to the most important economic, toxigenic Fusarium species occurring in cultivated high-yield crops (e.g. maize—F. verticillioides, wheat—F. graminearum, F. culmorum).
In related research, previously carried out by Rashmi et al. (2013), the researchers focused on diverse isolates (mainly toxigenic and non-toxigenic Fusarium, Aspergillus and Penicillium), demonstrating the applicability of multiplex PCR to detect ochratoxin-, fumonisin- and trichothecene-synthesizing isolates. However, Rashmi and co-workers did not attempt to provide a more detailed taxonomic identification of their cultures. In our approach, each pathogenic isolate was obtained by single-spore technique and its species assigned by both morphological and molecular methods. The test can efficiently detect the presence of the marker gene in five hundred picograms of template and about one infected kernel among hundred uninfected seeds and each obtained product can be validated by direct sequencing. Sensitivity on this level can significantly support the farmers for instance in the appropriate and rational use of fungicide treatments in the field. The developed diagnostic approach can directly be used in biological material obtained from the field (infected kernels) without the need for prior cultivation on artificial media. Unfortunately, such analysis is only possible in infected kernels. The DNA isolated from chaffs is not of sufficient quality to give reliable results, likely due to the presence of PCR inhibitors, such as polysaccharides (e.g. dextran sulphate, alginic acid—Demeke and Jenkins 2010). This could be alleviated by improvement in preparation procedures. There is a possibility of further extending the approach to direct quantitative studies of the mycotoxin-producing pathogens which (up to date) are typically focused on detection of specific fungal producers (F. graminearum, P. verrucosum, A. ochraceus) and not on assessing the toxigenic potential grounded in common genetic basis among related but distinct species (Vegi and Wolf-Hall 2013).
The multiplexed PCR assay used in the protocol allows for the detection of toxigenic potential in many species simultaneously and in a standardized way. The resulting quality of optimized PCRs allows for direct sequencing of amplification products. Additionally, the low cost (relative to HPLC analysis) of the assay allows easy coupling with simple, targeted techniques (e.g. ELISA) to quickly confirm presence of a specific toxin. Thus, the method can be easily adapted as early warning against mycotoxin contamination allowing much more effective application of fungicides and can serve as supplement conventional mycotoxin detection techniques. What is also very important is that, through the usage of the direct sequencing of the PCR products, the results from individually cultivated isolates should allow easy characterization of variability and phylogeny of infecting pathogen populations.
Acknowledgments
Research funded by LIDER/19/113/L-1/09/NCBiR/2010 ‘Modelling, prediction and verification of fungal toxin accumulation’ applied research grant.
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Fumonisin concentration and molecular detection of the toxigenicity in tested fungal isolates (fumonisin B chemotype).
Zearalenone concentration and molecular detection of the toxigenicity in tested fungal isolates (zearalenone chemotype).
Trichothecene concentration and molecular detection of the toxigenicity in tested fungal isolates (trichothecene B chemotype).
GenBank accessions of obtained PCR products.
References
- Baird R, Abbas HK, Windham G, Williams P, Baird S, Ma P, Kelley R, Hawkins L, et al. Identification of select fumonisin forming Fusarium species using PCR applications of the polyketide synthase gene and its relationship to fumonisin production in vitro. Int J Mol Sci. 2008;9:554–570. doi: 10.3390/ijms9040554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SE, Perrone G, Richardson NM, Gallo A, Kubicek CP. Phylogenomic analysis of polyketide synthase-encoding genes in Trichoderma. Microbiology. 2012;158:147–154. doi: 10.1099/mic.0.053462-0. [DOI] [PubMed] [Google Scholar]
- Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthiller F, Crews C, Dall'Asta C, Saeger SD, Haesaert G, Karlovsky P, Oswald IP, Seefelder W, et al. Masked mycotoxins: a review. Mol Nutr Food Res. 2013;57:165–186. doi: 10.1002/mnfr.201100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk L, Popiel D, Chelkowski J, Koczyk G, Samuels GJ, Sobieralski K, Siwulski M. Species diversity of Trichoderma in Poland. J Appl Genet. 2011;52:233–243. doi: 10.1007/s13353-011-0039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, McCormick SP, Alexander NJ, Proctor RH, Desjardins AE. A genetic and biochemical approach to study trichothecene diversity in Fusarium sporotrichioides and Fusarium graminearum. Fungal Genet Biol. 2001;32:121–133. doi: 10.1006/fgbi.2001.1256. [DOI] [PubMed] [Google Scholar]
- Cardoza RE, Malmierca MG, Hermosa MR, Alexander NJ, McCormick SP, Proctor RH, Tijerino AM, Rumbero A, et al. Identification of loci and functional characterization of trichothecene biosynthesis genes in filamentous fungi of the genus Trichoderma. Appl Environ Microbiol. 2011;77:4867–4877. doi: 10.1128/AEM.00595-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaverri P, Castlebury LA, Samuels GJ, Gaiser DM. Multilocus phylogenetic structure within the Trichoderma harzianumHypocrea lixii complex. Mol Phylogenet Evol. 2003;27:302–313. doi: 10.1016/s1055-7903(02)00400-1. [DOI] [PubMed] [Google Scholar]
- Chelkowski J, Golka L, Stepien L. Application of STS markers for leaf rust resistance genes in near-isogenic lines of spring wheat cv. Thatcher. J Appl Genet. 2003;44:323–338. [PubMed] [Google Scholar]
- Creppy EE, Traoré A, Baudrimont I, Cascante M, Carratú MR. Recent advances in the study of epigenetic effects induced by the phycotoxin okadaic acid. Toxicology. 2002;181–182:433–439. doi: 10.1016/s0300-483x(02)00489-4. [DOI] [PubMed] [Google Scholar]
- Dall'Erta A, Cirlini M, Dall'Asta M, Del Rio D, Galaverna G, Dall'Asta C. Masked mycotoxins are efficiently hydrolyzed by human colonic microbiota releasing their aglycones. Chem Res Toxicol. 2013;26:305–312. doi: 10.1021/tx300438c. [DOI] [PubMed] [Google Scholar]
- Dawidziuk A, Kaczmarek J, Jedryczka M. The effect of winter weather conditions on the ability of pseudothecia of Leptosphaeria maculans and L. biglobosa to release ascospores. Eur J Plant Pathol. 2012;134:329–343. [Google Scholar]
- Demeke T, Jenkins GR. Influence of DNA extraction methods, PCR inhibitors and quantification methods on real-time PCR assay of biotechnology-derived traits. Anal Bioanal Chem. 2010;396:1977–1990. doi: 10.1007/s00216-009-3150-9. [DOI] [PubMed] [Google Scholar]
- Desjardins AE, Proctor RH. Molecular biology of Fusarium mycotoxins. Int J Food Microbiol. 2007;119:47–50. doi: 10.1016/j.ijfoodmicro.2007.07.024. [DOI] [PubMed] [Google Scholar]
- D'Mello D, Mehta D, Pereira J, Rao CV. A toxicity study of simultaneous administration of Tamoxifen and Diazepam to female Wistar rats. Exp Toxicol Pathol. 1999;51:549–553. doi: 10.1016/S0940-2993(99)80139-0. [DOI] [PubMed] [Google Scholar]
- Doohan FM, Parry DW, Jenkinson P, Nicholson P. The use of species-specific PCR-based assays to analyze Fusarium ear blight of wheat. Plant Pathol. 1998;47:197–205. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans N, Baier A, Semenov MA, Gladders P, Fitt BDL. Range and severity of a plant disease increased by global warming. J R Soc Interface. 2008;5:525–531. doi: 10.1098/rsif.2007.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, et al. Ensembl. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser DM, del Mar Jiménez-Gasco M, Kang S, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, et al. FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. Eur J Plant Pathol. 2004;110:473–479. [Google Scholar]
- González-Jaén MT, Mirete S, Patiño B, López-Errasquín E, Vázquez C. Genetic markers for the analysis of variability and for production of specific diagnostic sequences in fumonisin-producing strains of Fusarium verticillioides. Eur J Plant Pathol. 2004;110:525–532. [Google Scholar]
- Gromadzka K, Chelkowski J, Popiel D, Kachlicki P, Kostecki M, Golinski P. Solid substrate bioassay to evaluate the effect of Trichoderma and Clonostachys on the production of zearalenone by Fusarium species. World Mycotoxin J. 2009;2:45–52. [Google Scholar]
- Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, et al. A higher-level phylogenetic classification of the Fungi. Mycol Res. 2007;111:509–547. doi: 10.1016/j.mycres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Jiao F, Kawakami A, Nakajima T. Effects of different carbon sources on trichothecene production and Tri gene expression by Fusarium graminearum in liquid culture. FEMS Microbiol Lett. 2008;285:212–219. doi: 10.1111/j.1574-6968.2008.01235.x. [DOI] [PubMed] [Google Scholar]
- Kaczmarek J, Jedryczka M, Fitt BDL, Lucas JA, Latunde-Dada AO. Analyses of air samples for ascospores of Leptosphaeria maculans and L. biglobosa with light microscopic and molecular techniques. J Appl Genet. 2009;50:411–419. doi: 10.1007/BF03195702. [DOI] [PubMed] [Google Scholar]
- Katoh K, Toh H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 2010;26:1899–1900. doi: 10.1093/bioinformatics/btq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Hutmacher RB, Davis RM. Characterization of California isolates of Fusarium oxysporum f. sp. vasinfectum. Plant Dis. 2005;4:366–372. doi: 10.1094/PD-89-0366. [DOI] [PubMed] [Google Scholar]
- Kimura M, Tokai T, O'Donnell K, Ward TJ, Fujimura M, Hamamoto H, Shibata T, Yamaguchi I. The trichothecene biosynthesis gene cluster of Fusarium graminearum F15 contains a limited number of essential pathway genes and expressed non-essential genes. FEBS Lett. 2003;539:105–110. doi: 10.1016/s0014-5793(03)00208-4. [DOI] [PubMed] [Google Scholar]
- Kimura M, Tokai T, Takahashi-Ando N, Ohsato S, Fujimura M. Molecular and genetic studies of Fusarium trichothecene biosynthesis: pathways, genes, and evolution. Biosci Biotechnol Biochem. 2007;71:2105–2123. doi: 10.1271/bbb.70183. [DOI] [PubMed] [Google Scholar]
- Kristensen R, Torp M, Kosiak B, Holst-Jensen A. Phylogeny and toxigenic potential is correlated in Fusarium species as revealed by partial translation elongation factor 1 alpha gene sequences. Mycol Res. 2005;109:173–186. doi: 10.1017/s0953756204002114. [DOI] [PubMed] [Google Scholar]
- Kuhn M. Building predictive models in R using the caret package. J Stat Softw. 2008;28:1–26. [Google Scholar]
- Leslie JF, Summerell BA. The Fusarium Laboratory Manual. Ames, IA: Blackwell Publishing; 2006. [Google Scholar]
- Li Y, Shen Y, Zhu X, Du L. Introduction of the AAL-toxin polyketide synthase gene ALT1 into FUM1-disrupted Fusarium verticillioides produces metabolites with the fumonisin methylation pattern. J Nat Prod. 2009;72:1328–1330. doi: 10.1021/np900193j. [DOI] [PubMed] [Google Scholar]
- Lysøe E, Bone KR, Klemsdal SS. Real-time quantitative expression studies of the zearalenone biosynthetic gene cluster in Fusarium graminearum. Phytopathology. 2009;99:176–184. doi: 10.1094/PHYTO-99-2-0176. [DOI] [PubMed] [Google Scholar]
- Ma Z, Michailides TJ. Approaches for eliminating PCR inhibitors and designing PCR primers for the detection of phytopathogenic fungi. Crop Prot. 2007;26:145–161. [Google Scholar]
- Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Susca A, Mulé G, Logrieco AF, Proctor RH. Molecular biodiversity of mycotoxigenic fungi that threaten food safety. Int J Food Microbiol. 2013;167:57–66. doi: 10.1016/j.ijfoodmicro.2013.06.033. [DOI] [PubMed] [Google Scholar]
- Mulè G, Susca A, Stea G, Moretti A. A species-specific PCR assay based on the calmodulin partial gene for identification of Fusarium verticillioides F. proliferatum and F. subglutinans. Eur J Plant Pathol. 2004;110:495–502. [Google Scholar]
- O'Donnell K, Rooney AP, Proctor RH, Brown DW, McCormick SP, Ward TJ, Frandsen RJN, Lysøe E, et al. Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet Biol. 2013;52:20–31. doi: 10.1016/j.fgb.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Owczarzy R, Moreira BG, You Y, Behlke MA, Walder JA. Predicting stability of DNA duplexes in solutions containing magnesium and monovalent cations. Biochemistry. 2008;47:5336–5353. doi: 10.1021/bi702363u. [DOI] [PubMed] [Google Scholar]
- Proctor RH, Busman M, Seo JA, Lee YW, Plattner RDA. Fumonisin biosynthetic gene cluster in Fusarium oxysporum strain O-1890 and the genetic basis for B versus C fumonisin production. Fungal Genet Biol. 2008;45:1016–1022. doi: 10.1016/j.fgb.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Proctor RH, McCormick SP, Alexander NJ, Desjardins AE. Evidence that a secondary metabolic biosynthetic gene cluster has grown by gene relocation during evolution of the filamentous fungus Fusarium. Mol Microbiol. 2009;74:1128–1142. doi: 10.1111/j.1365-2958.2009.06927.x. [DOI] [PubMed] [Google Scholar]
- Proctor RH, Van Hove F, Susca A, Stea G, Busman M, van der Lee T, Waalwijk C, Moretti A, et al. Birth, death and horizontal transfer of the fumonisin biosynthetic gene cluster during the evolutionary diversification of Fusarium. Mol Microbiol. 2013;90:290–306. doi: 10.1111/mmi.12362. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashmi R, Ramana MV, Shylaja R, Uppalapati SR, Murali HS, Batra HV. Evaluation of a multiplex PCR assay for concurrent detection of four major mycotoxigenic fungi from foods. J Appl Microbiol. 2013;114:819–827. doi: 10.1111/jam.12100. [DOI] [PubMed] [Google Scholar]
- Reeves CD, Hu Z, Reid R, Kealey JT. Genes for the biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl Environ Microbiol. 2008;74:5121–5129. doi: 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scauflaire J, Mahieu O, Louvieaux J, Foucart G, Renard F, Munaut F. Biodiversity of Fusarium species in ears and stalks of maize plants in Belgium. Eur J Plant Pathol. 2011;131:59–66. [Google Scholar]
- Stepien L, Koczyk G, Waskiewicz A. FUM cluster divergence in fumonisins-producing Fusarium species. Fungal Biol. 2011;115:112–123. doi: 10.1016/j.funbio.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Tag AG, Garifullina GF, Peplow AW, Ake C, Jr, Phillips TD, Hohn TM, Beremand MN. A novel regulatory gene, Tri10, controls trichothecene toxin production and gene expression. Appl Environ Microbiol. 2001;67:5294–5302. doi: 10.1128/AEM.67.11.5294-5302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegi A, Wolf-Hall CE. Multiplex real-time PCR method for detection and quantification of mycotoxigenic fungi belonging to three different genera. J Food Sci. 2013;78:M70–M76. doi: 10.1111/j.1750-3841.2012.03008.x. [DOI] [PubMed] [Google Scholar]
- Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer Publishing Company, Incorporated; 2009. [Google Scholar]
- Zhu X, Vogeler C, Du L. Functional complementation of fumonisin biosynthesis in FUM1-disrupted Fusarium verticillioides by the AAL-toxin polyketide synthase gene ALT1 from Alternaria alternata f. sp. lycopersici. J Nat Prod. 2008;71:957–960. doi: 10.1021/np8000514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fumonisin concentration and molecular detection of the toxigenicity in tested fungal isolates (fumonisin B chemotype).
Zearalenone concentration and molecular detection of the toxigenicity in tested fungal isolates (zearalenone chemotype).
Trichothecene concentration and molecular detection of the toxigenicity in tested fungal isolates (trichothecene B chemotype).
GenBank accessions of obtained PCR products.